WO2024130158A1 - Lipid nanoparticles and polynucleotides encoding extended serum half-life interleukin-22 for the treatment of metabolic disease - Google Patents
Lipid nanoparticles and polynucleotides encoding extended serum half-life interleukin-22 for the treatment of metabolic diseaseDownload PDFInfo
- Publication number
- WO2024130158A1 WO2024130158A1PCT/US2023/084348US2023084348WWO2024130158A1WO 2024130158 A1WO2024130158 A1WO 2024130158A1US 2023084348 WUS2023084348 WUS 2023084348WWO 2024130158 A1WO2024130158 A1WO 2024130158A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- lipid
- mol
- alkyl
- polypeptide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers
- A61K9/1272—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers comprising non-phosphatidyl surfactants as bilayer-forming substances, e.g. cationic lipids or non-phosphatidyl liposomes coated or grafted with polymers
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/5123—Organic compounds, e.g. fats, sugars
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
- A61K48/0025—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid
Definitions
- IL-22Interleukin-22
- IL-22R1IL-22 receptor 1
- IL-10R ⁇IL-10 receptor ⁇
- IL-22promotes the expression of downstream innate immune mediators, mitogenic modulators, and antiapoptotic modulators that promote survival, proliferation, and regeneration.
- IL-22exhibits a short serum half-life, which may limit the use of IL-22 in clinical applications.
- lipid nanoparticlecomprising: (a) an ionizable lipid of Formula (I): or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein R’ branched is: wherein denotes a point of attachme a ⁇ a ⁇ a ⁇ a ⁇ nt; wherein R , R , R , and R are each independently selected from the group consisting of H, C 2-12 alkyl, and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 5-14 alkyl and C 5-14 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and , wherein denotes a point of attachment; wherein R 10 is N(R)2;
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attac a ⁇ a ⁇ a ⁇ a ⁇ hment; and
- Ris C 2-12 alkyl;
- R , R , and Rare each H;
- R 2 and R 3are each C 5-14 alkyl;
- R 4is 10 ;
- Ris NH(C 1-6 alkyl);
- n2is 2; each R 5 is H; each R 6 is H;
- M and M’are each -C(O)O-;
- R’is a C 1-12 alkyl; l is 5; and m is 7.
- R’ais R’branched; R’branched is denotes a ⁇ a ⁇ a ⁇ a ⁇ 2 a point of attachment; R , R , R , and R are each H; R and R 3 are each C 5-14 alkyl; R 4 is -(CH 2 ) n OH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C 1-12 alkyl; l is 5; and m is 7.
- R’ais R’branched; R’branched is denotes a point of attachment; R a ⁇ and R a ⁇ are each H; R a ⁇ is C 2-12 alkyl; R 2 and R 3 are each C5-14 alkyl; R 4 is -(CH 2 ) n OH; n is 2; each R 5 is H; each R 6 is H; M and M’ are each -C(O)O-; R’ is a C 1-12 alkyl; l is 5; and m is 7.
- the ionizable lipidis: , or N-oxides, salts, or isomers thereof.
- the lipid nanoparticlefurther comprises: a phospholipid; a structural lipid; and a PEG-lipid.
- the lipid nanoparticlecomprises: 40-50 mol% of the ionizable lipid, 30-45 mol% of the structural lipid, 5-15 mol% of the phospholipid, and 1-5 mol% of the PEG-lipid.
- the lipid nanoparticlecomprises: 45-50 mol.% of the ionizable amino lipid, 10-15 mol.% of the phospholipid, 35-40 mol.% of the structural lipid, and 1-3 mol.% of the PEG-modified lipid.
- the phospholipidcomprises comprises 1,2-distearoyl-sn-glycero- 3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2- dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3- phosphocholine (DPPC), 1,2-diundecanoyl-sn-glycero-phosphocholine (DUPC), 1-palmitoyl-2- oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3- phosphocholine
- the phospholipidcomprises DSPC.
- the structural lipidis selected from the group consisting of: cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, derivatives thereof, and mixtures thereof.
- the structural lipidcomprises cholesterol or a derivative thereof.
- the PEG-lipidis selected from: 1,2-dimyristoyl-sn-glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N- [amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG- dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG-DAG), PEG- dipalmitoyl phosphatidylethanolamine (PEG-DPPE), PEG-l,2-dimyristyloxlpropyl-3-amine (PEG-c-DMA), or combinations thereof.
- PEG-DMG1,2-dimyristoyl-sn-glycerol methoxypolyethylene glycol
- PEG-DSPE1,2-distearoyl-sn
- the PEG-lipidcomprises PEG- DMG.
- the mRNA moleculewhen administered as a single dose to a human subject, is sufficient to increase serum IL-22 levels in the human subject to a level at least 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20-fold, 30-fold, 40-fold, or at least 50-fold higher than the level observed prior to the administration, for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120 hours, or at least one week post-administration.
- the mRNA moleculewhen administered as a single dose to a human subject, is sufficient to: (i) increase serum IL-22 levels in the human subject by at least about 1.1-fold, at least about 1.2-fold, at least about 1.5-fold, at least about 2.0-fold, at least 5.0- fold, or at least 10-fold, compared to the human subject’s baseline serum IL-22 levels for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120 hours, or at least one week post-administration; or (ii) increase IL-22 activity in the human subject by at least about 1.5-fold, at least bout 2.0-fold, or at least about 2.5-fold, compared to the human subject’s baseline IL-22 activity for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120, or at least one week post-administration.
- an mRNA moleculecomprises an open reading frame (ORF) encoding an Interleukin-22 (IL-22) polypeptide. In some embodiments, an mRNA molecule comprises an open reading frame (ORF) encoding an IL-22 polypeptide-containing fusion protein. In some embodiments, IL-22 polypeptide encoded by the mRNA molecule has an amino acid sequence identical to any one of SEQ ID NO:1-4. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to any one of SEQ ID NO:28-38. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to any one of SEQ ID NO:28-33.
- the IL-22 polypeptide-containing fusion proteinhas an amino acid sequence identical to any one of SEQ ID NO:34-38. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25, SEQ ID NO:26, SEQ ID NO:27, SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38.
- the ORFis at least 65% identical to SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328.
- the mRNA moleculefurther comprises a 5’UTR having a nucleic acid sequence according to SEQ ID NO:50, SEQ ID NO:55, or SEQ ID NO:56. In some embodiments, the mRNA molecule further comprises a 3’UTR having a nucleic acid sequence according to SEQ ID NO:108, SEQ ID NO:125, or SEQ ID NO:139. In some embodiments, the mRNA molecule comprises a poly-A region, wherein the poly-A region about 100 nucleotides in length. In some embodiments, the mRNA molecule comprises at least one chemically modified nucleobase, sugar, backbone, or any combination thereof.
- the at least one chemically modified nucleobaseis selected from the group consisting of pseudouracil ( ⁇ ), N1-methylpseudouracil (m1 ⁇ ), 1-ethylpseudouracil, 2- thiouracil (s2U), 4’-thiouracil, 5-methylcytosine, 5-methyluracil, 5-methoxyuracil, and any combination thereof.
- the mRNA moleculefurther comprises a 5′ terminal cap.
- the 5′ terminal capcomprises a m7G-ppp-Gm-AG, Cap0, Cap1, ARCA, inosine, N1-methyl-guanosine, 2′-fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino- guanosine, LNA-guanosine, 2-azidoguanosine, Cap2, Cap4, 5′ methylG cap, or an analog thereof.
- the 5’ terminal capcomprises m7G-ppp-Gm-AG or Cap1.
- the present disclosurerelates to a pharmaceutical composition
- a pharmaceutical compositioncomprising: the lipid nanoparticle according to any of the above embodiments; and a pharmaceutically acceptable carrier.
- the pharmaceutical compositionis for use in treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject.
- the pharmaceutical compositionis for use in reducing body weight, reducing total cholesterol levels, reducing total HDL cholesterol levels, reducing total LDL cholesterol levels, reducing total triglyceride levels, reducing liver steatosis, or reducing blood glucose levels in a subject.
- the lipid nanoparticle, IL-22 polypeptide, or mRNA molecule according to any of the above embodimentsis for use in the manufacture of a medicament or pharmaceutical composition for use in treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject.
- the lipid nanoparticle, IL-22 polypeptide, or mRNA molecule according to any of the above embodimentsis for use in the manufacture of a medicament or pharmaceutical composition for use in reducing body weight, reducing total cholesterol levels, reducing total HDL cholesterol levels, reducing total LDL cholesterol levels, reducing total triglyceride levels, reducing liver steatosis, or reducing blood glucose levels in a subject.
- the present disclosurerelates to a method of treating or delaying the onset and/or progression of metabolic disease in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of increasing serum IL-22 levels in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of increasing IL-22 activity in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of increasing one or more of serum IL-22 levels or IL-22 activity in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of reducing body weight of a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of reducing total cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of reducing total HDL cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of reducing total LDL cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of reducing total triglyceride levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of reducing liver steatosis in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the present disclosurerelates to a method of reducing blood glucose levels in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments.
- the administrationis a single administration.
- the administrationis a repeated administration.
- the administrationis about once per day, about once per week, about once per two weeks, or about once per month.
- the pharmaceutical compositionis administered intravenously, intramuscularly, or subcutaneously.
- the pharmaceutical compositionis administered intravenously.
- the pharmaceutical compositionis administered subcutaneously.
- the pharmaceutical compositionis administered intramuscularly.
- the present disclosurerelates to an IL-22 polypeptide-containing fusion protein having an amino acid sequence identical to SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25, SEQ ID NO:26, SEQ ID NO:27, SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38.
- the amino acid sequenceis identical to SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38.
- the present disclosurerelates to a pharmaceutical composition comprising: the IL-22 polypeptide according to any of the above embodiments; and a pharmaceutically acceptable carrier.
- the present disclosurerelates to a cell comprising the IL-22 polypeptide according to any of the above embodiments.
- the present disclosurerelates to a codon-optimized mRNA molecule encoding the IL-22 polypeptide according to any of the above embodiments.
- the codon-optimized mRNA moleculehas a nucleic acid sequence having at least about 60% identity to the nucleic acid sequence of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328.
- the codon-optimized mRNA moleculehas a sequence having at least about 60% identity to SEQ ID NO:350, SEQ ID NO:351, SEQ ID NO:352, SEQ ID NO:353, SEQ ID NO:354, SEQ ID NO:355, SEQ ID NO:356, SEQ ID NO:357, SEQ ID NO:358, SEQ ID NO:359, SEQ ID NO:360, SEQ ID NO:361, SEQ ID NO:362, SEQ ID NO:363, SEQ ID NO:364, SEQ ID NO:365, SEQ ID NO:366, SEQ ID NO:367, or SEQ ID NO:368.
- FIG.1Ais a graph showing levels of N- or C-terminally MSA-linked IL-22 in conditioned media determined by ELISA 24 hours after the transfection of an extended serum half-life IL-22 mRNA construct into HeLa cells.
- FIG.1Dis a graph showing IL-22 protein levels in sera obtained from mice administered with construct #1. Protein levels were determined by ELISA.
- FIG.2Ais a schematic illustrating a dose time course used in experiments described herein. Briefly, Male C57BL/6 from Taconic mice were fed on the AMLN diet from 6 weeks of age (Week -18).
- FIG.2Bis a set of graphs showing body weights of mice throughout the treatment period.
- FIG.2Cis a set of graphs showing total serum cholesterol, LDL cholesterol, HDL cholesterol, and triglyceride levels determined at Week -1 and 72 hours after the administration of each dose, expressed as % change above baseline.
- FIG.3Bshows representative images of H&E-stained liver and epididymal adipose tissue samples obtained from mice in the various treatment conditions described above.
- FIG 3Cis a set of graphs quantifying the scoring of levels of microvesicular and macrovesicular steatosis, inflammation, and liver and adipose hypertrophy in images of H&E-stained liver and epididymal adipose tissue samples obtained from mice in the various treatment conditions described above.
- FIG 3Dis a set of graphs showing differential expression levels of a select set of fibrosis and lipogenesis associated genes were determined relative to Rpl19 expression using qRT-PCR in MSA and construct #1 treated and untreated AMLN diet-fed mice and matched chow diet-fed control mice. Significance levels (*p ⁇ 0.05; **p ⁇ 0.01; ***p ⁇ 0.001) are shown relative to MSA treated mice and were determined by one-way ANOVA for histology scoring and gene expression. One construct #1 treated mouse was excluded from analyses due to pathology indicating an infection.
- FIG.4Ais a graph showing serum IL-22 levels measured weekly 72 hours after the administration of LNP-encapsulated construct #1 mRNA for 6 weeks in C57BL/6Tac mice fed the AMLN diet.
- FIG.4Bis a graph showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored every week 72 hours following the administration of construct #1 or MSA mRNA at 0.5 mg/kg to mice fed with the AMLN diet.
- FIG.4Cshows representative images of PSR and F4/80 stained liver tissue at the end of the treatment period in construct #1 treated, MSA treated, and untreated mice fed the AMLN diet and untreated mice fed a chow diet.
- FIG.4Dis a set of graphs quantifying and scoring F4/80-positive macrophages and liver fibrosis level using microscopic evaluation.
- FIG.5shows a set of graphs showing the expression of genes involved in fibrosis and lipogenesis in AMLN or chow fed C57BL/6Tac mice treated with weekly administration of LNP-encapsulated mRNA construct #1 or MSA mRNA over 6 weeks.
- FIG.6shows the results of a differential gene expression analysis in liver tissue of construct #1 treated mice.
- FIG.7Bis a graph showing levels of IL-22 in sera from treated mice measured 72 hours after each administration.
- FIG.7Cis a graph showing mouse body weight monitored through Day 38 without additional treatment.
- FIG.7Dis a graph showing mouse food intake monitored through Day 38 without additional treatment.
- FIG.7Eis a set of graphs showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels were measured after sera collection.
- FIG.8Ais a schematic illustrating a dose time course used in experiments described herein.
- Male C57BL/6mice from Taconicwere fed on the AMLN or chow diet from 6 weeks of age (Week -18).
- FIG.8Bis a graph showing levels of IL-22 in sera from treated mice were measured 72 h after each administration, on days 3, 17, and 31.
- FIG.8Cis a graph showing mouse body weight monitored through Day 38 without additional treatment. Mice were administered MSA or construct #1 via intravenous (IV) or subcutaneous (SC) administration.
- FIG.8Dis a set of graphs showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels measured after sera collection. Mice were administered MSA or construct #1 via intravenous (IV) or subcutaneous (SC) administration.
- FIG.8Eshows representative images of H&E stained liver and adipose tissue samples.
- FIG.8Fis a set of graphs showing differential expression levels determined relative to Rpl19 expression using qRT-PCR for a select set of fibrosis and lipogenesis associated genes in treated mice and untreated AMLN diet and chow diet fed mice. Significance levels (*p ⁇ 0.05; **p ⁇ 0.01; ***p ⁇ 0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test.
- FIG.9Ais a schematic illustrating a dose time course used in experiments described herein. Male CETP/APOB mice from Taconic were fed on the AMLN diet from Day -42.
- FIG.9Bis a set of graphs showing mouse body weight monitored through Day 39.
- FIG.9Cis a set of graphs showing mouse total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored through Day 39.
- FIG.9Dis a panel of representative images of H&E stained liver and adipose tissue samples obtained from MSA and construct #1 treated mice.
- FIG.9Eis a set of graphs quantifying and grading for microvesicular and macrovesicular steatosis, inflammation, and adipose hypertrophy.
- FIG.9Fis a set of graphs showing differential expression levels determined relative to Rpl19 expression using qRT-PCR for a select set of fibrosis and lipogenesis associated genes in MSA and construct #1 treated mice. Significance levels (*p ⁇ 0.05; **p ⁇ 0.01; ***p ⁇ 0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test or by t-test for the histology score and the gene expression analyses.
- FIG.10Ais a graph showing IL-22 serum levels in mice after weekly administration of construct #1 or MSA mRNA (0.5 mg/kg) in CETP/APOB mice fed an AMLN diet.
- FIG.10Bis a graph showing IL-22 serum levels in mice after weekly administration of construct #1 or MSA mRNA (0.5 mg/kg) in CETP/APOB mice fed a chow diet.
- FIG.10Cis a panel of representative images of H&E stained liver tissue samples from AMLN fed MSA and construct #1 treated mice.
- FIG.10Dis a graph showing a quantification of F4/80 stained liver macrophages in AMLN fed MSA and construct #1 treated mice.
- FIG.10Eis a set of graphs showing differential expression levels determined using qRT-PCR for a select set of genes of interest in AMLN-fed CETP/APOB mice treated with MSA or construct #1. Expression is expressed as fold change relative to Rpl19.
- FIG.10Fis a graph showing fasting PYY levels determined using ELISA. Serum PYY analyses excluded 3 mice in the MSA group and 1 in the construct #1 group due to insufficient volume after fastig samples. Significance levels (*p ⁇ 0.05; ***p ⁇ 0.001) were determined by one-way ANOVA and Dunnett’s post-hoc test.
- FIG.11Ais a schematic illustrating a dose time course used in experiments described herein.
- FIG.11Bis a graph showing mouse body weight monitored through Day 39.
- FIG.11Cis a graph showing mouse total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored through Day 39. Significance levels (*p ⁇ 0.05; ***p ⁇ 0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test. Not significant, ns.
- FIG.12Ais a schematic illustrating a dose time course used in experiments described herein.
- FIG.12Bis a graph showing mouse body weight measured on Days 1, 3, 7, 8, and 9.
- FIG.12Cis a set of graphs showing differential weight gain calculated on Day 9, food intake measured as the total intake throughout the duration of the study, energy intake calculated as food intake multiplied by metabolizable energy content in the diet, metabolic efficiency calculated as mg of body weight gain over energy intake (kcal), fasting and refeeding glucose levels determined using Abbott Alphatrak 2 glucometers, and Peptide YY (PYY) levels determined using ELISA. Significance levels (*p ⁇ 0.05; ***p ⁇ 0.001; Not significant, ns.) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test.
- FIG.13Ais a schematic illustrating a dose time course used in experiments described herein.
- FIG.13Bis a graph showing blood glucose levels in AMLN-fed animals during the course of the GTT.
- FIG.13Cis a graph showing blood glucose levels in chow diet-fed animals during the course of the GTT.
- FIG.14Ais a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various mouse IL-22 mRNA constructs, as measured by ELISA.
- FIG.14Bis a graph showing the bioactivity of mouse IL-22 mRNA constructs as a function of concentration, as measured in conditioned medium samples from HEK-Blue IL- 22 cells treated with IL-22 construct protein. The IL-22 bioactivity in the conditioned medium samples was quantified by measuring the sample OD at 655 nm following incubation in QUANTI-Blue solution.
- FIG.15Ais a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various human IL-22 mRNA constructs, as measured by ELISA.
- FIG.15Bis a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various human IL-22 mRNA constructs, as measured by ELISA. The IL-22 concentration is reported as a concentration in nM.
- FIG.16is a graph showing the bioactivity of human IL-22 mRNA constructs as a function of concentration, as measured in conditioned medium samples from HEK-Blue IL- 22 cells treated with IL-22 construct protein. The IL-22 bioactivity in the conditioned medium samples was quantified by measuring the sample OD at 655 nm following incubation in QUANTI-Blue solution.
- FIG.17is a schematic illustrating a dose time course used in experiments described herein.
- Male C57BL/6Tac micewere fed an AMLN diet starting from 6 weeks of age through 40 weeks of age, for a total of 34 weeks of AMLN feeding.
- Body weight measurements and serum collectionoccurred at the indicated time points.
- FIG.18is a graph showing the % change in mouse bodyweight (BW) over time as a function of treatment condition.
- FIG.19Ais a graph showing the daily caloric intake (kcal) per mouse over time as a function of treatment condition.
- FIG.19Bis a graph showing the daily food intake (g) per mouse over time as a function of treatment condition.
- FIG.19Cis a graph showing the cumulative caloric intake (kcal) per mouse over time as a function of treatment condition.
- FIG.20Ais a graph showing the weight gain (g) per mouse as a function of treatment condition.
- FIG.20Bis a graph showing the metabolic efficiency (g weight gain / MJ caloric intake) per mouse as a function of treatment condition.
- FIG.21Ais a graph showing the liver weight (g) per mouse as a function of treatment condition.
- FIG.21Bis a graph showing the epididymal white adipose tissue (eWAT) weight (g) per mouse as a function of treatment condition.
- FIG.21Cis a graph showing the liver weight (% bodyweight) per mouse as a function of treatment condition.
- FIG.21Dis a graph showing the the epididymal white adipose tissue (eWAT) weight (% bodyweight) per mouse as a function of treatment condition.
- FIG.22Ais a graph showing the total serum cholesterol level (mg/dL) per mouse as a function of treatment condition.
- FIG.22Bis a graph showing the serum HDL cholesterol level (mg/dL) per mouse as a function of treatment condition.
- FIG.22Cis a graph showing the serum LDL cholesterol level (mg/dL) per mouse as a function of treatment condition.
- FIG.22Dis a graph showing the serum triglyceride level (mg/dL) per mouse as a function of treatment condition.
- FIG.22Eis a graph showing the serum alanine aminotransferase (ALT) level (U/L) per mouse as a function of treatment condition.
- ALTserum alanine aminotransferase
- U/Lserum alanine aminotransferase
- Interleukin-22has been identified as one such cytokine with therapeutic potential due to its biological impacts on multiple metabolic disease factors.
- IL-22a member of the IL-10 cytokine family, mediates cellular responses by binding to the IL-22 receptor 1 (IL-22R1), inducing conformational changes that enable binding of the IL-22/IL-22R1 complex by the IL-10 receptor ⁇ (IL-10R ⁇ ).
- IL-22promotes the expression of downstream innate immune mediators, mitogenic modulators, and antiapoptotic modulators that promote survival andproliferation.
- IL-22does not target immune-modulatory cells but instead targets epithelial cell populations such as hepatocytes and pancreatic cells.
- IL-22 targeting of hepatocyteshas been implicated in protecting against acute hepatitis, inflammation, and alcohol-induced liver damage, and has been observed to play a potential role in liver regeneration, as well as in decreasing weight gain and improving lipid balance.
- IL-22has shown promise for the treatment of liver associated metabolic diseases with high tolerability in vivo, its short serum half-life ( ⁇ 2 hours) may limit the use of IL-22 in clinical applications.
- manipulations that serve to increase the serum half-life of IL-22are attractive strategies for the use of IL-22 in clinical applications.
- the present disclosureaddresses and utilizes these strategies.
- nucleic acid-based therapeuticse.g., mRNA therapeutics
- mRNA therapeuticse.g., mRNA therapeutics
- TLRstoll-like receptors
- ssRNAsingle-stranded RNA
- RAG-Iretinoic acid-inducible gene I
- Immune recognition of foreign mRNAscan result in unwanted cytokine effects including interleukin- 1 ⁇ (IL-1 ⁇ ) production, tumor necrosis factor- ⁇ (TNF- ⁇ ) distribution and a strong type I interferon (type I IFN) response.
- IL-1 ⁇interleukin- 1 ⁇
- TNF- ⁇tumor necrosis factor- ⁇
- type I IFNtype I interferon
- This disclosurefeatures the incorporation of different modified nucleotides within therapeutic mRNAs to minimize the immune activation and optimize the translation efficiency of mRNA to protein.
- Particular aspectsfeature a combination of nucleotide modification to reduce the innate immune response and sequence optimization, in particular, within the open reading frame (ORF) of therapeutic mRNAs encoding IL-22 to enhance protein expression.
- ORFopen reading frame
- Certain embodiments of the mRNA therapeutic technology of the instant disclosurealso feature delivery of mRNA encoding IL-22 via a lipid nanoparticle (LNP) delivery system.
- mRNAencodes an IL-22 polypeptide with an enhanced serum half-life (i.e., an extended serum half-life IL-22).
- LNPsare an ideal platform for the safe and effective delivery of mRNAs to target cells. LNPs have the unique ability to deliver nucleic acids by a mechanism involving cellular uptake, intracellular transport and endosomal release or endosomal escape.
- the instant inventionfeatures ionizable amino lipid-based LNPs combined with mRNA encoding extended serum half-life IL-22 which have improved properties when administered in vivo.
- the ionizable amino lipid-based LNP Formulations of this disclosurehave improved properties, for example, cellular uptake, intracellular transport and/or endosomal release or endosomal escape.
- LNPs administered by systemic routee.g., intravenous (IV) administration
- IVintravenous
- ABSCaccelerated blood clearance
- mRNA engineering and/or efficient delivery by LNPscan result in increased levels and or enhanced duration of protein (e.g., IL-22) being expressed following a first dose of administration, which in turn, can lengthen the time between first dose and subsequent dosing.
- LNPscan be engineered to avoid immune sensing and/or recognition and can thus further avoid ABC upon subsequent or repeat dosing.
- An exemplary aspect of the disclosurefeatures LNPs which have been engineered to have reduced ABC.
- the lipid nanoparticle compositions described hereinmay be used for the delivery of therapeutic and/or prophylactic agents, e.g., mRNAs, to mammalian cells or organs.
- the lipid nanoparticles described hereinhave little or no immunogenicity.
- the lipid compounds disclosed hereinhave a lower immunogenicity as compared to a reference lipid (e.g., MC3, KC2, or DLinDMA).
- a formulation comprising a lipid disclosed herein and a therapeutic or prophylactic agent, e.g., mRNAhas an increased therapeutic index as compared to a corresponding formulation which comprises a reference lipid (e.g., MC3, KC2, or DLinDMA) and the same therapeutic or prophylactic agent.
- a reference lipide.g., MC3, KC2, or DLinDMA
- the present applicationprovides pharmaceutical compositions comprising: (a) a delivery agent comprising a lipid nanoparticle; and (b) a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide. a.
- Lipid NanoparticlesIn some embodiments, polynucleotides of the present disclosure (e.g., IL-22 mRNA, e.g., extended serum half-life IL-22 mRNA) are included in a lipid nanoparticle (LNP).
- Lipid nanoparticles according to the present disclosuremay comprise: (i) an ionizable lipid (e.g., an ionizable amino lipid); (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-modified lipid.
- lipid nanoparticles according to the present disclosurefurther comprise one or more polynucleotides of the present disclosure (e.g., IL-22 mRNA, e.g., extended serum half-life IL-22 mRNA).
- the lipid nanoparticles according to the present disclosurecan be generated using components, compositions, and methods as are generally known in the art, see for example PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406; PCT/US2016000129; PCT/US2016/014280; PCT/US2016/014280; PCT/US2017/038426; PCT/US2014/027077; PCT/US2014/055394; PCT/US2016/52117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575 and PCT/US2016/069491 all of which
- the lipid nanoparticlecomprises an ioniziable cationic lipid (e.g., an ionizable amino lipid) at a content of 20-60 mol.%, 25-60 mol.%, 30-60 mol.%, 35- 60 mol.%, 40-60 mol.%, 45-60 mol.%, 20-55 mol.%, 25-55 mol.%, 30-55 mol.%, 35-55 mol.%, 40-55 mol.%, 45-55 mol.%, 20-50 mol.%, 25-50 mol.%, 30-50 mol.%, 35-50 mol.%, or 40-50 mol.%.
- an ioniziable cationic lipide.g., an ionizable amino lipid
- the lipid nanoparticlemay comprise an ionizable cationic lipid (e.g., an ionizable amino lipid) at a content of 40-50 mol.%, 45-50 mol.%, 45-46 mol.%, 46- 47 mol.%, 47-48 mol.%, 48-49 mol.%, or 49-50 mol.%, for example about 45 mol.%, about 45.5 mol.%, about 46 mol.%, about 46.5 mol.%, about 47 mol.%, about 47.5 mol.%, about 48 mol.%, about 48.5 mol.%, about 49 mol.%, or about 49.5 mol.% ionizable cationic lipid (e.g., an ionizable amino lipid).
- an ionizable cationic lipide.g., an ionizable amino lipid
- the lipid nanoparticlecomprises a non-cationic helper lipid or phospholipid at a content of 5-25 mol.%.
- the lipid nanoparticlemay comprise a non-cationic helper lipid or phospholipid at a content of molar ratio of 5-25 mol.%, 5-20 mol.%, 5-15 mol.%, 10-25 mol.%, 10-20 mol.%, 10-15 mol.%, 5-6 mol.%, 6-7 mol.%, 7-8 mol.%, 8-9 mol.%, 9-10 mol.%, 10-11 mol.%, 11-12 mol.%, 12-13 mol.%, 13-14 mol.%, 14- 15 mol.%, 10-14 mol.%, 10-13 mol.%, 10-12 mol.%, 10-11 mol.%, 9-15 mol.%, 9-14 mol.%, 9-13 mol.%, 9-12 mol.%, or 9-11 mol.% non-cationic lipid.
- the lipid nanoparticlecomprises a sterol or other structural lipid at a content molar ratio of 25-55 mol.%, 25-50 mol.%, 25-45 mol.%, 25-40 mol.%, 25- 35 mol.%, 30-55 mol.%, 30-50 mol.%, 30-45 mol.%, 30-40 mol.%, 30-35 mol.%, 35-55 mol.%, 35-50 mol.%, 35-45 mol.%, 35-40 mol.%, 25-30 mol.%, 30-35 mol.%, 25-28 mol.%, 28-30 mol.%, 30-33 mol.%, 35-38 mol.%, 38-40 mol.%, 36-40 mol.%, 37-40 mol.%, 38-40 mol.%, 38-39 mol.%, 36-40 mol.%, 37-40 mol.%, 36-39 mol.%, 36-39 mol.%, or 37-39 mol.%.
- the lipid nanoparticlemay comprise a sterol or other structural lipid at a content of about 30 mol.%, about 30.5 mol.%, about 31.0 mol.%, about 31.5 mol.%, about 32.0 mol.%, about 32.5 mol.%, about 33.0 mol.%, about 33.5 mol.%, about 34.0 mol.%, about 34.5 mol.%, about 35.0 mol.%, about 35.5 mol.%, about 36.0 mol.%, about 36.5 mol.%, about 37.0 mol.%, about 37.5 mol.%, about 38.0 mol.%, about 38.5 mol.%, about 39.0 mol.%, about 39.5 mol.%, about 40.0 mol.%, about 40.5 mol.%, about 41.0 mol.%, about 41.5 mol.%, about 42.0 mol.%, about 42.5 mol.%, about 43.0 mol.%, about 43.5 mol.%, about 44.0
- the lipid nanoparticlecomprises a PEG-modified lipid at a content of 0.5-15 mol.%, 1.0-15 mol.%, 1.5-15 mol.%, 2.0-15 mol.%, 2.5-15 mol.%, 3.0-15 mol.%, 3.5-15 mol.%, 4.0-15 mol.%, 4.5-15 mol.%, 5.0-15 mol.%, 10-15 mol.%, 0.5-10 mol.%, 0.5-5 mol.%, 0.5-4.5 mol.%, 0.5-4.0 mol.%, 0.5-3.5 mol.%, 0.5-3.0 mol.%, 0.5-2.5 mol.%, 0.5-2.0 mol.%, 0.5-1.5 mol.%, 0.5-1.0 mol.%, 1.0-10 mol.%, 1.0-5 mol.%, 1.0-4.5 mol.%, 1.0-4.0 mol.%, 1.0-3.5 mol.%, 1.0-3.0 mol.%, 1.0-2.5 mol.%
- the lipid nanoparticlemay comprise a PEG-modified lipid at a content of a about 0.5 mol.%, about 1.0 mol.%, about 1.5 mol.%, about 2.0 mol.%, about 2.5 mol.%, about 3.0 mol.%, about 3.5 mol.%, about 4.0 mol.%, about 4.5 mol.%, about 5.0 mol.%, about 6.0 mol.%, about 7.0 mol.%, about 8.0 mol.%, about 9.0 mol.%, about 10.0 mol.%, or about 15.0 mol.%.
- the lipid nanoparticlecomprises: (i) 20 to 60 mol.% ionizable cationic lipid (e.g.
- the lipid nanoparticlecomprises: (i) 40 to 50 mol.% ionizable cationic lipid (e.g. ionizable amino lipid), (ii) 30 to 45 mol.% sterol or other structural lipid, (iii) 5 to 15 mol.% non-cationic lipid (e.g., phospholipid), and (iv) 1 to 5 mol.% PEG- modified lipid.
- the lipid nanoparticlecomprises: (i) 45 to 50 mol.% ionizable cationic lipid (e.g. ionizable amino lipid), (ii) 35 to 45 mol.% sterol or other structural lipid, (iii) 8 to 12 mol.% non-cationic lipid (e.g., phospholipid), and (iv) 1.5 to 3.5 mol.% PEG- modified lipid.
- ionizable cationic lipide.g. ionizable amino lipid
- 35 to 45 mol.% sterol or other structural lipide.g. 8 to 12 mol.% non-cationic lipid (e.g., phospholipid)
- iv1.5 to 3.5 mol.% PEG- modified lipid.
- “Compounds” numbered with an “I-” prefixe.g., “Compound I-1,” “Compound I-2,” “Compound I-3,” “Compound I-VI,” etc., indicate specific
- the lipid nanoparticle of the present disclosurecomprises an ionizable cationic lipid (e.g., an ionizable amino lipid) that is a compound of Formula (I): (I) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein R’ branched is: ; wherein denotes a point of attachment; wherein R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇ are each independently selected from the group consisting of H, C 2-12 alkyl, and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 5-14 alkyl and C 5-14 alkenyl; R 4 is selected from the group
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇are each H;
- R 2 and R 3are each C 5-14 alkyl;
- R 4is -(CH 2 ) n OH; n is 2;
- each R 5is H;
- each R 6is H;
- M and M’are each -C(O)O-;
- R’is a C 1-12 alkyl; l is 5; and m is 7.
- R’ ais R’ branched ;
- R’ branchedis denotes a point of att a ⁇ a ⁇ a ⁇ a ⁇ achment;
- R , R , R , and Rare each H;
- R 2 and R 3are each C 5-14 alkyl;
- R 4is -(CH 2 ) n OH; n is 2;
- each R 5is H;
- each R 6is H;
- M and M’are each -C(O)O-;
- R’is a C 1-12 alkyl; l is 3; and m is 7.
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment;
- R a ⁇is C 2-12 alkyl;
- R a ⁇ , R a ⁇ , and R a ⁇are each H;
- R 2 and R 3are each C 5-14 alkyl;
- R 4is ;
- R 10is NH(C 1-6 alkyl);
- n2is 2;
- R 5is H; each R 6 is H;
- M and M’are each -C(O)O-;
- R’is a C1-12 alkyl; l is 5; and m is 7.
- R’ ais R’ branched ;
- R’ branched i a ⁇ a ⁇ a ⁇denotes a point of attachment;
- R , R , and Rare each H;
- R a ⁇is C 2-12 alkyl;
- R 2 and R 3are each C 5-14 alkyl;
- R 4is -(CH 2 ) n OH; n is 2;
- each R 5is H; each R 6 is H;
- M and M’are each -C(O)O-;
- R’is a C 1-12 alkyl; l is 5; and m is 7.
- the compound of Formula (I)is selected from:
- the compound of Formula (I)is: (Compound I-1).
- the compound of Formula (I)is: (Compound I-2). In some embodiments, the compound of Formula (I) is: (Compound I-3).
- the disclosurerelates to a compound of Formula (Ia): (Ia) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein R’ branched is: ; wherein denotes a point of attachment; wherein R a ⁇ , R a ⁇ , and R a ⁇ are each independently selected from the group consisting of H, C 2-12 alkyl, and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 5-14 alkyl and C 5-14 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein denotes a point of attachment; wherein R 10 is N(R) 2
- the disclosurerelates to a compound of Formula (Ib): (Ib) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein R’ branched is: ; wherein denotes a point of attachment; wherein R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇ are each independently selected from the group consisting of H, C 2-12 alkyl, and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 5-14 alkyl and C 5-14 alkenyl; R 4 is -(CH 2 ) n OH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; each R 5 is independently selected from the group consisting of C 1-3 alkyl, C 2-3 alkenyl, and H; each R 6 is independently selected from the group consisting of C 1-3 alkyl, C 2-3 alkenyl, and H; M and M’
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment a ⁇ a ⁇ a ⁇ 2 ;
- R , R , and Rare each H;
- R and R 3are each C 5-14 alkyl;
- R 4is -(CH 2 ) n OH; n is 2;
- each R 5is H;
- each R 6is H;
- M and M’are each -C(O)O-;
- R’is a C 1-12 alkyl; l is 5; and m is 7.
- R’ ais R’ branched ;
- R’ branchedis denotes a point of attachment; a ⁇ a ⁇ a ⁇ R and R are each H; R is C 2-12 alkyl; R 2 and R 3 are each C 5-14 alkyl; R 4 is -(CH 2 ) n OH; n is 2; each R 5 is H; each R 6 is H; M and M’ are each -C(O)O-; R’ is a C 1-12 alkyl; l is 5; and m is 7.
- the disclosurerelates to a compound of Formula (Ic): (Ic) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched ; wherein R’ branched is: ; wherein denotes a point of attachment; wherein R a ⁇ , R a ⁇ , R a ⁇ , and R a ⁇ are each independently selected from the group consisting of H, C 2-12 alkyl, and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 5-14 alkyl and C 5-14 alkenyl; R 4 is , wherein denotes a point of attachment; wherein R 10 is N(R) 2 ; each R is independently selected from the group consisting of C 1- 6 alkyl, C 2-3 alkenyl, and H; n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R 5 is independently selected from the group consisting of C 1-3 al
- R’ ais R’ branched ; R’ branched is ; denotes a point of attachment; R a ⁇ , R a ⁇ , and R a ⁇ are each H; R a ⁇ is C 2-12 alkyl; R 2 and R 3 are each C5-14 alkyl; R 4 is deno 10 tes a point of attachment; R is NH(C 1-6 alkyl); n2 is 2; each R 5 is H; each R 6 is H; M and M’ are each -C(O)O-; R’ is a C 1-12 alkyl; l is 5; and m is 7.
- the compound of Formula (Ic)is: (Compound I-2).
- the disclosurerelates to a compound of Formula (II): (II) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein wherein denotes a point of attachment; R a ⁇ and R a ⁇ are each independently selected from the group consisting of H, C 1-12 alkyl, and C 2-12 alkenyl, wherein at least one of R a ⁇ and R a ⁇ is selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R b ⁇ and R b ⁇ are each independently selected from the group consisting of H, C 1-12 alkyl, and C 2-12 alkenyl, wherein at least one of R b ⁇ and R b ⁇ is selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 1-14 alkyl and C 2-14 alkenyl; R 4
- the disclosurerelates to a compound of Formula (II-a): (II-a) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein R’ branched is: an b d R’ is: wherein denotes a point of attachment; R a ⁇ and R a ⁇ are each independently selected from the group consisting of H, C 1-12 alkyl, and C 2-12 alkenyl, wherein at least one of R a ⁇ and R a ⁇ is selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R b ⁇ and R b ⁇ are each independently selected from the group consisting of H, C 1-12 alkyl, and C 2-12 alkenyl, wherein at least one of R b ⁇ and R b ⁇ is selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group
- the disclosurerelates to a compound of Formula (II-b): (II-b) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein R’ branched is: and R’ b is: wherein denotes a point of attachment; R a ⁇ and R b ⁇ are each independently selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 1-14 alkyl and C 2-14 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein denotes a point of attachment; wherein R 10 is N(R) 2 ; each R is independently selected from the group consisting of C 1- 6 alkyl, C 2-3 alkenyl, and H; and n2 is selected from the group
- the disclosurerelates to a compound of Formula (II-c): (II-c) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein R’ branched is: b and R’ is: wherein denotes a point of attachment; wherein R a ⁇ is selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 1-14 alkyl and C 2-14 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein denotes a point of attachment; wherein R 10 is N(R) 2 ; each R is independently selected from the group consisting of C 1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1,
- the disclosurerelates to a compound of Formula (II-d): (II-d) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein R’ branched is: b and R’ is: wherein denotes a point of attachment; wherein R a ⁇ and R b ⁇ are each independently selected from the group consisting of C 1- 12 alkyl and C 2-12 alkenyl; R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and , wherein denotes a point of attachment; wherein R 10 is N(R) 2 ; each R is independently selected from the group consisting of C 1- 6 alkyl, C 2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C 1-12 al
- the disclosurerelates to a compound of Formula (II-e): (II-e) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein R’ branched is: and R’ b is: ; wherein denotes a point of attachment; wherein R a ⁇ is selected from the group consisting of C 1-12 alkyl and C 2-12 alkenyl; R 2 and R 3 are each independently selected from the group consisting of C 1-14 alkyl and C 2-14 alkenyl; R 4 is -(CH 2 ) n OH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C 1-12 alkyl or C 2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9.
- m and lare each independently selected from 4, 5, and 6. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), m and l are each 5. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), each R’ independently is a C 1-12 alkyl.
- each R’independently is a C 2-5 alkyl.
- R’ bis: R and R 2 and R 3 are each independently a C 1-14 alkyl.
- R’ bis: and R 2 and R 3 are each independently a C 6-10 alkyl.
- R’ bis: R and R 2 and R 3 are each a C 8 alkyl.
- R’ branchedis: and R’ b is: a ⁇ 2 3 R is a C 1-12 alkyl and R and R are each independently a C 6-10 alkyl.
- R’ branchedis: b a ⁇ and R’ is: , R is a C 2-6 alkyl and R 2 and R 3 are each independently a C 6-10 alkyl.
- R’ branchedis: and R’ b is: , a ⁇ 2 3 R is a C 2-6 alkyl, and R and R are each a C 8 alkyl.
- R’ branchedis: a ⁇ b ⁇ , and R and R are each a C 1-12 alkyl.
- R’ branchedis: a ⁇ b ⁇ , and R and R are each a C 2-6 alkyl.
- m and lare each independently selected from 4, 5, and 6 and each R’ independently is a C 1-12 alkyl.
- m and lare each 5 and each R’ independently is a C 2-5 alkyl.
- R’ branchedis: , R’ b is: , m and l are each independently selected from 4, 5, and 6, each R’ independently is a C 1-12 alkyl, and R a ⁇ and R b ⁇ are each a C 1-12 alkyl.
- R’ branchedis: b R’ is: , m and l are each 5, each R’ independently is a C 2-5 alkyl, and R a ⁇ and R b ⁇ are each a C 2-6 alkyl.
- R’ branchedis: and b R’ is: , m and l are each independently selected from 4, 5, and 6, R’ is a C 1-12 alkyl, R a ⁇ is a C 1-12 alkyl and R 2 and R 3 are each independently a C 6-10 alkyl.
- R’ branchedis: b and R’ is: , m and l are each 5, R’ is a C 2-5 alkyl, R a ⁇ is a C 2-6 alkyl, and R 2 and R 3 are each a C 8 alkyl.
- R 4is wherein R 10 is NH(C 1-6 alkyl) and n2 is 2.
- R 4is , wherei 10 n R is NH(CH 3 ) and n2 is 2.
- R’ branchedis: , R’ b is: , m and l are each independently selected from 4, 5, and 6, each R’ independently is a C 1-12 alkyl, R a ⁇ and R b ⁇ are each a C 1-12 alkyl, and R 4 is , wherein R 10 is NH(C 1-6 alkyl), and n2 is 2.
- R’ branchedis: , R’ b is: , m and l are each 5, each R’ independently is a C 2 alkyl, R a ⁇ and R b ⁇ are each a C alkyl, and R 4 is R - 5 2-6 wherein R 10 is NH(CH 3 ) and n2 is 2.
- R’ branchedis: and R’ b is: , m and l are each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, R 2 and R 3 are each independently a C6-10 alkyl, R a ⁇ is a C 1-12 alkyl, and R 4 is , wherein R 10 is NH(C 1-6 alkyl) and n2 is 2.
- R a ⁇ R’ branchedis: and R’ b is: , m and l are each 5, R’ is a C 2-5 alkyl, R a ⁇ is a C 2-6 alkyl, R 2 and R 3 are each a C 8 alkyl, and R 4 is , wherein R 10 is NH(CH 3 ) and n2 is 2.
- R 4is -(CH 2 ) n OH and n is 2, 3, or 4. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R 4 is -(CH 2 ) n OH and n is 2.
- R’ branchedis: b , R’ is: , m and l are each independently selected from 4, 5, and 6, each R’ independently is a C 1-12 alkyl, R a ⁇ and R b ⁇ are each a C 1-12 alkyl, R 4 is -(CH 2 ) n OH, and n is 2, 3, or 4.
- R’ branchedis: , R’ b is: , m and l are each 5, each R’ independently is a ⁇ a C2-5 alkyl, R and R b ⁇ are each a C 2-6 alkyl, R 4 is -(CH 2 ) n OH, and n is 2.
- the disclosurerelates to a compound of Formula (II-f): (II-f) or its N-oxide, or a salt or isomer thereof, wherein R’ a is R’ branched or R’ cyclic ; wherein R a ⁇ R’ branched is: a b nd R’ is: wherein denotes a point of attachment; R a ⁇ is a C 1-12 alkyl; R 2 and R 3 are each independently a C 1-14 alkyl; R 4 is -(CH 2 ) n OH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C 1-12 alkyl; m is selected from 4, 5, and 6; and l is selected from 4, 5, and 6.
- m and lare each 5, and n is 2, 3, or 4.
- R’is a C 2-5 alkyl, R a ⁇ is a C 2- 6 alkyl, and R 2 and R 3 are each a C 6-10 alkyl.
- m and lare each 5, n is 2, 3, or 4, R’ is a C 2-5 alkyl, R a ⁇ is a C 2-6 alkyl, and R 2 and R 3 are each a C 6-10 alkyl.
- the disclosurerelates to a compound of Formula (II-g): 4 (II-g), wherein R a ⁇ is a C 2-6 alkyl; R’ is a C 2-5 alkyl; and R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting of 3, 4, and 5, and wherein denotes a point of attachment, R 10 is NH(C 1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3.
- the disclosurerelates to a compound of Formula (II-h): (II-h), wherein R a ⁇ and R b ⁇ are each independently a C 2-6 alkyl; each R’ independently is a C 2-5 alkyl; and R 4 is selected from the group consisting of -(CH 2 ) n OH wherein n is selected from the group consisting of 3, 4, and 5, and wherein 1 denotes a point of attachment, R 0 is NH(C 1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3.
- R 4is , wherein R 10 is NH(CH 3 ) and n2 is 2.
- R 4is -(CH 2 ) 2 OH.
- the disclosurerelates to a compound having the Formula (III): or a salt or isomer thereof, wherein R 1 , R 2 , R 3 , R 4 , and R 5 are independently selected from the group consisting of C 5-20 alkyl, C 5-20 alkenyl, -R”MR’, -R*YR”, -YR”, and -R*OR”; each M is independently selected from the group consisting of -C(O)O-, -OC(O)-, -OC(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S) -, -CH(OH)-, -P(O)(OR’)O-,
- R 1 , R 2 , R 3 , R 4 , and R 5are each C 5-20 alkyl; X 1 is -CH 2 -; and X 2 and X 3 are each -C(O)-.
- the compound of Formula (III)is: (Compound I-VI), or a salt or isomer thereof.
- PhospholipidsThe lipid composition of the lipid nanoparticle composition disclosed herein can comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof. In general, phospholipids comprise a phospholipid moiety and one or more fatty acid moieties.
- a phospholipid moietycan be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin.
- a fatty acid moietycan be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid.
- Particular phospholipidscan facilitate fusion to a membrane.
- a cationic phospholipidcan interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue.
- elementse.g., a therapeutic agent
- a lipid-containing compositione.g., LNPs
- Non-natural phospholipid speciesincluding natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated.
- a phospholipidcan be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond).
- alkynese.g., an alkenyl group in which one or more double bonds is replaced with a triple bond.
- an alkyne groupcan undergo a copper- catalyzed cycloaddition upon exposure to an azide.
- Such reactionscan be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye).
- Phospholipidsinclude, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin.
- a phospholipid of the present disclosurecomprises 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2- dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-diundecanoyl-sn- glycero-phosphocholine (DUPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3-phosphochocholine (PO
- a phospholipid useful or potentially useful in the present disclosureis an analog or variant of DSPC.
- a phospholipid useful or potentially useful in the present disclosureis a compound of Formula (IV): (IV), or a salt thereof, wherein: each R 1 is independently optionally substituted alkyl; or optionally two R 1 are joined together with the intervening atoms to form optionally substituted monocyclic carbocyclyl or optionally substituted monocyclic heterocyclyl; or optionally three R 1 are joined together with the intervening atoms to form optionally substituted bicyclic carbocyclyl or optionally substitute bicyclic heterocyclyl; n is 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; A is of the Formula: each instance of L 2 is independently a bond or optionally substituted C 1-6 alkylene, wherein one methylene unit of the optionally substituted C 1-6 alkylene is optionally replaced with O, N(R N)
- the phospholipidsmay be one or more of the phospholipids described in U.S. Application No.62/520,530.
- a phospholipid useful or potentially useful in the present disclosurecomprises a modified phospholipid head (e.g., a modified choline group).
- a phospholipid with a modified headis DSPC, or analog thereof, with a modified quaternary amine.
- at least one of R 1is not methyl. In certain embodiments, at least one of R 1 is not hydrogen or methyl.
- the compound of Formula (IV)is of one of the following Formulae: , , , or a salt thereof, wherein: each t is independently 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; each u is independently 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; and each v is independently 1, 2, or 3.
- a compound of Formula (IV)is of Formula (IV-a): (IV-a), or a salt thereof.
- a phospholipid useful or potentially useful in the present disclosurecomprises a cyclic moiety in place of the glyceride moiety.
- a phospholipid useful in the present disclosureis DSPC, or analog thereof, with a cyclic moiety in place of the glyceride moiety.
- the compound of Formula (IV)is of Formula (IV-b): , (IV-b), or a salt thereof.
- a phospholipid useful or potentially useful in the present disclosurecomprises a modified tail.
- a phospholipid useful or potentially useful in the present disclosureis DSPC, or analog thereof, with a modified tail.
- a “modified tail”may be a tail with shorter or longer aliphatic chains, aliphatic chains with branching introduced, aliphatic chains with substituents introduced, aliphatic chains wherein one or more methylenes are replaced by cyclic or heteroatom groups, or any combination thereof.
- a phospholipid useful or potentially useful in the present disclosurecomprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful or potentially useful in the present disclosure is a compound of Formula (IV), wherein n is 1, 3, 4, 5, 6, 7, 8, 9, or 10.
- a compound of Formula (IV)is of one of the following Formulae: , , or a salt thereof.
- a phospholipid useful or potentially useful in the present disclosurecomprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful.
- an alternative lipidis used in place of a phospholipid of the present disclosure.
- an alternative lipid of the present disclosureis oleic acid.
- the alternative lipidis one of the following: , , , ,
- the lipid composition of a pharmaceutical composition disclosed hereincan comprise one or more structural lipids.
- structural lipidrefers to sterols and also to lipids containing sterol moieties. Incorporation of structural lipids in the lipid nanoparticle may help mitigate aggregation of other lipids in the particle.
- Structural lipidscan be selected from the group including but not limited to, cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, and mixtures thereof.
- the structural lipidis a sterol.
- sterolsare a subgroup of steroids consisting of steroid alcohols.
- the structural lipidis a steroid.
- the structural lipidis cholesterol.
- the structural lipidis an analog of cholesterol.
- the structural lipidis alpha-tocopherol.
- the structural lipidsmay be one or more of the structural lipids described in U.S. Application No.62/520,530. e. Polyethylene Glycol (PEG)-Lipids
- the lipid composition of a pharmaceutical composition disclosed hereincan comprise one or more a polyethylene glycol (PEG) lipids.
- PEG-lipidrefers to polyethylene glycol (PEG)-modified lipids.
- PEG-lipidsinclude PEG-modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG- CerC14 or PEG-CerC20), PEG-modified dialkylamines and PEG-modified 1,2- diacyloxypropan-3-amines.
- PEGylated lipidsPEGylated lipids.
- a PEG lipidcan be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
- the PEG-lipidincludes, but not limited to 1,2-dimyristoyl-sn- glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn-glycero-3- phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG- DAG), PEG-dipalmitoyl phosphatidylethanolamine (PEG-DPPE), or PEG-l,2- dimyristyloxlpropyl-3-amine (PEG-c-DMA).
- PEG-DMG1,2-dimyristoyl-sn- glycerol methoxypolyethylene glycol
- PEG-DSPE1,2-distearoyl-s
- the PEG-lipidis selected from the group consisting of a PEG- modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
- the lipid moiety of the PEG-lipidsincludes those having lengths of from about C 14 to about C 22 , preferably from about C 14 to about C 16 .
- a PEG moietyfor example an mPEG-NH 2 , has a size of about 1000, 2000, 5000, 10,000, 15,000 or 20,000 daltons.
- the PEG-lipidis PEG 2k - DMG.
- the lipid nanoparticles described hereincan comprise a PEG lipid which is a non-diffusible PEG.
- Non-limiting examples of non-diffusible PEGsinclude PEG-DSG and PEG-DSPE.
- PEG-lipidsare known in the art, such as those described in U.S. Patent No.8,158,601 and International Publ. No. WO 2015/130584 A2, which are incorporated herein by reference in their entirety.
- lipid component of a lipid nanoparticle compositionmay include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. Such species may be alternately referred to as PEGylated lipids.
- a PEG lipidis a lipid modified with polyethylene glycol.
- a PEG lipidmay be selected from the non-limiting group including PEG-modified phosphatidylethanolamines, PEG-modified phosphatidic acids, PEG-modified ceramides, PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, and mixtures thereof.
- a PEG lipidmay be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
- the PEG-modified lipidsare a modified form of PEG DMG.
- PEG-DMGhas the following structure:
- PEG lipids useful in the present disclosurecan be PEGylated lipids described in International Publication No. WO2012099755, the contents of which is herein incorporated by reference in its entirety. Any of these exemplary PEG lipids described herein may be modified to comprise a hydroxyl group on the PEG chain.
- the PEG lipidis a PEG-OH lipid.
- a “PEG-OH lipid”(also referred to herein as “hydroxy-PEGylated lipid”) is a PEGylated lipid having one or more hydroxyl (–OH) groups on the lipid.
- the PEG-OH lipidincludes one or more hydroxyl groups on the PEG chain.
- a PEG-OH or hydroxy-PEGylated lipidcomprises an –OH group at the terminus of the PEG chain.
- a PEG lipid useful in the present disclosureis a compound of Formula (V).
- R 3is –OR O ;
- R Ois hydrogen, optionally substituted alkyl, or an oxygen protecting group;
- ris an integer between 1 and 100, inclusive;
- L 1is optionally substituted C 1-10 alkylene, wherein at least one methylene of the optionally substituted C 1-10 alkylene is independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, O, N(R N ), S, C(O), C(O)N(R N ), NR N C(O), C(O)O, - OC(O), OC(O)O, OC(O)N(R N ), NR N C(O)O, or NR N C(O)N(R N );
- Dis a moiety obtained by click chemistry or a moiety cleavable under physiological conditions;
- mis 0, 1, 2, 3, 4, 5, 6, 7, 8,
- the compound of Fomula (V)is a PEG-OH lipid (i.e., R 3 is – OR O , and R O is hydrogen).
- the compound of Formula (V)is of Formula (V-OH): (V-OH), or a salt thereof.
- a PEG lipid useful in the present disclosureis a PEGylated fatty acid.
- a PEG lipid useful in the present disclosureis a compound of Formula (VI).
- R 3is–OR O ;
- R Ois hydrogen, optionally substituted alkyl or an oxygen protecting group;
- ris an integer between 1 and 100, inclusive;
- the compound of Formula (VI)is of Formula (VI-OH): (VI-OH), or a salt thereof.
- ris 45.
- the compound of Formula (VI)is: . or a salt thereof.
- ris 40-50.
- the compound of Formula (VI)is (Compound P-I).
- the lipid composition of the pharmaceutical compositions disclosed hereindoes not comprise a PEG-lipid.
- the PEG-lipidsmay be one or more of the PEG lipids described in U.S. Application No.62/520,530.
- a PEG lipid of the present disclosurecomprises a PEG- modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
- the PEG-modified lipidis PEG-DMG, PEG-c-DOMG (also referred to as PEG-DOMG), PEG-DSG and/or PEG-DPG.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of any of Formula I, II or III, a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising PEG-DMG. In some embodiments, a LNP of the present disclosure comprises an ionizable cationic lipid of any of Formula I, II or III, a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of Formula I, II or III, a phospholipid comprising a compound having Formula IV, a structural lipid, and the PEG lipid comprising a compound having Formula V or VI.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of Formula I, II or III, a phospholipid comprising a compound having Formula IV, a structural lipid, and the PEG lipid comprising a compound having Formula V or VI.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of Formula I, II or III, a phospholipid having Formula IV, a structural lipid, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of , and a PEG lipid comprising Formula VI.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of , and an alternative lipid comprising oleic acid.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of , an alternative lipid comprising oleic acid, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of a phospholipid comprising DOPE, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the present disclosurecomprises an ionizable cationic lipid of , a phospholipid comprising DOPE, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the present disclosurecomprises an N:P ratio of from about 2:1 to about 30:1. In some embodiments, a LNP of the present disclosure comprises an N:P ratio of about 6:1. In some embodiments, a LNP of the present disclosure comprises an N:P ratio of about 3:1. In some embodiments, a LNP of the present disclosure comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of from about 10:1 to about 100:1. In some embodiments, a LNP of the present disclosure comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of about 20:1.
- a LNP of the present disclosurecomprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of about 10:1. In some embodiments, a LNP of the present disclosure has a mean diameter from about 50nm to about 150nm. In some embodiments, a LNP of the present disclosure has a mean diameter from about 70nm to about 120nm.
- alkylAs used herein, the term “alkyl”, “alkyl group”, or “alkylene” means a linear or branched, saturated hydrocarbon including one or more carbon atoms (e.g., one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms), which is optionally substituted.
- C 1-14 alkylmeans an optionally substituted linear or branched, saturated hydrocarbon including 1-14 carbon atoms. Unless otherwise specified, an alkyl group described herein refers to both unsubstituted and substituted alkyl groups.
- alkenylmeans a linear or branched hydrocarbon including two or more carbon atoms (e.g., two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms) and at least one double bond, which is optionally substituted.
- C 2 - 14 alkenylmeans an optionally substituted linear or branched hydrocarbon including 2-14 carbon atoms and at least one carbon-carbon double bond.
- An alkenyl groupmay include one, two, three, four, or more carbon-carbon double bonds.
- C 18 alkenylmay include one or more double bonds.
- a C 18 alkenyl group including two double bondsmay be a linoleyl group.
- an alkenyl group described hereinrefers to both unsubstituted and substituted alkenyl groups.
- alkynylmeans a linear or branched hydrocarbon including two or more carbon atoms (e.g., two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms) and at least one carbon-carbon triple bond, which is optionally substituted.
- C 2-14 alkynylmeans an optionally substituted linear or branched hydrocarbon including 2-14 carbon atoms and at least one carbon-carbon triple bond.
- An alkynyl groupmay include one, two, three, four, or more carbon-carbon triple bonds.
- C 18 alkynylmay include one or more carbon-carbon triple bonds.
- an alkynyl group described hereinrefers to both unsubstituted and substituted alkynyl groups.
- the term "carbocycle” or “carbocyclic group”means an optionally substituted mono- or multi-cyclic system including one or more rings of carbon atoms. Rings may be three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, or twenty membered rings.
- the notation "C 3-6 carbocycle”means a carbocycle including a single ring having 3-6 carbon atoms.
- Carbocyclesmay include one or more carbon-carbon double or triple bonds and may be non- aromatic or aromatic (e.g., cycloalkyl or aryl groups). Examples of carbocycles include cyclopropyl, cyclopentyl, cyclohexyl, phenyl, naphthyl, and 1,2 dihydronaphthyl groups.
- cycloalkylas used herein means a non-aromatic carbocycle and may or may not include any double or triple bond.
- carbocycles described hereinrefers to both unsubstituted and substituted carbocycle groups, i.e., optionally substituted carbocycles.
- heterocycleor “heterocyclic group” means an optionally substituted mono- or multi-cyclic system including one or more rings, where at least one ring includes at least one heteroatom.
- Heteroatomsmay be, for example, nitrogen, oxygen, or sulfur atoms. Rings may be three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, or fourteen membered rings.
- Heterocyclesmay include one or more double or triple bonds and may be non-aromatic or aromatic (e.g., heterocycloalkyl or heteroaryl groups).
- heterocyclesinclude imidazolyl, imidazolidinyl, oxazolyl, oxazolidinyl, thiazolyl, thiazolidinyl, pyrazolidinyl, pyrazolyl, isoxazolidinyl, isoxazolyl, isothiazolidinyl, isothiazolyl, morpholinyl, pyrrolyl, pyrrolidinyl, furyl, tetrahydrofuryl, thiophenyl, pyridinyl, piperidinyl, quinolyl, and isoquinolyl groups.
- heterocycloalkylas used herein means a non-aromatic heterocycle and may or may not include any double or triple bond. Unless otherwise specified, heterocycles described herein refers to both unsubstituted and substituted heterocycle groups, i.e., optionally substituted heterocycles.
- heteroalkylrefers respectively to an alkyl, alkenyl, alkynyl group, as defined herein, which further comprises one or more (e.g., 1, 2, 3, or 4) heteroatoms (e.g., oxygen, sulfur, nitrogen, boron, silicon, phosphorus) wherein the one or more heteroatoms is inserted between adjacent carbon atoms within the parent carbon chain and/or one or more heteroatoms is inserted between a carbon atom and the parent molecule, i.e., between the point of attachment.
- heteroatomse.g., oxygen, sulfur, nitrogen, boron, silicon, phosphorus
- heteroalkyls, heteroalkenyls, or heteroalkynyls described hereinrefers to both unsubstituted and substituted heteroalkyls, heteroalkenyls, or heteroalkynyls, i.e., optionally substituted heteroalkyls, heteroalkenyls, or heteroalkynyls.
- a "biodegradable group”is a group that may facilitate faster metabolism of a lipid in a mammalian entity.
- a biodegradable groupmay be selected from the group consisting of, but is not limited to, -C(O)O-, -OC(O)-, -C(O)N(R')-, -N(R')C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR')O-, -S(O)2-, an aryl group, and a heteroaryl group.
- an "aryl group”is an optionally substituted carbocyclic group including one or more aromatic rings. Examples of aryl groups include phenyl and naphthyl groups.
- heteroaryl groupis an optionally substituted heterocyclic group including one or more aromatic rings.
- heteroaryl groupsinclude pyrrolyl, furyl, thiophenyl, imidazolyl, oxazolyl, and thiazolyl. Both aryl and heteroaryl groups may be optionally substituted.
- M and M'can be selected from the non-limiting group consisting of optionally substituted phenyl, oxazole, and thiazole. In the Formulas herein, M and M' can be independently selected from the list of biodegradable groups above.
- aryl or heteroaryl groups described hereinrefers to both unsubstituted and substituted groups, i.e., optionally substituted aryl or heteroaryl groups.
- Alkyl, alkenyl, and cyclyl (e.g., carbocyclyl and heterocyclyl) groupsmay be optionally substituted unless otherwise specified.
- Ris an alkyl or alkenyl group, as defined herein.
- the substituent groupsthemselves may be further substituted with, for example, one, two, three, four, five, or six substituents as defined herein.
- a C 1-6 alkyl groupmay be further substituted with one, two, three, four, five, or six substituents as described herein.
- Compounds of the disclosure that contain nitrogenscan be converted to N-oxides by treatment with an oxidizing agent (e.g., 3-chloroperoxybenzoic acid (mCPBA) and/or hydrogen peroxides) to afford other compounds of the disclosure.
- an oxidizing agente.g., 3-chloroperoxybenzoic acid (mCPBA) and/or hydrogen peroxides
- N-hydroxy compoundscan be prepared by oxidation of the parent amine by an oxidizing agent such as m CPBA.
- lipid composition of a pharmaceutical composition disclosed hereincan include one or more components in addition to those described above.
- the lipid compositioncan include one or more permeability enhancer molecules, carbohydrates, polymers, surface altering agents (e.g., surfactants), or other components.
- a permeability enhancer moleculecan be a molecule described by U.S. Patent Application Publication No.2005/0222064.
- Carbohydratescan include simple sugars (e.g., glucose) and polysaccharides (e.g., glycogen and derivatives and analogs thereof).
- a polymercan be included in and/or used to encapsulate or partially encapsulate a pharmaceutical composition disclosed herein (e.g., a pharmaceutical composition in lipid nanoparticle form).
- a polymercan be biodegradable and/or biocompatible.
- a polymercan be selected from, but is not limited to, polyamines, polyethers, polyamides, polyesters, polycarbamates, polyureas, polycarbonates, polystyrenes, polyimides, polysulfones, polyurethanes, polyacetylenes, polyethylenes, polyethyleneimines, polyisocyanates, polyacrylates, polymethacrylates, polyacrylonitriles, and polyarylates.
- the ratio between the lipid composition and the polynucleotide rangecan be from about 10:1 to about 60:1 (wt/wt).
- the ratio between the lipid composition and the polynucleotidecan be about 10:1, 11:1, 12:1, 13:1, 14:1, 15:1, 16:1, 17:1, 18:1, 19:1, 20:1, 21:1, 22:1, 23:1, 24:1, 25:1, 26:1, 27:1, 28:1, 29:1, 30:1, 31:1, 32:1, 33:1, 34:1, 35:1, 36:1, 37:1, 38:1, 39:1, 40:1, 41:1, 42:1, 43:1, 44:1, 45:1, 46:1, 47:1, 48:1, 49:1, 50:1, 51:1, 52:1, 53:1, 54:1, 55:1, 56:1, 57:1, 58:1, 59:1 or 60:1 (wt/wt).
- the wt/wt ratio of the lipid composition to the polynucleotide encoding a therapeutic agentis about 20:1 or about 15:1.
- the pharmaceutical composition disclosed hereincan contain more than one polynucleotides.
- a pharmaceutical composition disclosed hereincan contain two or more polynucleotides (e.g., RNA, e.g., mRNA).
- the lipid nanoparticles described hereincan comprise polynucleotides (e.g., mRNA) in a lipid:polynucleotide weight ratio of 5:1, 10:1, 15:1, 20:1, 25:1, 30:1, 35:1, 40:1, 45:1, 50:1, 55:1, 60:1 or 70:1, or a range or any of these ratios such as, but not limited to, 5:1 to about 10:1, from about 5:1 to about 15:1, from about 5:1 to about 20:1, from about 5:1 to about 25:1, from about 5:1 to about 30:1, from about 5:1 to about 35:1, from about 5:1 to about 40:1, from about 5:1 to about 45:1, from about 5:1 to about 50:1, from about 5:1 to about 55:1, from about 5:1 to about 60:1, from about 5:1 to about 70:1, from about 10:1 to about 15:1, from about 10:1 to about 20:1, from about 10:1 to about 25
- the lipid nanoparticles described hereincan comprise the polynucleotide in a concentration from approximately 0.1 mg/ml to 2 mg/ml such as, but not limited to, 0.1 mg/ml, 0.2 mg/ml, 0.3 mg/ml, 0.4 mg/ml, 0.5 mg/ml, 0.6 mg/ml, 0.7 mg/ml, 0.8 mg/ml, 0.9 mg/ml, 1.0 mg/ml, 1.1 mg/ml, 1.2 mg/ml, 1.3 mg/ml, 1.4 mg/ml, 1.5 mg/ml, 1.6 mg/ml, 1.7 mg/ml, 1.8 mg/ml, 1.9 mg/ml, 2.0 mg/ml or greater than 2.0 mg/ml.
- the pharmaceutical compositions disclosed hereinare Formulated as lipid nanoparticles (LNPs). Accordingly, the present disclosure also provides nanoparticle compositions comprising (i) a lipid composition comprising a delivery agent such as compound as described herein, and (ii) a polynucleotide encoding an IL-22 polypeptide. In such nanoparticle composition, the lipid composition disclosed herein can encapsulate the polynucleotide encoding an IL-22 polypeptide. Nanoparticle compositions are typically sized on the order of micrometers or smaller and can include a lipid bilayer.
- Nanoparticle compositionsencompass lipid nanoparticles (LNPs), liposomes (e.g., lipid vesicles), and lipoplexes.
- a nanoparticle compositioncan be a liposome having a lipid bilayer with a diameter of 500 nm or less.
- Nanoparticle compositionsinclude, for example, lipid nanoparticles (LNPs), liposomes, and lipoplexes.
- nanoparticle compositionsare vesicles including one or more lipid bilayers.
- a nanoparticle compositionincludes two or more concentric bilayers separated by aqueous compartments. Lipid bilayers can be functionalized and/or crosslinked to one another.
- Lipid bilayerscan include one or more ligands, proteins, or channels.
- a lipid nanoparticlecomprises an ionizable amino lipid, a structural lipid, a phospholipid, and mRNA.
- the LNPcomprises an ionizable amino lipid, a PEG-modified lipid, a sterol and a structural lipid.
- the LNPhas a molar ratio of about 40-50% ionizable amino lipid; about 5-15% structural lipid; about 30-45% sterol; and about 1-5% PEG-modified lipid.
- the lipid nanoparticlecomprises 47-49 mol.% ionizable cationic lipid (e.g.
- ionizable amino lipide.g., Compound I-1, Compound I-2, or Compound I-3
- 10-12 mol.% non-cationic lipide.g., phospholipid, e.g., DSPC
- 38-40 mol.% sterole.g., cholesterol
- 1-3 mol.% PEG-modified lipide.g., PEG- DMG or Compound P-I
- the lipid nanoparticle (“LNP-1”)may comprise the following components at the following molar ratios: (i) 45-50 mol.% Compound I-1 (ii) 35-45 mol.% sterol (e.g., cholesterol); (iii) 8-12 mol.% phospholipid (e.g., DSPC or DOPE); and (iv) 1.5-3.5 mol.% PEG-lipid (e.g., Compound P-I or PEG-DMG).
- the lipid nanoparticle (“LNP-1A”)may comprise the following components at the following molar ratios: (i) 45-50 mol.% Compound I-1 (ii) 35-45 mol.% Cholesterol; (iii) 8-12 mol.% DSPC; and (iv) 1.5-3.5 mol.% PEG-DMG.
- the lipid nanoparticle (“LNP-1B”)may comprise the following components at the following molar ratios: (i) 45-50 mol.% Compound I-1 (ii) 35-45 mol.% Cholesterol; (iii) 8-12 mol.% DSPC; and (iv) 1.5-3.5 mol.% Compound P-I.
- the lipid nanoparticle (“LNP-2”)may comprise the following: (i) 45-50 mol.% Compound I-2; (ii) 35-45 mol.% sterol (e.g., Cholesterol); (iii) 8-12 mol.% phospholipid (e.g., DSPC or DOPE); and (iv) 1.5-3.5 mol.% PEG-lipid (e.g., Compound P-I or PEG-DMG).
- sterole.g., Cholesterol
- 8-12 mol.% phospholipide.g., DSPC or DOPE
- PEG-lipide.g., Compound P-I or PEG-DMG
- the lipid nanoparticle (“LNP-2A”)may comprise the following: (i) 45-50 mol.% Compound I-2; (ii) 35-45 mol.% Cholesterol; (iii) 8-12 mol.% DSPC; and (iv) 1.5-3.5 mol.% PEG-DMG.
- the lipid nanoparticle (“LNP-2B”)may comprise the following components at the following molar ratios: (i) 45-50 mol.% Compound I-2; (ii) 35-45 mol.% Cholesterol; (iii) 8-12 mol.% DSPC; and (iv) 1.5-3.5 mol.% Compound P-I.
- the lipid nanoparticle (“LNP-3”)may comprise the following: (i) 45-50 mol.% Compound I-3; (ii) 35-45 mol.% sterol (e.g., Cholesterol); (iii) 8-12 mol.% phospholipid (e.g., DSPC or DOPE); and (iv) 1.5-3.5 mol.% PEG-lipid (e.g., Compound P-I or PEG-DMG).
- sterole.g., Cholesterol
- 8-12 mol.% phospholipide.g., DSPC or DOPE
- PEG-lipide.g., Compound P-I or PEG-DMG
- the lipid nanoparticle (“LNP-3A”)may comprise the following: (i) 45-50 mol.% Compound I-3; (ii) 35-45 mol.% Cholesterol; (iii) 8-12 mol.% DSPC; and (iv) 1.5-3.5 mol.% PEG-DMG.
- the lipid nanoparticle (“LNP-3B”)may comprise the following: (i) 45-50 mol.% Compound I-3; (ii) 35-45 mol.% Cholesterol; (iii) 8-12 mol.% DSPC; and (iv) 1.5-3.5 mol.% Compound P-I.
- the LNPhas a polydispersity value of less than 0.4.
- the LNPhas a net neutral charge at a neutral pH. In some embodiments, the LNP has a mean diameter of 50-150 nm. In some embodiments, the LNP has a mean diameter of 80-100 nm.
- the term “lipid”refers to a small molecule that has hydrophobic or amphiphilic properties. Lipids may be naturally occurring or synthetic. Examples of classes of lipids include, but are not limited to, fats, waxes, sterol-containing metabolites, vitamins, fatty acids, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, and polyketides, and prenol lipids.
- a lipid nanoparticlemay comprise an ionizable amino lipid.
- ionizable amino lipidhas its ordinary meaning in the art and may refer to a lipid comprising one or more charged moieties.
- an ionizable amino lipidmay be positively charged or negatively charged.
- An ionizable amino lipidmay be positively charged, in which case it can be referred to as “cationic lipid”.
- an ionizable amino lipid moleculemay comprise an amine group, and can be referred to as an ionizable amino lipid.
- a “charged moiety”is a chemical moiety that carries a formal electronic charge, e.g., monovalent (+1, or -1), divalent (+2, or -2), trivalent (+3, or -3), etc.
- the charged moietymay be anionic (i.e., negatively charged) or cationic (i.e., positively charged).
- positively-charged moietiesinclude amine groups (e.g., primary, secondary, and/or tertiary amines), ammonium groups, pyridinium group, guanidine groups, and imidizolium groups.
- the charged moietiescomprise amine groups.
- negatively- charged groups or precursors thereofinclude carboxylate groups, sulfonate groups, sulfate groups, phosphonate groups, phosphate groups, hydroxyl groups, and the like.
- the charge of the charged moietymay vary, in some cases, with the environmental conditions, for example, changes in pH may alter the charge of the moiety, and/or cause the moiety to become charged or uncharged. In general, the charge density of the molecule may be selected as desired. It should be understood that the terms “charged” or “charged moiety” does not refer to a “partial negative charge” or “partial positive charge” on a molecule.
- partial negative chargeand “partial positive charge” are given its ordinary meaning in the art.
- a “partial negative charge”may result when a functional group comprises a bond that becomes polarized such that electron density is pulled toward one atom of the bond, creating a partial negative charge on the atom.
- the ionizable amino lipidis sometimes referred to in the art as an “ionizable cationic lipid”.
- the ionizable amino lipidmay have a positively charged hydrophilic head and a hydrophobic tail that are connected via a linker structure.
- an ionizable amino lipidmay also be a lipid including a cyclic amine group.
- the ionizable amino lipidmay be selected from, but not limited to, an ionizable amino lipid described in International Publication Nos. WO2013086354 and WO2013116126; the contents of each of which are herein incorporated by reference in their entirety.
- the ionizable amino lipidmay be selected from, but not limited to, Formula CLI-CLXXXXII of US Patent No.7,404,969; each of which is herein incorporated by reference in their entirety.
- the lipidmay be a cleavable lipid such as those described in International Publication No.
- Nanoparticle compositionscan be characterized by a variety of methods. For example, microscopy (e.g., transmission electron microscopy or scanning electron microscopy) can be used to examine the morphology and size distribution of a nanoparticle composition. Dynamic light scattering or potentiometry (e.g., potentiometric titrations) can be used to measure zeta potentials. Dynamic light scattering can also be utilized to determine particle sizes.
- microscopye.g., transmission electron microscopy or scanning electron microscopy
- Dynamic light scattering or potentiometrye.g., potentiometric titrations
- Dynamic light scatteringcan also be utilized to determine particle sizes.
- Nanoparticle compositionssuch as the Zetasizer Nano ZS (Malvern Instruments Ltd, Malvern, Worcestershire, UK) can also be used to measure multiple characteristics of a nanoparticle composition, such as particle size, polydispersity index, and zeta potential.
- the size of the nanoparticlescan help counter biological reactions such as, but not limited to, inflammation, or can increase the biological effect of the polynucleotide.
- size or “mean size” in the context of nanoparticle compositionsrefers to the mean diameter of a nanoparticle composition.
- a nanoparticle compositioncan be relatively homogenous.
- a polydispersity indexcan be used to indicate the homogeneity of a nanoparticle composition, e.g., the particle size distribution of the nanoparticle composition.
- a small (e.g., less than 0.3) polydispersity indexgenerally indicates a narrow particle size distribution.
- a nanoparticle compositioncan have a polydispersity index from about 0 to about 0.25, such as 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, or 0.25.
- the polydispersity index of a nanoparticle composition disclosed hereincan be from about 0.10 to about 0.20.
- the zeta potential of a nanoparticle compositioncan be used to indicate the electrokinetic potential of the composition.
- the zeta potentialcan describe the surface charge of a nanoparticle composition. Nanoparticle compositions with relatively low charges, positive or negative, are generally desirable, as more highly charged species can interact undesirably with cells, tissues, and other elements in the body.
- the zeta potential of a nanoparticle composition disclosed hereincan be from about -10 mV to about +20 mV, from about -10 mV to about +15 mV, from about 10 mV to about +10 mV, from about -10 mV to about +5 mV, from about -10 mV to about 0 mV, from about -10 mV to about -5 mV, from about -5 mV to about +20 mV, from about -5 mV to about +15 mV, from about -5 mV to about +10 mV, from about -5 mV to about +5 mV, from about -5 mV to about 0 mV, from about 0 mV to about +20 mV, from about 0 mV to about +15 mV, from about 0 mV to about +10 mV, from about 0 mV to about +5 mV, from about 0 mV to about +20
- the zeta potential of the lipid nanoparticlescan be from about 0 mV to about 100 mV, from about 0 mV to about 90 mV, from about 0 mV to about 80 mV, from about 0 mV to about 70 mV, from about 0 mV to about 60 mV, from about 0 mV to about 50 mV, from about 0 mV to about 40 mV, from about 0 mV to about 30 mV, from about 0 mV to about 20 mV, from about 0 mV to about 10 mV, from about 10 mV to about 100 mV, from about 10 mV to about 90 mV, from about 10 mV to about 80 mV, from about 10 mV to about 70 mV, from about 10 mV to about 60 mV, from about 10 mV to about 50 mV, from about 10 mV to about 40 mV, from about 10
- the zeta potential of the lipid nanoparticlescan be from about 10 mV to about 50 mV, from about 15 mV to about 45 mV, from about 20 mV to about 40 mV, and from about 25 mV to about 35 mV. In some embodiments, the zeta potential of the lipid nanoparticles can be about 10 mV, about 20 mV, about 30 mV, about 40 mV, about 50 mV, about 60 mV, about 70 mV, about 80 mV, about 90 mV, and about 100 mV.
- encapsulation efficiency of a polynucleotidedescribes the amount of the polynucleotide that is encapsulated by or otherwise associated with a nanoparticle composition after preparation, relative to the initial amount provided.
- encapsulationcan refer to complete, substantial, or partial enclosure, confinement, surrounding, or encasement. Encapsulation efficiency is desirably high (e.g., close to 100%). The encapsulation efficiency can be measured, for example, by comparing the amount of the polynucleotide in a solution containing the nanoparticle composition before and after breaking up the nanoparticle composition with one or more organic solvents or detergents. Fluorescence can be used to measure the amount of free polynucleotide in a solution.
- the encapsulation efficiency of a polynucleotidecan be at least 50%, for example 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100%. In some embodiments, the encapsulation efficiency can be at least 80%. In certain embodiments, the encapsulation efficiency can be at least 90%.
- the amount of a polynucleotide present in a pharmaceutical composition disclosed hereincan depend on multiple factors such as the size of the polynucleotide, desired target and/or application, or other properties of the nanoparticle composition as well as on the properties of the polynucleotide.
- the amount of an mRNA useful in a nanoparticle compositioncan depend on the size (expressed as length, or molecular mass), sequence, and other characteristics of the mRNA.
- the relative amounts of a polynucleotide in a nanoparticle compositioncan also vary.
- the relative amounts of the lipid composition and the polynucleotide present in a lipid nanoparticle composition of the present disclosurecan be optimized according to considerations of efficacy and tolerability.
- the N:P ratiocan serve as a useful metric.
- the N:P ratio of a nanoparticle compositioncontrols both expression and tolerability, nanoparticle compositions with low N:P ratios and strong expression are desirable.
- N:P ratiosvary according to the ratio of lipids to RNA in a nanoparticle composition. In general, a lower N:P ratio is preferred.
- the one or more RNA, lipids, and amounts thereofcan be selected to provide an N:P ratio from about 2:1 to about 30:1, such as 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 12:1, 14:1, 16:1, 18:1, 20:1, 22:1, 24:1, 26:1, 28:1, or 30:1.
- the N:P ratiocan be from about 2:1 to about 8:1.
- the N:P ratiois from about 5:1 to about 8:1.
- the N:P ratiois between 5:1 and 6:1.
- the N:P ratiois about is about 5.67:1.
- the present disclosurealso provides methods of producing lipid nanoparticles comprising encapsulating a polynucleotide.
- Such methodcomprises using any of the pharmaceutical compositions disclosed herein and producing lipid nanoparticles in accordance with methods of production of lipid nanoparticles known in the art. See, e.g., Wang et al. (2015) “Delivery of oligonucleotides with lipid nanoparticles” Adv. Drug Deliv. Rev.87:68-80; Silva et al. (2015) “Delivery Systems for Biopharmaceuticals. Part I: Nanoparticles and Microparticles” Curr. Pharm. Technol.16: 940-954; Naseri et al.
- the LNP formulations described hereincan additionally comprise a permeability enhancer molecule.
- Non-limiting permeability enhancer moleculesare described in U.S. Pub. No. US20050222064, herein incorporated by reference in its entirety.
- the LNP formulationscan comprise a carbohydrate carrier.
- the carbohydrate carriercan include, but is not limited to, an anhydride-modified phytoglycogen or glycogen-type material, phytoglycogen octenyl succinate, phytoglycogen beta-dextrin, anhydride-modified phytoglycogen beta-dextrin (e.g., Intl. Pub. No. WO2012109121, herein incorporated by reference in its entirety).
- the LNP formulationscan be coated with a surfactant or polymer to improve the delivery of the particle.
- the LNPcan be coated with a hydrophilic coating such as, but not limited to, PEG coatings and/or coatings that have a neutral surface charge as described in U.S. Pub. No.
- the LNP formulationscan be engineered to alter the surface properties of particles so that the lipid nanoparticles can penetrate the mucosal barrier as described in U.S. Pat. No. 8,241,670 or Intl. Pub. No. WO2013110028, each of which is herein incorporated by reference in its entirety.
- the LNP engineered to penetrate mucuscan comprise a polymeric material (i.e., a polymeric core) and/or a polymer-vitamin conjugate and/or a tri-block co-polymer.
- the polymeric materialcan include, but is not limited to, polyamines, polyethers, polyamides, polyesters, polycarbamates, polyureas, polycarbonates, poly(styrenes), polyimides, polysulfones, polyurethanes, polyacetylenes, polyethylenes, polyethyeneimines, polyisocyanates, polyacrylates, polymethacrylates, polyacrylonitriles, and polyarylates.
- LNP engineered to penetrate mucuscan also include surface altering agents such as, but not limited to, polynucleotides, anionic proteins (e.g., bovine serum albumin), surfactants (e.g., cationic surfactants such as for example dimethyldioctadecyl-ammonium bromide), sugars or sugar derivatives (e.g., cyclodextrin), nucleic acids, polymers (e.g., heparin, polyethylene glycol and poloxamer), mucolytic agents (e.g., N-acetylcysteine, mugwort, bromelain, papain, clerodendrum, acetylcysteine, bromhexine, carbocisteine, eprazinone, mesna, ambroxol, sobrerol, domiodol, letosteine, stepronin, tiopronin, gelsolin, thymosin ⁇ 4 do
- the mucus penetrating LNPcan be a hypotonic formulation comprising a mucosal penetration enhancing coating.
- the formulationcan be hypotonic for the epithelium to which it is being delivered.
- hypotonic formulationscan be found in, e.g., Intl. Pub. No. WO2013110028, herein incorporated by reference in its entirety.
- the polynucleotide described hereinis Formulated as a lipoplex, such as, without limitation, the ATUPLEXTM system, the DACC system, the DBTC system and other siRNA-lipoplex technology from Silence Therapeutics (London, United Kingdom), STEMFECTTM from STEMGENT® (Cambridge, MA), and polyethylenimine (PEI) or protamine-based targeted and non-targeted delivery of nucleic acids (Aleku et al. Cancer Res.200868:9788-9798; Strumberg et al.
- a lipoplexsuch as, without limitation, the ATUPLEXTM system, the DACC system, the DBTC system and other siRNA-lipoplex technology from Silence Therapeutics (London, United Kingdom), STEMFECTTM from STEMGENT® (Cambridge, MA), and polyethylenimine (PEI) or protamine-based targeted and non-targeted delivery of nucleic acids (Aleku et al. Cancer Res.200868:9788
- the polynucleotides described hereinare Formulated as a solid lipid nanoparticle (SLN), which can be spherical with an average diameter between 10 to 1000 nm.
- SLNpossess a solid lipid core matrix that can solubilize lipophilic molecules and can be stabilized with surfactants and/or emulsifiers.
- Exemplary SLNcan be those as described in Intl. Pub. No. WO2013105101, herein incorporated by reference in its entirety.
- the polynucleotides described hereincan be Formulated for controlled release and/or targeted delivery.
- controlled releaserefers to a pharmaceutical composition or compound release profile that conforms to a particular pattern of release to effect a therapeutic outcome.
- the polynucleotidescan be encapsulated into a delivery agent described herein and/or known in the art for controlled release and/or targeted delivery.
- the term “encapsulate”means to enclose, surround or encase.
- encapsulationcan be substantial, complete or partial.
- substantially encapsulatedmeans that at least greater than 50, 60, 70, 80, 85, 90, 95, 96, 97, 98, 99, or greater than 99% of the pharmaceutical composition or compound of the present disclosure can be enclosed, surrounded or encased within the delivery agent.
- Partial encapsulationor “partially encapsulate” means that less than 10, 10, 20, 30, 4050 or less of the pharmaceutical composition or compound of the present disclosure can be enclosed, surrounded or encased within the delivery agent.
- encapsulationcan be determined by measuring the escape or the activity of the pharmaceutical composition or compound of the present disclosure using fluorescence and/or electron micrograph. For example, at least 1, 5, 10, 20, 30, 40, 50, 60, 70, 80, 85, 90, 95, 96, 97, 98, 99, 99.9, or greater than 99% of the pharmaceutical composition or compound of the present disclosure are encapsulated in the delivery agent.
- the polynucleotides described hereincan be encapsulated in a therapeutic nanoparticle, referred to herein as "therapeutic nanoparticle polynucleotides.”
- Therapeutic nanoparticlescan be Formulated by methods described in, e.g., Intl. Pub. Nos.
- the therapeutic nanoparticle polynucleotidecan be Formulated for sustained release.
- sustained releaserefers to a pharmaceutical composition or compound that conforms to a release rate over a specific period of time.
- the period of timecan include, but is not limited to, hours, days, weeks, months and years.
- the sustained release nanoparticle of the polynucleotides described hereincan be Formulated as disclosed in Intl. Pub. No. WO2010075072 and U.S. Pub. Nos. US20100216804, US20110217377, US20120201859 and US20130150295, each of which is herein incorporated by reference in their entirety.
- the therapeutic nanoparticle polynucleotidecan be Formulated to be target specific, such as those described in Intl. Pub.
- LNPscan be prepared using microfluidic mixers or micromixers.
- Exemplary microfluidic mixerscan include, but are not limited to, a slit interdigital micromixer including, but not limited to those manufactured by Microinnova (Allerheiligen bei Wildon, Austria) and/or a staggered herringbone micromixer (SHM) (see Zhigaltsevet al., "Bottom-up design and synthesis of limit size lipid nanoparticle systems with aqueous and triglyceride cores using millisecond microfluidic mixing," Langmuir 28:3633-40 (2012); Belliveau et al., “Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA," Molecular Therapy-Nucleic Acids.1:e37 (2012); Chen et al., “Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation," J.
- SHMherringbone micromixer
- micromixersinclude Slit Interdigital Microstructured Mixer (SIMM-V2) or a Standard Slit Interdigital Micro Mixer (SSIMM) or Caterpillar (CPMM) or Impinging-jet (IJMM,) from the Institut für Mikrotechnik Mainz GmbH, Mainz Germany.
- methods of making LNP using SHMfurther comprise mixing at least two input streams wherein mixing occurs by microstructure-induced chaotic advection (MICA).
- MICAmicrostructure-induced chaotic advection
- This methodcan also comprise a surface for fluid mixing wherein the surface changes orientations during fluid cycling.
- Methods of generating LNPs using SHMinclude those disclosed in U.S. Pub. Nos. US20040262223 and US20120276209, each of which is incorporated herein by reference in their entirety.
- the polynucleotides described hereincan be Formulated in lipid nanoparticles using microfluidic technology (see Whitesides, George M., "The Origins and the Future of Microfluidics," Nature 442: 368-373 (2006); and Abraham et al., "Chaotic Mixer for Microchannels," Science 295: 647-651 (2002); each of which is herein incorporated by reference in its entirety).
- the polynucleotidescan be Formulated in lipid nanoparticles using a micromixer chip such as, but not limited to, those from Harvard Apparatus (Holliston, MA) or Dolomite Microfluidics (Royston, UK).
- a micromixer chipcan be used for rapid mixing of two or more fluid streams with a split and recombine mechanism.
- the polynucleotides described hereincan be Formulated in lipid nanoparticles having a diameter from about 1 nm to about 100 nm such as, but not limited to, about 1 nm to about 20 nm, from about 1 nm to about 30 nm, from about 1 nm to about 40 nm, from about 1 nm to about 50 nm, from about 1 nm to about 60 nm, from about 1 nm to about 70 nm, from about 1 nm to about 80 nm, from about 1 nm to about 90 nm, from about 5 nm to about from 100 nm, from about 5 nm to about 10 nm, about 5 nm to about 20 nm, from about 5 nm to about 30 nm, from about 5 nm to about 40 nm, from about 5 nm to about 50 nm, from about 5 nm to about 60 nm, from about 5 nm to about 70
- the lipid nanoparticlescan have a diameter from about 10 to 500 nm. In some embodiments, the lipid nanoparticle can have a diameter greater than 100 nm, greater than 150 nm, greater than 200 nm, greater than 250 nm, greater than 300 nm, greater than 350 nm, greater than 400 nm, greater than 450 nm, greater than 500 nm, greater than 550 nm, greater than 600 nm, greater than 650 nm, greater than 700 nm, greater than 750 nm, greater than 800 nm, greater than 850 nm, greater than 900 nm, greater than 950 nm or greater than 1000 nm.
- the polynucleotidescan be delivered using smaller LNPs.
- Such particlescan comprise a diameter from below 0.1 ⁇ m up to 100 nm such as, but not limited to, less than 0.1 ⁇ m, less than 1.0 ⁇ m, less than 5 ⁇ m, less than 10 ⁇ m, less than 15 um, less than 20 um, less than 25 um, less than 30 um, less than 35 um, less than 40 um, less than 50 um, less than 55 um, less than 60 um, less than 65 um, less than 70 um, less than 75 um, less than 80 um, less than 85 um, less than 90 um, less than 95 um, less than 100 um, less than 125 um, less than 150 um, less than 175 um, less than 200 um, less than 225 um, less than 250 um, less than 275 um, less than 300 um, less than 325 um, less than 350 um, less than 375 um, less than 400 um, less than 425 um, less than 450 um, less than 475 um, less than 500 um, less than 0.1
- the nanoparticles and microparticles described hereincan be geometrically engineered to modulate macrophage and/or the immune response.
- the geometrically engineered particlescan have varied shapes, sizes and/or surface charges to incorporate the polynucleotides described herein for targeted delivery such as, but not limited to, pulmonary delivery (see, e.g., Intl. Pub. No. WO2013082111, herein incorporated by reference in its entirety).
- Other physical features the geometrically engineering particlescan include, but are not limited to, fenestrations, angled arms, asymmetry and surface roughness, charge that can alter the interactions with cells and tissues.
- the nanoparticles described hereinare stealth nanoparticles or target-specific stealth nanoparticles such as, but not limited to, those described in U.S. Pub. No. US20130172406, herein incorporated by reference in its entirety.
- the stealth or target-specific stealth nanoparticlescan comprise a polymeric matrix, which can comprise two or more polymers such as, but not limited to, polyethylenes, polycarbonates, polyanhydrides, polyhydroxyacids, polypropylfumerates, polycaprolactones, polyamides, polyacetals, polyethers, polyesters, poly(orthoesters), polycyanoacrylates, polyvinyl alcohols, polyurethanes, polyphosphazenes, polyacrylates, polymethacrylates, polycyanoacrylates, polyureas, polystyrenes, polyamines, polyesters, polyanhydrides, polyethers, polyurethanes, polymethacrylates, polyacrylates, polycyanoacrylates, or combinations thereof.
- polymerssuch as, but not limited to, polyethylenes, polycarbonates, polyanhydrides, polyhydroxyacids, polypropylfumerates, polycaprolactones, polyamides, polyacetals, polyethers, polyester
- ionizable lipidsare susceptible to the formation of lipid-polynucleotide adducts.
- ionizable lipids that comprise a tertiary amine groupmay decompose into one or both of a secondary amine and a reactive aldehyde species capable of interacting with polynucleotides (such as mRNA) to form an ionizable lipid-polynucleotide adduct impurity that can be detected by reverse phase ion pair chromatography (RP-IP HPLC).
- RP-IP HPLCreverse phase ion pair chromatography
- the ionizable lipid-polynucleotide adduct impurityis an aldehyde-mRNA adduct impurity. It also has been determined that such adducts may disrupt mRNA translation and impact the activity of lipid nanoparticle (LNP) formulated mRNA products.
- LNPlipid nanoparticle
- an LNP compositionwherein less than about 10%, less than about 5%, or less than about 1%, of the mRNA is in the form of ionizable lipid-polynucleotide adduct impurity, including less than 10%, less than 5%, or less than 1%, as may be measured by RP-IP HPLC.
- an amount of lipid aldehydes in the compositionis less than about 50 ppm, including less than 50 ppm.
- an amount of N-oxide compounds in the compositionis less than about 50 ppm, including less than 50 ppm.
- an amount of transition metals, such as Fe, in the compositionis less than about 50 ppm, including less than 50 ppm. Additionally or alternatively, in some aspects an amount of alkyl halide compounds in the composition is less than about 50 ppm, including less than 50 ppm. Additionally or alternatively, in some aspects an amount of anhydride compounds in the composition is less than about 50 ppm, including less than 50 ppm. Additionally or alternatively, in some aspects an amount of ketone compounds in the composition is less than about 50 ppm, including less than 50 ppm. Additionally or alternatively, in some aspects an amount of conjugated diene compounds in the composition is less than about 50 ppm, including less than 50 ppm.
- the compositionis stable against the formation of ionizable lipid- polynucleotide adduct impurity.
- an amount of ionizable lipid-polynucleotide adduct impurity in the compositionincreases at an average rate of less than about 2% per day when stored at a temperature of about 25 °C or below, including at an average rate of less than 2% per day.
- an amount of ionizable lipid-polynucleotide adduct impurity in the compositionincreases at an average rate of less than about 0.5% per day when stored at a temperature of about 5 °C or below, including at an average rate of less than 0.5% per day.
- an amount of ionizable lipid-polynucleotide adduct impurity in the compositionincreases at an average rate of less than about 0.5% per day when stored at a refrigerated temperature, optionally wherein the refrigerated temperature is about 5 °C.
- Lipid vehicle (e.g., LNP) compositions with a reduced content of ionizable lipid- polynucleotide adduct impuritycan be prepared by methods that inhibit formation of one or both of N-oxides and aldehydes.
- Such methodsmay comprise treating a composition comprising an ionizable lipid comprising a tertiary amine group to inhibit formation of one or both of N-oxides and aldehydes, such as by treating the composition with a reducing agent; treating the composition with a chelating agent; adjusting the pH of the composition; adjusting the temperature of the composition; and adjusting the buffer in the composition.
- Such methodsmay comprise, prior to combining the ionizable lipid with a polynucleotide, one or more of treating the ionizable lipid with a scavenging agent; treating the ionizable lipid with a reductive treatment agent; treating the ionizable lipid with a reducing agent; treating the ionizable lipid with a chelating agent; treating the polynucleotide with a reducing agent; and treating the polynucleotide with a chelating agent.
- the scavenging agent, reductive treatment agent, and/or reducing agentmay be an agent that reacts with aldehyde, ketone, anhydride and/or diene compounds.
- a scavenging agentmay comprise one or more selected from (O- (2,3,4,5,6-Pentafluorobenzyl)hydroxylamine hydrochloride) (PFBHA), methoxyamine (e.g., methoxyamine hydrochloride), benzyloxyamine (e.g., benzyloxyamine hydrochloride), ethoxyamine (e.g., ethoxyamine hydrochloride), 4-[2-(aminooxy)ethyl]morpholine dihydrochloride, butoxyamine (e.g., tert-butoxyamine hydrochloride), 4- Dimethylaminopyridine (DMAP), 1,4-diazabicyclo[2.2.2]octane (DABCO), Triethylamine
- DMAP1,4-d
- a reductive treatment agentmay comprise a boron compound (e.g., sodium borohydride and/or bis(pinacolato)diboron).
- a reductive treatment agentmay comprise a boron compound, such as one or both of sodium borohydride and bis(pinacolato)diboron).
- a chelating agentmay comprise immobilized iminodiacetic acid.
- a reducing agentmay comprise an immobilized reducing agent, such as immobilized diphenylphosphine on silica (Si-DPP), immobilized thiol on agarose (Ag-Thiol), immobilized cysteine on silica (Si-Cysteine), immobilized thiol on silica (Si-Thiol), or a combination thereof.
- an immobilized reducing agentsuch as immobilized diphenylphosphine on silica (Si-DPP), immobilized thiol on agarose (Ag-Thiol), immobilized cysteine on silica (Si-Cysteine), immobilized thiol on silica (Si-Thiol), or a combination thereof.
- a buffermay be selected from sodium phosphate, sodium citrate, sodium succinate, histidine, histidine-HCl, sodium malate, sodium carbonate, and TRIS (tris(hydroxymethyl)aminomethane).
- a buffermay be TRIS and may be, or adjusted to be, from about 20 mM to about 150 mM TRIS.
- the temperature of the compositionmay be, or adjusted to be, 25 0C or less.
- the compositionmay also comprise a free reducing agent or antioxidant. 2.
- Interleukin-22IL-22
- IL-22R1IL-22 receptor 1
- IL-10R ⁇IL-10 receptor ⁇
- ERKextracellular signal-regulated kinases
- JNKc-Jun N-terminal kinase
- MAPKmitogen-activated protein kinase
- IL-22is an ⁇ -helical cytokine.
- IL-22binds to a heterodimeric cell surface receptor composed of IL-10R2 and IL-22R1 subunits.
- IL-22Ris expressed on tissue cells, and it is absent on immune cells.
- IL-22is encoded by the IL22 gene in humans.
- IL22is found on chromosome 12 at 12q15.
- IL-22is produced by several populations of immune cells at a site of inflammation. Producers are ⁇ T cells classes Th1, Th22 and Th17 along with ⁇ T cells, NKT, ILC3, neutrophils and macrophages.
- IL-22takes effect on non-hematopoietic cells – mainly stromal and epithelial cells.
- IL-22thus participates in both wound healing and in protection against microbes.
- IL-22 dysregulationtakes part in pathogenesis of several autoimmune diseases, such as systemic lupus erythematosus, rheumatoid arthritis, and psoriasis.
- IL-22 biological activityis initiated by binding to a cell-surface complex composed of IL-22R1 and IL-10R2 receptor chains and further regulated by interactions with a soluble binding protein, IL-22BP, which shares sequence similarity with an extracellular region of IL-22R1 (sIL-22R1).
- IL-22 and IL-10 receptor chainsplay a role in cellular targeting and signal transduction to selectively initiate and regulate immune responses.
- IL-22can contribute to immune disease through the stimulation of inflammatory responses, S100s, and defensins.
- IL-22also promotes hepatocyte survival in the liver and epithelial cells in the lung and gut similar to IL-10.
- the pro-inflammatory versus tissue-protective functions of IL-22are regulated by the often co-expressed cytokine IL-17A.
- the production and secretion of IL-22is primarily facilitated through activated T cells, and unlike other IL-10 members, IL-22 does not target immune-modulatory cells but instead targets epithelial cell populations such as hepatocytes and pancreatic cells.
- IL-22 targeting of hepatocyteshas been indicated to protect against acute hepatitis, inflammation, and alcohol-induced liver damage, and has demonstrated roles in liver regeneration as well as decreasing weight gain and improving lipid parameters.
- the wild type human IL-22 canonical mRNA sequenceis described at the NCBI Reference Sequence database (RefSeq) under accession number NM_020525.5 ("Homo sapiens interleukin 22 (IL22), mRNA").
- the wild type IL-22 canonical protein sequenceis described at the RefSeq database under accession number NP_065386.1 ("interleukin-22 precursor [Homo sapiens]").
- the IL-22 proteinis 179 amino acids long, and has a molecular weight of 20 kDa.
- nucleic acid sequences encoding the reference protein sequence in the Ref Seq sequencesare the coding sequence as indicated in the respective RefSeq database entry.
- An amino acid sequence of human IL-22is provided in SEQ ID NO: 1: MAALQKSVSSFLMGTLATSCLLLLALLVQGGAAAPISSHCRLDKSNFQQPYITNRTFMLAKE ASLADNNTDVRLIGEKLFHGVSMSERCYLMKQVLNFTLEEVLFPQSDRFQPYMQEVVPFLAR LSNRLSTCHIEGDDLHIQRNVQKLKDTVKKLGESGEIKAIGELDLLFMSLRNACI (SEQ ID NO:1).
- the IL-22 polypeptide of this disclosureis a wild type full length human IL-22 protein. In some embodiments, the IL-22 polypeptide of this disclosure is a wild type full length mouse IL-22 protein. In some embodiments, the IL-22 polypeptide of this disclosure is a variant, a peptide or a polypeptide containing a substitution, and insertion and/or an addition, a deletion and/or a covalent modification with respect to a wild-type IL-22 sequence. In some embodiments, sequence tags or amino acids, can be added to the sequences encoded by the polynucleotides of this disclosure (e.g., at the N-terminal or C-terminal ends), e.g., for localization.
- amino acid residues located at the carboxy, amino terminal, or internal regions of a polypeptide of this disclosurecan optionally be deleted providing for fragments.
- the polynucleotidee.g., a RNA, e.g., an mRNA
- a nucleotide sequencee.g., an ORF
- the polynucleotidee.g., a RNA, e.g., an mRNA
- a nucleotide sequencee.g., an ORF
- the substitutional variantcan comprise one or more conservative amino acids substitutions.
- the variantis an insertional variant.
- the variantis a deletional variant.
- IL-22 protein fragments, functional protein domains, variants, and homologous proteins (orthologs)are also within the scope of the IL-22 polypeptides of the disclosure.
- modifying an IL-22 polypeptide in order to increase its serum half-lifee.g., by fusing an IL-22 polypeptide to another protein known to have a longer serum half-life (e.g., immunoglobulins, serum albumin, etc.), represents a strategy for increasing the serum half-life of IL-22, for example, in the treatment of metabolic disease.
- a longer serum half-lifee.g., immunoglobulins, serum albumin, etc.
- Certain compositions and methods presented in this disclosurerefer to the protein or polynucleotide sequences of wild type human IL-22. Such disclosures are equally applicable to any other variants of IL-22 known in the art or described herein. 3.
- Serum half-life extendersa. Serum half-life extension domain 1.
- a fusion proteincomprises a serum half-life extension domain.
- the serum half-life extension domaincomprises a molecule selected from the group consisting of a polypeptide capable of binding albumin, serum albumin, an antibody or antibody fragment (i.e., a single chain variable fragment),an Fc domain of an antibody, a polyethylene glycol moiety (PEG), a poly(lactic- co-glycolic acid) (PLGA) polymer, a polymeric hydrogel, a nanoparticle, a fatty acid chain, an acyl group, a myristic acid group, a palmitoylated group, and a steryl group.
- PEGpolyethylene glycol moiety
- PLGApoly(lactic- co-glycolic acid)
- the serum half-life extension domainbinds serum albumin (e.g., human serum albumin (HSA) or mouse serum albumin (MSA) or an antigen-binding portion of an anti- HSA antibody.
- serum albumine.g., human serum albumin (HSA) or mouse serum albumin (MSA) or an antigen-binding portion of an anti- HSA antibody.
- a polypeptide that enhances serum half-life in vivois a polypeptide that occurs naturally in vivo and/or that can resist degradation or removal by endogenous mechanisms which remove unwanted material from the organism (e.g., a mammal, or a human).
- a polypeptide that enhances serum half-life in vivocan be selected from proteins from the extracellular matrix, proteins found in blood, proteins found at the blood brain barrier or in neural tissue, proteins localized to the kidney, liver, lung, heart, skin or bone, stress proteins, disease-specific proteins, or proteins involved in Fc transport.
- suitable polypeptides comprising a serum half-life extension domain that enhances serum half-life in vivoinclude, for example, transferrin receptor specific ligand-neuropharmaceutical agent fusion proteins (see U.S. Pat.
- brain capillary endothelial cell receptortransferrin, transferrin receptor (e.g., soluble transferrin receptor), insulin, insulin- like growth factor 1 (IGF 1) receptor, insulin-like growth factor 2 (IGF 2) receptor, insulin receptor, blood coagulation factor X, ⁇ 1-antitrypsin and HNF 1 ⁇ .
- transferrintransferrin receptor
- IGF 1insulin-like growth factor 1
- IGF 2insulin-like growth factor 2
- HNF 1 ⁇blood coagulation factor X
- HNF 1 ⁇blood coagulation factor 1 ⁇
- Suitable polypeptides that enhance serum half-lifealso include alpha-1 glycoprotein (orosomucoid; AAG), alpha-1 antichymotrypsin (ACT), alpha-1 microglobulin (protein HC; AIM), antithrombin III (AT III), apolipoprotein A-1 (Apo A-1), apolipoprotein B (Apo B), ceruloplasmin (Cp), complement component C3 (C3), complement component C4 (C4), C1 esterase inhibitor (C1 INH), C-reactive protein (CRP), ferritin (FER), hemopexin (HPX), lipoprotein(a) (Lp(a)), mannose-binding protein (MBP), myoglobin (Myo), prealbumin (transthyretin; PAL), retinol- binding protein (RBP), and rheumatoid factor (RF).
- alpha-1 glycoproteinorosomucoid
- AAGalpha-1 antichymotrypsin
- suitable polypeptides comprising a serum half-life extension domain that enhances serum half-life in vivoinclude proteins from the extracellular matrix include, for example, collagens, laminins, integrins and fibronectin.
- Collagensare the major proteins of the extracellular matrix.
- about 15 types of collagen moleculesare currently known, found in different parts of the body, e.g. type I collagen (accounting for 90% of body collagen) found in bone, skin, tendon, ligaments, cornea, internal organs or type II collagen found in cartilage, vertebral disc, notochord, and vitreous humor of the eye.
- suitable polypeptides comprising a serum half-life extension domain that enhances serum half-life in vivoinclude proteins from the blood include, for example, plasma proteins (e.g., fibrin, ⁇ -2 macroglobulin, serum albumin, fibrinogen (e.g., fibrinogen A, fibrinogen B), serum amyloid protein A, haptoglobin, profilin, ubiquitin, uteroglobulin and ⁇ -2-microglobulin), enzymes and enzyme inhibitors (e.g., plasminogen, lysozyme, cystatin C, alpha-1-antitrypsin and pancreatic trypsin inhibitor), proteins of the immune system, such as immunoglobulin proteins (e.g., IgA, IgD, IgE, IgG, IgM, immunoglobulin light chains (kappa/lambda)), transport proteins (e.g., retinol binding protein, ⁇ -1 microglobulin), defensins
- plasma proteins
- Suitable proteins found at the blood brain barrier or in neural tissueinclude, for example, melanocortin receptor, myelin, ascorbate transporter and the like.
- suitable polypeptides comprising a half-life extension domain that enhances serum half-life in vivoinclude proteins localized to the kidney (e.g., polycystin, type IV collagen, organic anion transporter K1, Heymann's antigen), proteins localized to the liver (e.g., alcohol dehydrogenase, G250), proteins localized to the lung (e.g., secretory component, which binds IgA), proteins localized to the heart (e.g., HSP 27, which is associated with dilated cardiomyopathy), proteins localized to the skin (e.g., keratin), bone specific proteins such as morphogenic proteins (BMPs), which are a subset of the transforming growth factor ⁇ superfamily of proteins that demonstrate osteogenic activity (e.g., BMP-2, BMP-4, BMP-5
- disease-specific proteins that can extend the serum half-life of a proteininclude, for example, antigens expressed only on activated T-cells, including LAG- 3 (lymphocyte activation gene), osteoprotegerin ligand (OPGL; see Nature 402, 304-309 (1999)), OX40 (a member of the TNF receptor family, expressed on activated T cells and specifically up-regulated in human T cell leukemia virus type-I (HTLV-I)-producing cells; see Immunol.165 (1):263-70 (2000)).
- LAG- 3lymphocyte activation gene
- osteoprotegerin ligandOPGL
- OX40a member of the TNF receptor family, expressed on activated T cells and specifically up-regulated in human T cell leukemia virus type-I (HTLV-I)-producing cells; see Immunol.165 (1):263-70 (2000)).
- Suitable disease-specific proteinsalso include, for example, metalloproteases (associated with arthritis/cancers) including CG6512 Drosophila, human paraplegin, human FtsH, human AFG3L2, murine ftsH; and angiogenic growth factors, including acidic fibroblast growth factor (FGF-1), basic fibroblast growth factor (FGF-2), vascular endothelial growth factor/vascular permeability factor (VEGF/VPF), transforming growth factor- ⁇ (TGF ⁇ ), tumor necrosis factor-alpha (TNF- ⁇ ), angiogenin, interleukin-3 (IL-3), interleukin-8 (IL-8), platelet-derived endothelial growth factor (PD- ECGF), placental growth factor (P1GF), midkine platelet-derived growth factor-BB (PDGF), and fractalkine.
- metalloproteasesassociated with arthritis/cancers
- FGF-1acidic fibroblast growth factor
- FGF-2basic fibroblast growth factor
- suitable polypeptides comprising a half-life extension domain that enhances serum half-life in vivoinclude stress proteins such as heat shock proteins (HSPs).
- HSPsare normally found intracellularly. When they are found extracellularly, it is an indicator that a cell has died and spilled out its contents. This unprogrammed cell death (necrosis) occurs when as a result of trauma, disease or injury, extracellular HSPs trigger a response from the immune system. Binding to extracellular HSP can result in localizing the compositions of the disclosure to a disease site.
- the serum half-life extension domaincomprises serum albumin or a molecule capable of binding serum albumin.
- Serum albuminmay be human serum albumin (HSA), mouse serum albumin (MSA), or serum albumin from another mammalian species.
- HSAmolecular mass ⁇ 67 kDa
- a mature form of HSAcomprises about 585 amino acids.
- HSAserves to maintain plasma pH, contributes to colloidal blood pressure, functions as carrier of many metabolites and fatty acids, and serves as a major drug transport protein in plasma.
- HSAis responsible for a significant proportion of the osmotic pressure of serum and also functions as a carrier of endogenous and exogenous ligands.
- HSAis present at a concentration of about 50 mg/ml (600 ⁇ M) in plasma and has a half-life of around 20 days in humans.
- albuminas a carrier molecule and its inert nature are desirable properties for use as a carrier and transporter of polypeptides in vivo.
- the use of albumin as a component of an albumin fusion protein as a carrier for various proteinsis known to those of skill in the art. See e.g., WO 1993/15199, WO 1993/15200, and EP 413622.
- a fragment of serum albumine.g., HSA or MSA
- the serum albumin fragmentcan be a N-terminal fragment.
- genetically or chemically fusing or conjugating the fusion protein described herein to human albuminwould stabilize or extend the serum half-life, and/or retain the molecule's activity for extended periods of time in solution, in vitro and/or in vivo.
- An amino acid sequence of wild-type HSAis set forth in SEQ ID NO: 5.
- a nonlimiting example of a polypeptide encoded by the polynucleotides of this disclosureis shown in SEQ ID NO:5-6.
- An amino acid sequence of a fragment of MSA proteinis provided in SEQ ID NO:8.
- AHKSEIAHRYNDLGEQHFKGLVLIAFSQYLQKCSYDEHAKLVQEVTDFAKTCVADESAANCDKSLHTLFGDKLCAIPNLRENYGELADCCTKQEPERNECFLQHKDDNPSLPPFERPEAEAMCT SFKENPTTFMGHYLHEVARRHPYFYAPELLYYAEQYNEILTQCCAEADKESCLTPKLDGVKE KALVSSVRQRMKCSSMQKFGERAFKAWAVARLSQTFPNADFAEITKLATDLTKVNKECCHGD LLECADDRAELAKYMCENQATISSKLQTCCDKPLLKKAHCLSEVEHDTMPADLPAIAADFVE DQEVCKNYAEAKDVFLGTFLYEYSRRHP
- a serum albumin fusion protein Fusion of albumin to another proteinmay be achieved by genetic manipulation, such that the DNA coding for HSA or MSA, or a fragment or variant thereof.
- a suitable hostis then transformed or transfected with the fused nucleotide sequences, so arranged on a suitable plasmid as to express a fusion polypeptide.
- the expressionmay be effected in vitro from, for example, prokaryotic or eukaryotic cells, or in vivo (e.g. from a transgenic organism). Additional methods pertaining to HSA fusions can be found, for example, in WO 2001/077137 and WO 2003/06007, incorporated herein by reference.
- Noncovalent association between a protein of interest and serum albuminextends the elimination half-time of short lived proteins.
- the half-life of the fusion proteinis enhanced by at least about 2-fold, at least about 3-fold, at least about 4-fold, at least about 5-fold, at least about 6-fold, at least about 8-fold, at least about 10-fold, at least about 15-fold, at least about 16-fold, at least about 18-fold, at least about 20-fold, or at least about 30-fold when compared to a protein lacking the serum half-life extension domain.
- the half-life extensionis based on the mean plasma residence of the fusion protein.
- Serum clearance rates of the fusion protein comprising the HSAcan be optimized by testing conjugates containing a truncated wild-type HSA.
- the serum half-life extension domaincomprises a molecule capable of binding serum albumin.
- a peptide ligand that binds a human serum albumine.g., an albumin domain antibody or fragment thereof
- a peptide ligand that binds a mammalian serum albumincan be identified by its ability to compete for binding of human serum albumin in an in vitro assay with peptide ligands.
- a peptide ligand that binds a human serum albumincomprises the amino acid sequence having at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 99% or 100% sequence identity to SEQ ID NO:10.
- An amino acid sequence of an albumin domain antibody fragmentis set forth in SEQ ID NO:10.
- the serum half-life extension domaincomprises a fatty acid chain conjugated polypeptide.
- the fatty acid chainmay be selected from a C-16 fatty acid chain or a C-18 fatty acid chain.
- the fatty acid chainis a C-16 fatty acid conjugated molecule.
- the serum half-life extension domaincomprises an antibody fragment that selectively binds serum albumin.
- the antibody fragmentis a single domain antibody, a Complementarity-Determining Region (CDR) of a single domain antibody, or a single-chain variable fragment (scFv).
- CDRComplementarity-Determining Region
- scFvsingle-chain variable fragment
- Native antibody moleculesconsist of two identical heavy chains, and two identical light chains.
- the heavy chain constant regionincludes CH1, a hinge region, CH2, and CH3.
- Papain digestion of antibodiesproduces two fragments, Fab and Fc.
- the Fc fragmentconsists of CH2, CH3, and part of the hinge region. It has been recognized that the Fc region is critical for maintaining the serum half-life of an antibody of class IgG (Ward and Ghetie, Ther. Immunol.2:77-94 (1995)).
- FcRnneonatal Fc receptor
- FcRnis a heterodimer consisting of a transmembrane a chain and a soluble ⁇ chain ( ⁇ 2-microglobulin).
- Igimmunoglobulin
- Fcrefers to a polypeptide comprising the CH3, CH2 and at least a portion of the hinge region of a constant domain of an antibody.
- an Fc regionmay include a CH4 domain, present in some antibody classes.
- An Fcmay comprise the entire hinge region of a constant domain of an antibody.
- the fusion proteincomprises an Fc and a CH1 region of an antibody.
- the fusion proteincomprises an Fc and CH3 region of an antibody.
- the fusion proteincomprises an Fc, a CH1 region and a kappa/lambda region from the constant domain of an antibody.
- Exemplary modificationsinclude additions, deletions or substitutions of one or more amino acids in one or more domains.
- the fusion protein of the present disclosurecomprising an Fc region/domain as a half- life extension domain may be produced by standard recombinant DNA techniques or by protein synthetic techniques, e.g., by use of a peptide synthesizer.
- a nucleic acid molecule encoding a Fc fusion proteincan be synthesized by conventional techniques including automated DNA synthesizers.
- PCR amplification of gene fragmentscan be carried out using anchor primers which give rise to complementary overhangs between two consecutive gene fragments which can subsequently be annealed and re- amplified to generate a chimeric gene sequence (see, e.g., “Current Protocols in Molecular Biology”, Ausubel et al., eds., John Wiley & Sons, (1992)).
- a nucleic acid encoding the fusion protein disclosed hereincan be cloned into an expression vector containing the Fc domain or a fragment thereof such that the the fusion protein disclosed herein is linked in-frame to the constant domain or fragment thereof.
- suitable polypeptides comprising a serum half-life extension domain that enhances serum half-life in vivoinclude proteins involved in Fc transport. These proteins include, for example, Brambell receptor (also known as FcRB). This Fc receptor has two functions, both of which are potentially useful for delivery. The functions are (1) transport of IgG from mother to child across the placenta (2) protection of IgG from degradation thereby prolonging its serum half-life. It is thought that the receptor recycles IgG from endosomes. (See, Holliger et al, Nat Biotechnol 15(7):632-6 (1997).) 4.
- FcRBBrambell receptor
- a polynucleotide of this disclosuree.g., a RNA, e.g., an mRNA
- a polynucleotide of this disclosurecan comprise more than one nucleic acid sequence (e.g., an ORF) encoding a polypeptide of interest.
- polynucleotides of this disclosurecomprise a single ORF encoding an IL-22 polypeptide, a functional fragment, or a variant thereof.
- the polynucleotide of this disclosurecan comprise more than one ORF, for example, a first ORF encoding an IL-22 polypeptide (a first polypeptide of interest), a functional fragment, or a variant thereof, and a second ORF expressing a second polypeptide of interest, a functional fragment, or a variant thereof.
- two or more polypeptides of interestcan be genetically fused, i.e., two or more polypeptides can be encoded by the same ORF.
- the polynucleotidecan comprise a nucleic acid sequence encoding a linker (e.g., a G 4 S (SEQ ID NO: 200), peptide linker, or another linker known in the art) between two or more polypeptides of interest.
- a linkere.g., a G 4 S (SEQ ID NO: 200), peptide linker, or another linker known in the art
- a polynucleotide of this disclosuree.g., a RNA, e.g., an mRNA
- ORFseach expressing a polypeptide of interest.
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) can comprise a first nucleic acid sequence (e.g., a first ORF) encoding an IL-22 polypeptide and a second nucleic acid sequence (e.g., a second ORF) encoding a second polypeptide of interest.
- a first nucleic acid sequencee.g., a first ORF
- a second nucleic acid sequencee.g., a second ORF
- the mRNAs of the disclosureencode more than one polypeptide (e.g., an IL-22 polypeptide and a second polypeptide, such as a serum half-life extender polypeptide), referred to herein as multimer constructs.
- the mRNAfurther encodes a linker located between each domain.
- the linkeris a GGGS (SEQ ID NO: 201) linker.
- the multimer constructcontains three domains with intervening linkers, having the structure: domain-linker-domain-linker-domain e.g., IL-22 domain-linker-serum half-life extension domain-linker-serum half-life extension domain.
- a fusion proteincomprises an IL-22 polypeptide fused to a serum half-life extension domain.
- a fusion proteincomprises an IL-22 polypeptide fused to a serum albumin polypeptide.
- a fusion proteincomprises an IL-22 polypeptide fused to an albumin domain antibody domain.
- a fusion proteine.g., a polypeptide, e.g., a fusion polypeptide
- a fusion protein of the present disclosurecomprises an amino acid sequence encoding an IL-22 polypeptide (e.g., an IL-22 polypeptide comprising the IL-22 wild-type sequence, functional fragment, or variant thereof) and an serum albumin polypeptide (e.g., an serum albumin polypeptide comprising a serum albumin wild-type sequence, functional fragment, or variant thereof), wherein the amino acid sequence has at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO
- the instant inventionfeatures mRNAs for use in treating or preventing metabolic disease.
- the mRNAs featured for use in this disclosureare administered to subjects and encode human IL-22 and/or human serum albumin protein in vivo.
- this disclosurerelates to polynucleotides, e.g., mRNA, comprising an open reading frame of linked nucleosides encoding IL-22, isoforms thereof, variants thereof, functional fragments thereof, and fusion proteins comprising IL-22.
- sequence-optimized polynucleotidescomprising nucleotides encoding the polypeptide sequence of IL-22 (e.g., human IL-22 or mouse IL-22) (or a variant thereof), or sequence having high sequence identity with those sequence optimized polynucleotides.
- An exemplary polynucleotide encoding wild type human IL-22is set forth in SEQ ID NO: 300.
- sequence-optimized polynucleotidescomprising nucleotides encoding the polypeptide sequence of human serum albumin (or a variant thereof), or sequence having high sequence identity with those sequence optimized polynucleotides.
- An exemplary polynucleotide encoding wild type HSAis set forth in SEQ ID NO: 304.
- An exemplary polynucleotide encoding wild type MSAis set forth in SEQ ID NO: 306.
- the encoded IL-22 polypeptide of this disclosurecan be selected from: (i) a full length IL-22 polypeptide (e.g., having the same or essentially the same length as wild-type IL-22; e.g., SEQ ID NO:1-2); (ii) a functional fragment of IL-22 described herein (e.g., a truncated (e.g., deletion of carboxy, amino terminal, or internal regions) sequence shorter than IL-22; but still retaining IL-22 function) (e.g., SEQ ID NO:3-4); (iii) a variant thereof (e.g., full length or truncated IL-22 protein in which one or more amino acids have been replaced, e.g., variants that retain all or most of the IL-22 activity of the polypeptide with respect to a reference protein (e.g., any natural or artificial variants known in the art or described herein)); or (iv) a fusion protein comprising (i) a
- the encoded IL-22 polypeptideis a mammalian IL-22 polypeptide, such as a human IL-22 polypeptide, a functional fragment or a variant thereof.
- the encoded serum albumin polypeptideis a mammalian serum albumin polypeptide, such as a human serum albumin polypeptide or a mouse serum albumin polypeptide, a functional fragment, or a variant thereof.
- the fusion proteincomprises a mammalian IL-22 polypeptide and a mammalian serum albumin polypeptide, such as a human IL-22 polypeptide or functional fragment or a variant thereof, and a human serum albumin polypeptide functional fragment or a variant thereof.
- the fusion proteincomprises an amino acid sequence as set forth in one of SEQ ID NO: 20-38.
- the polynucleotidee.g., a RNA, e.g., an mRNA
- the polynucleotideis introduced to the cells in vitro. In some embodiments, the polynucleotide is introduced to the cells in vivo. In some embodiments, the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosure comprise a nucleotide sequence (e.g., an ORF) that encodes a wild-type IL-22 polypeptide, e.g., SEQ ID NO:1-2, or variant, fragment, or isoform thereof.
- a nucleotide sequencee.g., an ORF
- the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosurecomprise a nucleotide sequence (e.g., an ORF) that encodes a wild-type IL-22, e.g., SEQ ID NO:3-4, or a fragment, variant, or isoform thereof.
- the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosurecomprise a nucleotide sequence (e.g., an ORF) that encodes a wild-type serum albumin, e.g., SEQ ID NO:5-8, or a fragment, variant, or isoform thereof.
- the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosurecomprise a nucleotide sequence (e.g., an ORF) that encodes a variant human IL- 22, or an isoform thereof.
- the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosurecomprise a nucleotide sequence (e.g., an ORF) that encodes a variant human serum albumin, or an isoform thereof.
- the polynucleotides(e.g., a RNA, e.g., an mRNA) of this disclosure comprise a nucleotide sequence that encodes a fusion protein comprising an IL-22 polypeptide and a serum albumin polypeptide.
- the polynucleotidecomprises a nucleotide sequence that encodes a fusion protein comprising a wild-type IL-22 protein, (e.g., SEQ ID NO:1-2), or a variant, isoform, or fragment thereof (e.g., SEQ ID NO:3-4).
- the polynucleotidecomprises a nucleotide sequence that encodes a fusion protein comprising a wild-type serum albumin protein, (e.g., SEQ ID NO:5- 8), or a variant, isoform, or fragment thereof.
- the polynucleotidecomprises a nucleotide sequence that encodes a linker, such as a linker as described herein.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of the present disclosurecomprises a nucleotide sequence (e.g., an ORF) encoding an IL-22- containing fusion protein (e.g., an IL-22-containing fusion protein comprising the IL-22 wild- type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has 65% to 100%, 70% to 100%, 75% to 100%, 80% to 100%, 85% to 100%, 90% to 100%, 95% to 100%, 97% to 100%, 98% to 100%, 99% to 10)%, 90% to 95%, 90% to 97%, 90% to 98%, 95% to 97%, 95% to 98%, or 95% to 99% sequence identity to the sequence of of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of the present disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22- containing fusion protein (e.g., an IL-22-containing fusion protein comprising the IL-22 wild- type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence is at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% identical to the sequence of of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:32
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of the present disclosurecomprises a nucleotide sequence (e.g., an ORF) encoding an IL-22- containing fusion protein (e.g., an IL-22-containing fusion protein comprising the IL-22 wild- type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence is 65% to 100%, 70% to 100%, 75% to 100%, 80% to 100%, 85% to 100%, 90% to 100%, 95% to 100%, 97% to 100%, 98% to 100%, 99% to 100%, 90% to 95%, 90% to 97%, 90% to 98%, 95% to 97%, 95% to 98%, or 95% to 99% identical to the sequence of of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:
- a polynucleotidee.g., a RNA, e.g., an mRNA
- a codon optimized nucleic acid sequencewherein the open reading frame (ORF) of the codon optimized nucleic acid sequence is derived from a wild-type IL-22 sequence (e.g., wild-type human IL-22).
- ORFopen reading frame
- the corresponding wild type sequenceis the native human IL-22.
- the corresponding wild type sequenceis the corresponding fragment from human IL-22.
- the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosurecomprise a nucleotide sequence encoding IL-22 having the full-length sequence of human IL-22 (i.e., including the initiator methionine).
- the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosurecomprise a nucleotide sequence encoding IL-22 having the full-length sequence of mouse IL-22 (i.e., including the initiator methionine).
- the polynucleotidese.g., a RNA, e.g., an mRNA
- a nucleotide sequencee.g., an ORF
- the polynucleotides of this disclosurecomprise an ORF encoding an IL-22 polypeptide that comprises at least one point mutation in the IL-22 amino acid sequence and retains IL-22 signaling activity.
- the mutant IL-22 polypeptidehas an IL-22 activity which is at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100% of the IL-22 activity of the corresponding wild-type IL-22 (e.g., SEQ ID NO:1).
- the polynucleotidee.g., a RNA, e.g., an mRNA
- this disclosurecomprising an ORF encoding a mutant IL-22 polypeptide is sequence optimized.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) that encodes an IL-22 polypeptide with mutations that do not alter IL-22 signaling activity. Such mutant IL-22 polypeptides can be referred to as function-neutral.
- the polynucleotidecomprises an ORF that encodes a mutant IL-22 polypeptide comprising one or more function-neutral point mutations.
- the mutant IL-22 polypeptidehas higher IL-22 enzymatic activity than the corresponding wild-type IL-22.
- the mutant IL-22 polypeptidehas an IL-22 activity that is at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100% higher than the activity of the corresponding wild-type IL-22 (i.e., the same IL-22 protein but without the mutation(s)).
- the polynucleotidese.g., a RNA, e.g., an mRNA
- a nucleotide sequencee.g., an ORF
- a functional IL-22 fragmente.g., where one or more fragments correspond to a polypeptide subsequence of a wild type IL-22 polypeptide and retain IL-22 enzymatic activity.
- the IL-22 fragmenthas an IL-22 activity which is at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100% of the IL-22 activity of the corresponding full length IL-22.
- the polynucleotidese.g., a RNA, e.g., an mRNA
- the polynucleotides of this disclosurecomprising an ORF encoding a functional IL-22 fragment is sequence optimized.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 fragment that has higher IL-22 signaling activity than the corresponding full length IL-22.
- a nucleotide sequencee.g., an ORF
- the IL-22 fragmenthas an IL-22 activity which is at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100% higher than the IL-22 activity of the corresponding full length IL-22.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 fragment that is at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24% or 25% shorter than wild-type IL-22.
- a nucleotide sequencee.g., an ORF
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence encodes an amino acid sequence that is at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% identical to the sequence of any of SEQ ID NO:1-4.
- a nucleotide sequencee.g., an ORF
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding a serum albumin polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence encodes an amino acid sequence that is at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% identical to the sequence of any of SEQ ID NO:5-8.
- a nucleotide sequencee.g., an ORF
- serum albumin polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of any of SEQ ID NO:300-303.
- a nucleotide sequencee.g., an ORF
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding a serum albumin polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of any of SEQ ID NO:304-307.
- a nucleotide sequencee.g., an ORF
- serum albumin polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has 90% to 100%, 95% to 100%, 97% to 100%, 98% to 100%, 90% to 95%, 90% to 97%, 90% to 98%, 95% to 97%, 95% to 98%, or 95% to 99% sequence identity to the sequence of any of SEQ ID NO:300-303.
- a nucleotide sequencee.g., an ORF
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding a serum albumin polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has 90% to 100%, 95% to 100%, 97% to 100%, 98% to 100%, 90% to 95%, 90% to 97%, 90% to 98%, 95% to 97%, 95% to 98%, or 95% to 99% sequence identity to the sequence of any of SEQ ID NO:304-307.
- a nucleotide sequencee.g., an ORF
- serum albumin polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of any of SEQ ID NO:300-303.
- a nucleotide sequencee.g., an ORF
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding a serum albumin polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of any of SEQ ID NO:304-307.
- a nucleotide sequencee.g., an ORF
- serum albumin polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has 90% to 100%, 95% to 100%, 97% to 100%, 98% to 100%, 90% to 95%, 90% to 97%, 90% to 98%, 95% to 97%, 95% to 98%, or 95% to 99%, sequence identity to the sequence of any of SEQ ID NO:300-303.
- a nucleotide sequencee.g., an ORF
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding a serum albumin polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has 90% to 100%, 95% to 100%, 97% to 100%, 98% to 100%, 90% to 95%, 90% to 97%, 90% to 98%, 95% to 97%, 95% to 98%, or 95% to 99%, sequence identity to the sequence of any of SEQ ID NO:304-307.
- a nucleotide sequencee.g., an ORF
- serum albumin polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence is between 90% and 100% identical; between 91% and 99% identical; between 92% and 98% identical; between 93% and 97% identical, or between 94% and 96% identical to the sequence of any of SEQ ID NO:300-303.
- a nucleotide sequencee.g., an ORF
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding a serum albumin polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence is between 90% and 100% identical; between 91% and 99% identical; between 92% and 98% identical; between 93% and 97% identical, or between 94% and 96% identical to the sequence of any of SEQ ID NO:304-307.
- a nucleotide sequencee.g., an ORF
- serum albumin polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises from about 1,000 to about 100,000 nucleotides (e.g., from 1,000 to 2,500, from 1,000 to 2,600, from 1,000 to 2,700, from 1,000 to 2,800, from 1,000 to 2,900, from 1,000 to 3,000, from 1,000 to 5,000, from 1,000 to 10,000, from 1,000 to 25,000, from 1,000 to 50,000, from 1,000 to 70,000, or from 1,000 to 100,000).
- a RNAe.g., an mRNA
- the polynucleotide of this disclosurecomprises from about 1,000 to about 100,000 nucleotides (e.g., from 1,000 to 2,500, from 1,000 to 2,600, from 1,000 to 2,700, from 1,000 to 2,800, from 1,000 to 2,900, from 1,000 to 3,000, from 1,000 to 5,000, from 1,000 to 10,000, from 1,000 to 25,000, from 1,000 to 50,000, from 1,000 to 70,000, or from 1,000 to 100,000).
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the length of the nucleotide sequence (e.g., an ORF) is at least 500 nucleotides in length (e.g., at least or greater than about 500, 600, 700, 800, 900, 1,000, 1,050, 1,100, 1,200, 1,300, 1,400, 1,500, 1,600, 1,700, 1,800, 1,900, 2,000, 2,100, 2,200, 2,300, 2,400, 2,500, 2,600, 2,700, 2,800, 2,900, 3,000, 3,100, 3,200, 3,300, 3,400, 3,500, 3,600, 3,700, 3,800, 3,900, 4,000, 4,100, 4,200, 4,300, 4,400, 4,500, 4,600
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof) further comprises a 5′-UTR (e.g., SEQ ID NO:56) and a 3′-UTR (e.g., SEQ ID NO: 108).
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of any one of SEQ ID NO:300-303.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of any one of SEQ ID NO:304-307.
- the polynucleotide (e.g., a RNA, e.g., an mRNA)comprises a 5′ terminal cap (e.g., m 7 Gp-ppGm-A, Cap0, Cap1, ARCA, inosine, N1-methyl-guanosine, 2′-fluoro- guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino-guanosine, LNA-guanosine, 2- azidoguanosine, Cap2, Cap4, 5′ methylG cap, or an analog thereof) and a poly-A-tail region (e.g., about 100 nucleotides in length).
- the mRNAcomprises a polyA tail.
- the poly A tailis 50-150 (SEQ ID NO:197), 75-150 (SEQ ID NO:198), 85-150 (SEQ ID NO:199), 90-120 (SEQ ID NO:193), 90-130 (SEQ ID NO:194), or 90-150 (SEQ ID NO:192) nucleotides in length.
- the poly A tailis 100 nucleotides in length (SEQ ID NO:195).
- the poly A tailis protected (e.g., with an inverted deoxy-thymidine).
- the poly A tailcomprises A100- UCUAG-A20-inverted deoxy-thymidine.
- the poly A tailis A100- UCUAG-A20-inverted deoxy-thymidine.
- the polynucleotide of this disclosuree.g., a RNA, e.g., an mRNA
- a nucleotide sequencee.g., an ORF
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- at least one nucleic acid sequence that is noncodinge.g., a microRNA binding site.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurefurther comprises a 5′-UTR (e.g., SEQ ID NO: 56) and a 3′ UTR (e.g., SEQ ID NO: 108).
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of any one of SEQ ID NO:300-303.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of any one of SEQ ID NO:304-307.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of any of SEQ ID NO: 310-328.
- the polynucleotide (e.g., a RNA, e.g., an mRNA)comprises a 5′ terminal cap (e.g., m 7 Gp-ppGm-A, Cap0, Cap1, ARCA, inosine, N1-methyl-guanosine, 2′-fluoro- guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino-guanosine, LNA-guanosine, 2- azidoguanosine, Cap2, Cap4, 5′ methylG cap, or an analog thereof) and a poly-A-tail region (e.g., about 100 nucleotides in length, e.g., A100-UCUAG-A20-
- the mRNAcomprises a polyA tail.
- the poly A tailis 50-150 (SEQ ID NO:197), 75-150 (SEQ ID NO:198), 85-150 (SEQ ID NO:199), 90-120 (SEQ ID NO:193), 90-130 (SEQ ID NO:194), or 90-150 (SEQ ID NO:192) nucleotides in length.
- the poly A tailis 100 nucleotides in length (SEQ ID NO:195).
- the poly A tailis protected (e.g., with an inverted deoxy-thymidine).
- the poly A tailcomprises A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
- the poly A tailis A100-UCUAG-A20-inverted deoxy- thymidine (SEQ ID NO:211).
- the polynucleotide of this disclosuree.g., a RNA, e.g., an mRNA
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurefurther comprises a 5′-UTR (e.g., selected from the sequences of SEQ ID NOs: 50-79) and a 3′UTR (e.g., selected from the sequences of SEQ ID NOs: 100-147).
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of SEQ ID NO:300.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of SEQ ID NO:301. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of SEQ ID NO:302. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of SEQ ID NO:303.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of SEQ ID NO:304. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of SEQ ID NO:306. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of SEQ ID NO:307.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of any of SEQ ID NO:310-328.
- the polynucleotide (e.g., a RNA, e.g., an mRNA)comprises a 5′ terminal cap (e.g., m 7 Gp-ppGm-A, Cap0, Cap1, ARCA, inosine, N1-methyl-guanosine, 2′-fluoro- guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino-guanosine, LNA-guanosine, 2- azidoguanosine, Cap2, Cap4, 5′ methylG cap, or an analog thereof) and a poly-A-tail region (e.g., about 100 nucleotides in length, e.g., SEQ ID NO:195).
- a poly-A-tail regione
- the mRNAcomprises a 3′ UTR comprising a nucleic acid sequence of SEQ ID NO:108.
- the mRNAcomprises a polyA tail.
- the poly A tailis 50-150 (SEQ ID NO:197), 75-150 (SEQ ID NO:198), 85-150 (SEQ ID NO:199), 90-120 (SEQ ID NO:193), 90-130 (SEQ ID NO:194), or 90-150 (SEQ ID NO:192) nucleotides in length.
- the poly A tailis 100 nucleotides in length (SEQ ID NO:195).
- the poly A tailis protected (e.g., with an inverted deoxy-thymidine).
- the poly A tailcomprises A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211). In some instances, the poly A tail is A100-UCUAG-A20-inverted deoxy- thymidine (SEQ ID NO:211).
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence (e.g., an ORF, e.g., the sequence of any of SEQ ID NO:300-303) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof) further comprises a 5′-UTR (e.g., selected from the sequences of SEQ ID NOs:50-79) and a 3′-UTR (e.g., selected from the sequences of SEQ ID NOs: 100- 147).
- a nucleotide sequencee.g., an ORF, e.g., the sequence of any of SEQ ID NO:300-303
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, or variant thereof
- a 5′-UTRe.g., selected from the sequences of SEQ ID NOs
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of SEQ ID NO:300. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of SEQ ID NO:301. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of SEQ ID NO:302.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of SEQ ID NO:303. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of SEQ ID NO:304. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of SEQ ID NO:306.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosurecomprises the sequence of SEQ ID NO:307. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises the sequence of any of SEQ ID NO:310-328.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) comprises a 5′ terminal cap (e.g., m 7 Gp-ppGm-A, Cap0, Cap1, ARCA, inosine, N1-methyl-guanosine, 2′-fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino- guanosine, LNA-guanosine, 2-azidoguanosine, Cap2, Cap4, 5′ methylG cap, or an analog thereof) and a poly-A-tail region (e.g., about 100 nucleotides in length).
- a 5′ terminal cape.g., m 7 Gp-ppGm-A, Cap0, Cap1, ARCA, inosine, N1-methyl-guanosine, 2′-fluoro-guanosine, 7-deaza-guanosine, 8-o
- the mRNAcomprises a polyA tail.
- the poly A tailis 50-150 (SEQ ID NO:197), 75-150 (SEQ ID NO:198), 85-150 (SEQ ID NO:199), 90-120 (SEQ ID NO:193), 90-130 (SEQ ID NO:194), or 90-150 (SEQ ID NO:192) nucleotides in length.
- the poly A tailis 100 nucleotides in length (SEQ ID NO:195).
- the poly A tailis protected (e.g., with an inverted deoxy-thymidine).
- the poly A tailcomprises A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
- the poly A tailis A100-UCUAG-A20-inverted deoxy- thymidine (SEQ ID NO:211).
- the polynucleotide of this disclosuree.g., a RNA, e.g., an mRNA
- the polynucleotide of this disclosurecomprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO:56 and a nucleotide sequence (e.g., an ORF) encoding the IL-22 polypeptide of SEQ ID NO:1 or SEQ ID NO:34.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO:56, a nucleotide sequence (e.g., an ORF) encoding the IL-22 polypeptide of SEQ ID NO:1 or SEQ ID NO:34, and a 3' UTR comprising the nucleotide sequence of SEQ ID NO:108.
- a RNAe.g., an mRNA
- a 5′ UTRcomprising the nucleotide sequence of SEQ ID NO:56
- a nucleotide sequencee.g., an ORF
- a 3' UTRcomprising the nucleotide sequence of SEQ ID NO:108.
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO:56 and a nucleotide sequence (e.g., an ORF) encoding the serum albumin polypeptide of SEQ ID NO:2.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO:55, a nucleotide sequence (e.g., an ORF) encoding the serum albumin polypeptide of SEQ ID NO:2, and a 3' UTR comprising the nucleotide sequence of SEQ ID NO:108.
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO:56 and a nucleotide sequence (e.g., an ORF) encoding the IL-22 polypeptide-containing fusion protein of any of SEQ ID NO:3-9.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) comprises a 5′ UTR comprising the nucleotide sequence of SEQ ID NO: 50 or SEQ ID NO:56, a nucleotide sequence (e.g., an ORF) encoding the IL-22 polypeptide-containing fusion protein of any of SEQ ID NO:3-9, and a 3' UTR comprising the nucleotide sequence of SEQ ID NO:108.
- a RNAe.g., an mRNA
- a 5′ UTRcomprising the nucleotide sequence of SEQ ID NO: 50 or SEQ ID NO:56
- a nucleotide sequencee.g., an ORF
- a 3' UTRcomprising the nucleotide sequence of SEQ ID NO:108.
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide, and serum albumin polypeptide, or a serum albumin-IL-22 fusion protein is single stranded or double stranded.
- the polynucleotide of this disclosurecomprising a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof) is DNA or RNA.
- the polynucleotide of this disclosureis RNA.
- the polynucleotide of this disclosureis, or functions as, a mRNA.
- the mRNAcomprises a nucleotide sequence (e.g., an ORF) that encodes at least one IL-22 polypeptide, and is capable of being translated to produce the encoded IL-22 polypeptide in vitro, in vivo, in situ or ex vivo.
- a nucleotide sequencee.g., an ORF
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) comprises a sequence-optimized nucleotide sequence (e.g., an ORF) encoding an IL- 22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof, see e.g., SEQ ID NO:12), wherein the polynucleotide comprises at least one chemically modified nucleobase, e.g., N1-methylpseudouracil or 5-methoxyuracil. In certain embodiments, all uracils in the polynucleotide are N1-methylpseudouracils.
- all uracils in the polynucleotideare 5-methoxyuracils.
- the polynucleotidefurther comprises a miRNA binding site, e.g., a miRNA binding site that binds to miR-142 and/or a miRNA binding site that binds to miR-126.
- the polynucleotide(e.g., a RNA, e.g., a mRNA) disclosed herein is Formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., Compound II or Compound B; a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound VI or Compound I, or any combination thereof.
- a delivery agentcomprising, e.g., a compound having the Formula (I), e.g., Compound II or Compound B; a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound VI or Compound I, or any combination thereof.
- the delivery agentcomprises an ionizable amino lipid (e.g., Compound II, VI, or B), a helper lipid (e.g., DSPC), a sterol (e.g., Cholesterol), and a PEG lipid (e.g., Compound I or PEG- DMG), e.g., with a mole ratio in the range of about (i) 40-50 mol% ionizable amino lipid (e.g., Compound II, VI, or B), optionally 45-50 mol% ionizable amino lipid, for example, 45- 46 mol%, 46-47 mol%, 47-48 mol%, 48-49 mol%, or 49-50 mol% for example about 45 mol%, 45.5 mol%, 46 mol%, 46.5 mol%, 47 mol%, 47.5 mol%, 48 mol%, 48.5 mol%, 49 mol%, or 49.5 mol%; (ii) 30-45
- the delivery agentcomprises Compound B, Cholesterol, DSPC, and Compound I with a mole ratio of 47:39:11:3.
- the polynucleotidee.g., a RNA, e.g., a mRNA
- the polynucleotide disclosed hereinis Formulated with a delivery agent comprising LNP-1A, LNP 1-B, LNP-2A, LNP-2B, LNP-3A, or LNP-3B.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-1A.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-1B.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-2A.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-2B.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-3A. In some embodiments, the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosure is Formulated with LNP-3B.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NO:50-79), an ORF sequence of SEQ ID NO:310-328, a 3′UTR (e.g., any one of SEQ ID NO:100-147), and a poly A tail (e.g., about 100 nt in length, e.g., SEQ ID NO:195), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g., Cap1, e.g., m 7 Gp-ppGm-A
- a 5′UTRe.g., any one of SEQ ID NO:50-79
- the polynucleotideis included in a delivery agent comprising LNP-1A, LNP- 1B, LNP-2A, LNP-2B, LNP-3A, or LNP-3B.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NOs:50-79), an ORF sequence of any of SEQ ID NO:300-303, a 3′UTR (e.g., any one of SEQ ID NOs:100-147), and a poly A tail (e.g., about 100 nt in length, e.g., SEQ ID NO:195), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g
- the polynucleotideis included in a delivery agent comprising LNP-1A, LNP-1B, LNP-2A, LNP-2B, LNP-3A, or LNP-3B.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NOs:50-79), an ORF sequence of any of SEQ ID NO:304-307, a 3′UTR (e.g., any one of SEQ ID NOs:100-147), and a poly A tail (e.g., about 100 nt in length, e.g., SEQ ID NO:195), wherein all uracils in the polynucleotide are N1 methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g.
- the polynucleotideis included in a delivery agent comprising LNP-1A, LNP-1B, LNP-2A, LNP-2B, LNP-3A, or LNP-3B.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NOs:50-79), an ORF sequence of any one of SEQ ID NO:310-328, a 3′UTR (e.g., any one of SEQ ID NOs:100-147), and a poly A tail (e.g., about 100 nt in length, e.g., SEQ ID NO:195), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5- methoxyuracil.
- a 5′-terminal cape.g
- the polynucleotideis included in a delivery agent comprising LNP-1A, LNP-1B, LNP-2A, LNP-2B, LNP-3A, or LNP-3B.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap 1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NO:50-79), an ORF sequence of any one of SEQ ID NO: 310-328, a 3′UTR (e.g., any one of SEQ ID NO:100-147), and a poly A tail (e.g., about 100 nt in length), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g., Cap 1, e.g., m 7 G
- the delivery agentcomprises Compound II or Compound VI as the ionizable amino lipid and PEG-DMG or Compound I as the PEG lipid.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap 1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NO:50-79), an ORF sequence of any one of SEQ ID NO: 310-328, a 3′UTR (e.g., any one of SEQ ID NO:100-147), and a poly A tail (e.g., about 100 nt in length), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g., Cap 1, e.g., m 7 Gp-ppGm-A
- the delivery agentcomprises Compound II or Compound VI as the ionizable amino lipid and PEG-DMG or Compound I as the PEG lipid.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap 1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NO:50-79), an ORF sequence of any one of SEQ ID NO: 310-328, a 3′UTR (e.g., any one of SEQ ID NO:100-147), and a poly A tail (e.g., about 100 nt in length), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g., Cap 1, e.g., m 7 Gp-ppGm-A
- the delivery agentcomprises Compound B as the ionizable amino lipid and PEG-DMG or Compound I as the PEG lipid.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap 1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NO:50-79), an ORF sequence of SEQ ID NO: 310-328, a 3′UTR (e.g., any one of SEQ ID NO: 100-147), and a poly A tail (e.g., about 100 nt in length), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g., Cap 1, e.g., m 7 Gp-ppGm-A
- a 5′UTRe
- the delivery agentcomprises Compound B as the ionizable amino lipid and PEG-DMG or Compound I as the PEG lipid.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap 1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NOs:50-79), an ORF sequence of any one of SEQ ID NO: 310-328, a 3′UTR (e.g., any one of SEQ ID NOs: 100-147), and a poly A tail (e.g., about 100 nt in length), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g., Cap 1, e.g., m 7 Gp-ppGm-A
- the delivery agentcomprises Compound II or Compound VI as the ionizable amino lipid and PEG-DMG or Compound I as the PEG lipid.
- the polynucleotide of the disclosureis an mRNA that comprises a 5′-terminal cap (e.g., Cap 1, e.g., m 7 Gp-ppGm-A), a 5′UTR (e.g., any one of SEQ ID NOs:50-79), an ORF sequence of any one of SEQ ID NO: 310-328, a 3′UTR (e.g., any one of SEQ ID NOs: 100-147), and a poly A tail (e.g., about 100 nt in length), wherein all uracils in the polynucleotide are N1-methylpseudouracils or 5-methoxyuracil.
- a 5′-terminal cape.g., Cap 1, e.g., m 7 Gp-ppGm-A
- the delivery agentcomprises Compound B as the ionizable amino lipid and PEG-DMG or Compound I as the PEG lipid.
- the polynucleotidese.g., a RNA, e.g., an mRNA
- the polynucleotides of this disclosurecan also comprise nucleotide sequences that encode additional features that facilitate trafficking of the encoded polypeptides to therapeutically relevant sites.
- One such feature that aids in protein traffickingis the signal sequence, or targeting sequence.
- the peptides encoded by these signal sequencesare known by a variety of names, including targeting peptides, transit peptides, and signal peptides.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence (e.g., an ORF) that encodes a signal peptide operably linked to a nucleotide sequence that encodes an IL-22 polypeptide described herein.
- a nucleotide sequencee.g., an ORF
- the "signal sequence” or “signal peptide”is a polynucleotide or polypeptide, respectively, which is from about 30-210, e.g., about 45-80 or 15-60 nucleotides (e.g., about 20, 30, 40, 50, 60, or 70 amino acids) in length that, optionally, is incorporated at the 5′ (or N-terminus) of the coding region or the polypeptide, respectively. Addition of these sequences results in trafficking the encoded polypeptide to a desired site, such as the endoplasmic reticulum or the mitochondria through one or more targeting pathways.
- a desired sitesuch as the endoplasmic reticulum or the mitochondria through one or more targeting pathways.
- the polynucleotide of this disclosurecomprises a nucleotide sequence encoding an IL-22 polypeptide, wherein the nucleotide sequence further comprises a 5′ nucleic acid sequence encoding a heterologous signal peptide. 7. Sequence Optimization of Nucleotide Sequence Encoding an IL-22 Polypeptide or an IL-22-containing fusion protein
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosureis sequence optimized.
- the polynucleotide(e.g., a RNA, e.g., an mRNA) of this disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide or an IL-22-containing fusion protein (e.g., a serum albumin-IL-22 fusion protein), optionally, a nucleotide sequence (e.g, an ORF) encoding another polypeptide of interest, a 5′-UTR, a 3′- UTR, the 5′ UTR or 3′ UTR optionally comprising at least one microRNA binding site, optionally a nucleotide sequence encoding a linker, a polyA tail, or any combination thereof), in which the ORF(s) are sequence optimized.
- a nucleotide sequencee.g., an ORF
- a sequence-optimized nucleotide sequencee.g., a codon-optimized mRNA sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein (e.g., a serum albumin- IL-22 fusion protein)
- a reference sequencee.g., a wild type nucleotide sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein (e.g., a serum albumin- IL-22 fusion protein)
- a sequence-optimized nucleotide sequencecan be partially or completely different in sequence from the reference sequence.
- a reference sequence encoding polyserine uniformly encoded by UCU codonscan be sequence-optimized by having 100% of its nucleobases substituted (for each codon, U in position 1 replaced by A, C in position 2 replaced by G, and U in position 3 replaced by C) to yield a sequence encoding polyserine which would be uniformly encoded by AGC codons.
- the percentage of sequence identity obtained from a global pairwise alignment between the reference polyserine nucleic acid sequence and the sequence-optimized polyserine nucleic acid sequencewould be 0%.
- the protein products from both sequenceswould be 100% identical.
- sequence optimizationalso sometimes referred to codon optimization
- resultscan include, e.g., matching codon frequencies in certain tissue targets and/or host organisms to ensure proper folding; biasing G/C content to increase mRNA stability or reduce secondary structures; minimizing tandem repeat codons or base runs that can impair gene construction or expression; customizing transcriptional and translational control regions; inserting or removing protein trafficking sequences; removing/adding post translation modification sites in an encoded protein (e.g., glycosylation sites); adding, removing or shuffling protein domains; inserting or deleting restriction sites; modifying ribosome binding sites and mRNA degradation sites; adjusting translational rates to allow the various domains of the protein to fold properly; and/or reducing or eliminating problem secondary structures within the polynucleotide.
- Sequence optimization tools, algorithms and servicesare known in the art, non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (
- a polynucleotidee.g., a RNA, e.g., an mRNA
- a sequence-optimized nucleotide sequencee.g., an ORF
- an IL-22 polypeptide, a functional fragment, or a variant thereof, or an IL-22-containing fusion proteine.g., a serum albumin-IL-22 fusion protein
- the IL-22 polypeptide, functional fragment, or a variant thereof or fusion protein encoded by the sequence-optimized nucleotide sequencehas improved properties (e.g., compared to an IL-22 polypeptide, functional fragment, or a variant thereof or fusion protein encoded by a reference nucleotide sequence that is not sequence optimized), e.g., improved properties related to expression efficacy after administration in vivo.
- Such propertiesinclude, but are not limited to, improving nucleic acid stability (e.g., mRNA stability), increasing translation efficacy in the target tissue, reducing the number of truncated proteins expressed, improving the folding or prevent misfolding of the expressed proteins, reducing toxicity of the expressed products, reducing cell death caused by the expressed products, increasing and/or decreasing protein aggregation.
- nucleic acid stabilitye.g., mRNA stability
- increasing translation efficacy in the target tissuereducing the number of truncated proteins expressed, improving the folding or prevent misfolding of the expressed proteins, reducing toxicity of the expressed products, reducing cell death caused by the expressed products, increasing and/or decreasing protein aggregation.
- sequence-optimized nucleotide sequence(e.g., an ORF) is codon optimized for expression in human subjects, having structural and/or chemical features that avoid one or more of the problems in the art, for example, features which are useful for optimizing Formulation and delivery of nucleic acid-based therapeutics while retaining structural and functional integrity; overcoming a threshold of expression; improving expression rates; half-life and/or protein concentrations; optimizing protein localization; and avoiding deleterious bio-responses such as the immune response and/or degradation pathways.
- an ORFcodon optimized for expression in human subjects, having structural and/or chemical features that avoid one or more of the problems in the art, for example, features which are useful for optimizing Formulation and delivery of nucleic acid-based therapeutics while retaining structural and functional integrity; overcoming a threshold of expression; improving expression rates; half-life and/or protein concentrations; optimizing protein localization; and avoiding deleterious bio-responses such as the immune response and/or degradation pathways.
- the polynucleotides of this disclosurecomprise a nucleotide sequence (e.g., a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide, a nucleotide sequence (e.g., an ORF) encoding another polypeptide of interest (e.g., a serum albumin polypeptide), a nucleotide sequence encoding a serum albumin-IL-22 fusion protein, a 5′-UTR, a 3′-UTR, a microRNA binding site, a nucleic acid sequence encoding a linker, or any combination thereof) that is sequence-optimized according to a method comprising: (i) substituting at least one codon in a reference nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) with an alternative codon to increase or decrease uridine content to generate a uridine-modified sequence; (ii) substituting at least one codon
- the sequence-optimized nucleotide sequence(e.g., an ORF encoding an IL-22 polypeptide) has at least one improved property with respect to the reference nucleotide sequence.
- the sequence optimization methodis multiparametric and comprises one, two, three, four, or more methods disclosed herein and/or other optimization methods known in the art.
- Features, which can be considered beneficial in some embodiments of this disclosure,can be encoded by or within regions of the polynucleotide and such regions can be upstream (5′) to, downstream (3′) to, or within the region that encodes the IL-22 polypeptide or IL-22- containing fusion protein.
- these regionscan be incorporated into the polynucleotide before and/or after sequence-optimization of the protein encoding region or open reading frame (ORF).
- ORFopen reading frame
- examples of such featuresinclude, but are not limited to, untranslated regions (UTRs), microRNA sequences, Kozak sequences, oligo(dT) sequences, poly-A tail, and detectable tags and can include multiple cloning sites that can have XbaI recognition.
- the polynucleotide of this disclosurecomprises a 5′ UTR, a 3′ UTR and/or a microRNA binding site.
- the polynucleotidecomprises two or more 5′ UTRs and/or 3′ UTRs, which can be the same or different sequences.
- the polynucleotidecomprises two or more microRNA binding sites, which can be the same or different sequences. Any portion of the 5′ UTR, 3′ UTR, and/or microRNA binding site, including none, can be sequence-optimized and can independently contain one or more different structural or chemical modifications, before and/or after sequence optimization.
- the polynucleotideis reconstituted and transformed into a vector such as, but not limited to, plasmids, viruses, cosmids, and artificial chromosomes.
- the optimized polynucleotidecan be reconstituted and transformed into chemically competent E. coli, yeast, neurospora, maize, drosophila, etc.
- the polynucleotide of this disclosurecomprises a sequence- optimized nucleotide sequence encoding an IL-22 polypeptide or IL-22-containing fusion protein disclosed herein.
- the polynucleotide of this disclosurecomprises an open reading frame (ORF) encoding an IL-22 polypeptide or IL-22-containing fusion protein, wherein the ORF has been sequence optimized.
- sequence-optimized nucleotide sequence encoding full length IL-22is set forth as SEQ ID NO:300-303.
- An exemplary sequence-optimized nucleotide sequence encoding full length serum albuminis set forth as SEQ ID NO:304-307.
- Exemplary sequence-optimized nucleotide sequences encoding serum albumin-IL-22 fusion proteinsare set forth as SEQ ID NO:310-328 and SEQ ID NO:350-368.
- sequence optimized IL-22 sequence, fragment, variant, and fusion protein thereofare used to practice the methods disclosed herein.
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding an IL-22 polypeptide, comprises from 5′ to 3′ end: (i) a 5′ cap such as provided herein, for example, m 7 Gp-ppGm-A; (ii) a 5′ UTR, such as the sequences provided herein, for example, SEQ ID NO:50 or SEQ ID NO:56; (iii) an open reading frame encoding an IL-22 polypeptide, e.g., a sequence optimized nucleic acid sequence encoding IL-22 set forth as SEQ ID NO:300-303, or a sequence optimized nucleic acid sequence encoding an IL-22-containing fusion protein as set forth in any one of SEQ ID NO:310-328; (iv) at least one stop codon (if not present at 5′ terminus of 3′UTR); (v) a 3′ UTR, such
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding an IL-22 polypeptide, comprises from 5′ to 3′ end: (i) a 5′ cap such as provided herein, for example, m 7 Gp-ppGm-A; (ii) a 5′ UTR, such as the sequences provided herein, for example, one of SEQ ID NOs:50-79; (iii) an open reading frame encoding an IL-22 polypeptide, e.g., a sequence optimized nucleic acid sequence encoding IL-22 set forth as SEQ ID NO:300-303 or a sequence optimized nucleic acid sequence encoding an IL-22-containing fusion protein as set forth in any one of SEQ ID NO:310-328; (iv) at least one stop codon (if not present at 5′ terminus of 3′UTR); (v) a 3′ UTR, such as the sequence
- all uracils in the polynucleotideare N1-methylpseudouracil (G5). In certain embodiments, all uracils in the polynucleotide are 5-methoxyuracil (G6).
- the sequence-optimized nucleotide sequences disclosed hereinare distinct from the corresponding wild type nucleotide acid sequences and from other known sequence- optimized nucleotide sequences, e.g., these sequence-optimized nucleic acids have unique compositional characteristics.
- the percentage of uracil or thymine nucleobases in a sequence- optimized nucleotide sequenceis modified (e.g., reduced) with respect to the percentage of uracil or thymine nucleobases in the reference wild-type nucleotide sequence.
- a sequence- optimized nucleotide sequencee.g., encoding an IL-22 polypeptide, a functional fragment, or a variant thereof
- is modifiede.g., reduced
- a sequenceis referred to as a uracil-modified or thymine-modified sequence.
- the percentage of uracil or thymine content in a nucleotide sequencecan be determined by dividing the number of uracils or thymines in a sequence by the total number of nucleotides and multiplying by 100.
- the sequence-optimized nucleotide sequencehas a lower uracil or thymine content than the uracil or thymine content in the reference wild-type sequence.
- the uracil or thymine content in a sequence-optimized nucleotide sequence of this disclosureis greater than the uracil or thymine content in the reference wild- type sequence and still maintain beneficial effects, e.g., increased expression and/or reduced Toll-Like Receptor (TLR) response when compared to the reference wild-type sequence.
- TLRToll-Like Receptor
- Codon optimizationmay be used to match codon frequencies in target and host organisms to ensure proper folding; bias GC content to increase mRNA stability or reduce secondary structures; minimize tandem repeat codons or base runs that may impair gene construction or expression; customize transcriptional and translational control regions; insert or remove protein trafficking sequences; remove/add post translation modification sites in encoded protein (e.g., glycosylation sites); add, remove or shuffle protein domains; insert or delete restriction sites; modify ribosome binding sites and mRNA degradation sites; adjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide.
- encoded proteine.g., glycosylation sites
- add, remove or shuffle protein domainsadd or delete restriction sites
- modify ribosome binding sites and mRNA degradation sitesadjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide.
- Codon optimization tools, algorithms and servicesare known in the art - non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (Menlo Park CA) and/or proprietary methods.
- the open reading frame (ORF) sequenceis optimized using optimization algorithms. 9.
- the polynucleotidee.g., a RNA, e.g., an mRNA
- a sequence optimized nucleic acid disclosed herein encoding an IL-22 polypeptide or an IL-22-containing fusion proteincan be tested to determine whether at least one nucleic acid sequence property (e.g., stability when exposed to nucleases) or expression property has been improved with respect to the non-sequence optimized nucleic acid.
- expression propertyrefers to a property of a nucleic acid sequence either in vivo (e.g., translation efficacy of a synthetic mRNA after administration to a subject in need thereof) or in vitro (e.g., translation efficacy of a synthetic mRNA tested in an in vitro model system).
- Expression propertiesinclude but are not limited to the amount of protein produced by an mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein after administration, and the amount of soluble or otherwise functional protein produced.
- sequence optimized nucleic acids disclosed hereincan be evaluated according to the viability of the cells expressing a protein encoded by a sequence optimized nucleic acid sequence (e.g., a RNA, e.g., an mRNA) encoding an IL-22 polypeptide or an IL- 22-containing fusion protein disclosed herein.
- a plurality of sequence optimized nucleic acids disclosed hereine.g., a RNA, e.g., an mRNA
- a property of interestfor example an expression property in an in vitro model system, or in vivo in a target tissue or cell.
- the desired property of the polynucleotideis an intrinsic property of the nucleic acid sequence.
- the nucleotide sequencee.g., a RNA, e.g., an mRNA
- the nucleotide sequencecan be sequence optimized for expression in a given target tissue or cell.
- the nucleic acid sequenceis sequence optimized to increase its plasma half-life by preventing its degradation by endo and exonucleases.
- the nucleic acid sequenceis sequence optimized to increase its resistance to hydrolysis in solution, for example, to lengthen the time that the sequence optimized nucleic acid or a pharmaceutical composition comprising the sequence optimized nucleic acid can be stored under aqueous conditions with minimal degradation.
- the sequence optimized nucleic acidcan be optimized to increase its resistance to hydrolysis in dry storage conditions, for example, to lengthen the time that the sequence optimized nucleic acid can be stored after lyophilization with minimal degradation.
- the desired property of the polynucleotideis the level of expression of an IL-22 polypeptide encoded by a sequence optimized sequence disclosed herein.
- Protein expression levelscan be measured using one or more expression systems.
- expressioncan be measured in cell culture systems, e.g., CHO cells or HEK293 cells.
- expressioncan be measured using in vitro expression systems prepared from extracts of living cells, e.g., rabbit reticulocyte lysates, or in vitro expression systems prepared by assembly of purified individual components.
- the protein expressionis measured in an in vivo system, e.g., mouse, rabbit, monkey, etc.
- protein expression in solution formcan be desirable.
- a reference sequencecan be sequence optimized to yield a sequence optimized nucleic acid sequence having optimized levels of expressed proteins in soluble form.
- Levels of protein expression and other properties such as solubility, levels of aggregation, and the presence of truncation productscan be measured according to methods known in the art, for example, using electrophoresis (e.g., native or SDS-PAGE) or chromatographic methods (e.g., HPLC, size exclusion chromatography, etc.).
- electrophoresise.g., native or SDS-PAGE
- chromatographic methodse.g., HPLC, size exclusion chromatography, etc.
- the expression of heterologous therapeutic proteins encoded by a nucleic acid sequencecan have deleterious effects in the target tissue or cell, reducing protein yield, or reducing the quality of the expressed product (e.g., due to the presence of protein fragments or precipitation of the expressed protein in inclusion bodies), or causing toxicity.
- the sequence optimization of a nucleic acid sequence disclosed hereine.g., a nucleic acid sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein, can be used to increase the viability of target cells expressing the protein encoded by the sequence optimized nucleic acid.
- Heterologous protein expressioncan also be deleterious to cells transfected with a nucleic acid sequence for autologous or heterologous transplantation. Accordingly, in some embodiments of the present disclosure the sequence optimization of a nucleic acid sequence disclosed herein can be used to increase the viability of target cells expressing the protein encoded by the sequence optimized nucleic acid sequence. Changes in cell or tissue viability, toxicity, and other physiological reaction can be measured according to methods known in the art. d.
- a sequence optimized nucleic acid encoding IL- 22 polypeptide, a functional fragment thereof, or a fusion protein comprising an IL-22 polypeptide or fragment thereofcan trigger an immune response, which could be caused by (i) the therapeutic agent (e.g., an mRNA encoding an IL-22 polypeptide or fragment thereof), or (ii) the expression product of such therapeutic agent (e.g., the IL-22 polypeptide or fragment thereof encoded by the mRNA), or (iv) a combination thereof.
- the therapeutic agente.g., an mRNA encoding an IL-22 polypeptide or fragment thereof
- the expression product of such therapeutic agente.g., the IL-22 polypeptide or fragment thereof encoded by the mRNA
- nucleic acid sequencee.g., an mRNA
- sequence optimization of nucleic acid sequencecan be used to decrease an immune or inflammatory response triggered by the administration of a nucleic acid encoding an IL-22 polypeptide or by the expression product of IL-22 encoded by such nucleic acid.
- an inflammatory responsecan be measured by detecting increased levels of one or more inflammatory cytokines using methods known in the art, e.g., ELISA.
- inflammatory cytokinerefers to cytokines that are elevated in an inflammatory response.
- inflammatory cytokinesexamples include interleukin-6 (IL-6), CXCL1 (chemokine (C-X-C motif) ligand 1; also known as GRO ⁇ , interferon- ⁇ (IFN ⁇ ), tumor necrosis factor ⁇ (TNF ⁇ ), interferon ⁇ -induced protein 10 (IP-10), or granulocyte-colony stimulating factor (G-CSF).
- IL-6interleukin-6
- CXCL1chemokine (C-X-C motif) ligand 1
- IFN ⁇interferon- ⁇
- TNF ⁇tumor necrosis factor ⁇
- IP-10interferon ⁇ -induced protein 10
- G-CSFgranulocyte-colony stimulating factor
- the term inflammatory cytokinesincludes also other cytokines associated with inflammatory responses known in the art, e.g., interleukin-1 (IL-1), interleukin-8 (IL-8), interleukin-12 (IL-12), interleukin-13 (Il-13), interferon
- the polynucleotidee.g., a RNA, e.g., an mRNA
- the polynucleotide of this disclosurecomprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1-methylpseudouracil, 5-methoxyuracil, or the like.
- the mRNAis a uracil-modified sequence comprising an ORF encoding an IL- 22 polypeptide or an IL-22-containing fusion protein, wherein the mRNA comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1-methylpseudouracil, or 5-methoxyuracil.
- a chemically modified uracile.g., pseudouracil, N1-methylpseudouracil, or 5-methoxyuracil.
- uracil in the polynucleotideis at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least 90%, at least 95%, at least 99%, or about 100% modified uracil. In one embodiment, uracil in the polynucleotide is at least 95% modified uracil. In another embodiment, uracil in the polynucleotide is 100% modified uracil. In embodiments where uracil in the polynucleotide is at least 95% modified uracil overall uracil content can be adjusted such that an mRNA provides suitable protein expression levels while inducing little to no immune response.
- the uracil content of the ORFis between about 100% and about 150%, between about 100% and about 110%, between about 105% and about 115%, between about 110% and about 120%, between about 115% and about 125%, between about 120% and about 130%, between about 125% and about 135%, between about 130% and about 140%, between about 135% and about 145%, between about 140% and about 150% of the theoretical minimum uracil content in the corresponding wild-type ORF (%U TM ). In other embodiments, the uracil content of the ORF is between about 121% and about 136% or between 123% and 134% of the %U TM .
- the uracil content of the ORF encoding an IL-22 polypeptide or an IL- 22-containing fusion proteinis about 115%, about 120%, about 125%, about 130%, about 135%, about 140%, about 145%, or about 150% of the %U TM .
- the term "uracil”can refer to modified uracil and/or naturally occurring uracil.
- the uracil content in the ORF of the mRNA encoding an IL-22 polypeptide of this disclosureis less than about 30%, about 25%, about 20%, about 15%, or about 10% of the total nucleobase content in the ORF.
- the uracil content in the ORFis between about 10% and about 20% of the total nucleobase content in the ORF. In other embodiments, the uracil content in the ORF is between about 10% and about 25% of the total nucleobase content in the ORF. In one embodiment, the uracil content in the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein is less than about 20% of the total nucleobase content in the open reading frame.
- the term "uracil”can refer to modified uracil and/or naturally occurring uracil.
- the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein having modified uracil and adjusted uracil contenthas increased Cytosine (C), Guanine (G), or Guanine/Cytosine (G/C) content (absolute or relative).
- the overall increase in C, G, or G/C content (absolute or relative) of the ORFis at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 10%, at least about 15%, at least about 20%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 100% relative to the G/C content (absolute or relative) of the wild-type ORF.
- the G, the C, or the G/C content in the ORFis less than about 100%, less than about 90%, less than about 85%, or less than about 80% of the theoretical maximum G, C, or G/C content of the corresponding wild type nucleotide sequence encoding the IL-22 polypeptide (%G TMX ; %C TMX , or %G/C TMX ).
- the increases in G and/or C content (absolute or relative) described hereincan be conducted by replacing synonymous codons with low G, C, or G/C content with synonymous codons having higher G, C, or G/C content.
- the increase in G and/or C contentis conducted by replacing a codon ending with U with a synonymous codon ending with G or C.
- the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein of this disclosurecomprises modified uracil and has an adjusted uracil content containing less uracil pairs (UU) and/or uracil triplets (UUU) and/or uracil quadruplets (UUUU) than the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide.
- the ORF of the mRNA encoding an IL-22 polypeptide of this disclosurecontains no uracil pairs and/or uracil triplets and/or uracil quadruplets. In some embodiments, uracil pairs and/or uracil triplets and/or uracil quadruplets are reduced below a certain threshold, e.g., no more than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 occurrences in the ORF of the mRNA encoding the IL-22 polypeptide.
- a certain thresholde.g., no more than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 occurrences in the ORF of the mRNA encoding the IL-22 polypeptide.
- the ORF of the mRNA encoding the IL- 22 polypeptide of this disclosurecontains less than 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 non-phenylalanine uracil pairs and/or triplets. In another embodiment, the ORF of the mRNA encoding the IL-22 polypeptide contains no non- phenylalanine uracil pairs and/or triplets.
- the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein of this disclosurecomprises modified uracil and has an adjusted uracil content containing less uracil-rich clusters than the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide.
- the ORF of the mRNA encoding the IL-22 polypeptide of this disclosurecontains uracil-rich clusters that are shorter in length than corresponding uracil-rich clusters in the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide.
- alternative lower frequency codonsare employed.
- the ORFalso has adjusted uracil content, as described above.
- at least one codon in the ORF of the mRNA encoding the IL-22 polypeptideis substituted with an alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set.
- the adjusted uracil content, IL-22 polypeptide-encoding ORF of the modified uracil-comprising mRNAexhibits expression levels of IL-22 when administered to a mammalian cell that are higher than expression levels of IL-22 from the corresponding wild-type mRNA.
- the mammalian cellis a mouse cell, a rat cell, or a rabbit cell.
- the mammalian cellis a monkey cell or a human cell.
- the human cellis a HeLa cell, a BJ fibroblast cell, or a peripheral blood mononuclear cell (PBMC).
- PBMCperipheral blood mononuclear cell
- IL-22is expressed at a level higher than expression levels of IL-22 from the corresponding wild-type mRNA when the mRNA is administered to a mammalian cell in vivo.
- the mRNAis administered to mice, rabbits, rats, monkeys, or humans. In one embodiment, mice are null mice.
- the mRNAis administered to mice in an amount of about 0.01 mg/kg, about 0.05 mg/kg, about 0.1 mg/kg, or 0.2 mg/kg or about 0.5 mg/kg.
- the mRNAis administered intravenously or intramuscularly.
- the IL-22 polypeptideis expressed when the mRNA is administered to a mammalian cell in vitro. In some embodiments, the expression is increased by at least about 2-fold, at least about 5-fold, at least about 10-fold, at least about 50-fold, at least about 500- fold, at least about 1500-fold, or at least about 3000-fold.
- the expressionis increased by at least about 10%, about 20%, about 30%, about 40%, about 50%, 60%, about 70%, about 80%, about 90%, or about 100%.
- adjusted uracil content, IL-22 polypeptide-encoding ORF of the modified uracil-comprising mRNAexhibits increased stability.
- the mRNAexhibits increased stability in a cell relative to the stability of a corresponding wild-type mRNA under the same conditions.
- the mRNAexhibits increased stability including resistance to nucleases, thermal stability, and/or increased stabilization of secondary structure.
- increased stability exhibited by the mRNAis measured by determining the half-life of the mRNA (e.g., in a plasma, serum, cell, or tissue sample) and/or determining the area under the curve (AUC) of the protein expression by the mRNA over time (e.g., in vitro or in vivo).
- An mRNAis identified as having increased stability if the half-life and/or the AUC is greater than the half-life and/or the AUC of a corresponding wild-type mRNA under the same conditions.
- the mRNA of the present inventioninduces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by a corresponding wild-type mRNA under the same conditions.
- the mRNA of the present disclosureinduces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by an mRNA that encodes an IL-22 polypeptide but does not comprise modified uracil under the same conditions, or relative to the immune response induced by an mRNA that encodes for an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content under the same conditions.
- the innate immune responsecan be manifested by increased expression of pro- inflammatory cytokines, activation of intracellular PRRs (RIG-I, MDA5, etc), cell death, and/or termination or reduction in protein translation.
- a reduction in the innate immune responsecan be measured by expression or activity level of Type 1 interferons (e.g., IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , and IFN- ⁇ ) or the expression of interferon-regulated genes such as the toll-like receptors (e.g., TLR7 and TLR8), and/or by decreased cell death following one or more administrations of the mRNA of this disclosure into a cell.
- Type 1 interferonse.g., IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , and IFN- ⁇
- interferon-regulated genese.g., TLR7 and TLR8
- the expression of Type-1 interferons by a mammalian cell in response to the mRNA of the present disclosureis reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99%, 99.9%, or greater than 99.9% relative to a corresponding wild-type mRNA, to an mRNA that encodes an IL-22 polypeptide but does not comprise modified uracil, or to an mRNA that encodes an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content.
- the interferonis IFN- ⁇ .
- cell death frequency caused by administration of mRNA of the present disclosure to a mammalian cellis 10%, 25%, 50%, 75%, 85%, 90%, 95%, or over 95% less than the cell death frequency observed with a corresponding wild-type mRNA, an mRNA that encodes for an IL-22 polypeptide but does not comprise modified uracil, or an mRNA that encodes for an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content.
- the mammalian cellis a BJ fibroblast cell. In other embodiments, the mammalian cell is a splenocyte.
- the mammalian cellis that of a mouse or a rat. In other embodiments, the mammalian cell is that of a human. In one embodiment, the mRNA of the present disclosure does not substantially induce an innate immune response of a mammalian cell into which the mRNA is introduced. 11. Methods for Modifying Polynucleotides The disclosure includes modified polynucleotides comprising a polynucleotide described herein (e.g., a polynucleotide, e.g. mRNA, comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein).
- a polynucleotide described hereine.g., a polynucleotide, e.g. mRNA, comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein.
- modified polynucleotidescan be chemically modified and/or structurally modified.
- modified polynucleotidescan be referred to as "modified polynucleotides.”
- the present disclosureprovides for modified nucleosides and nucleotides of a polynucleotide (e.g., RNA polynucleotides, such as mRNA polynucleotides) encoding an IL- 22 polypeptide.
- nucleosiderefers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as "nucleobase").
- organic basee.g., a purine or pyrimidine
- nucleobasealso referred to herein as “nucleobase”
- nucleotiderefers to a nucleoside including a phosphate group. Modified nucleotides can be synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Polynucleotides can comprise a region or regions of linked nucleosides.
- Such regionscan have variable backbone linkages.
- the linkagescan be standard phosphodiester linkages, in which case the polynucleotides would comprise regions of nucleotides.
- the modified polynucleotides disclosed hereincan comprise various distinct modifications.
- the modified polynucleotidescontain one, two, or more (optionally different) nucleoside or nucleotide modifications.
- a modified polynucleotide, introduced to a cellcan exhibit one or more desirable properties, e.g., improved protein expression, reduced immunogenicity, or reduced degradation in the cell, as compared to an unmodified polynucleotide.
- a polynucleotide of the present inventione.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22- containing fusion protein
- a "structural" modificationis one in which two or more linked nucleosides are inserted, deleted, duplicated, inverted or randomized in a polynucleotide without significant chemical modification to the nucleotides themselves. Because chemical bonds will necessarily be broken and reformed to effect a structural modification, structural modifications are of a chemical nature and hence are chemical modifications. However, structural modifications will result in a different sequence of nucleotides.
- compositions of the present disclosurecomprise, in some embodiments, at least one nucleic acid (e.g., RNA) having an open reading frame encoding IL-22 (e.g., SEQ ID NO: 12), wherein the nucleic acid comprises nucleotides and/or nucleosides that can be standard (unmodified) or modified as is known in the art.
- nucleic acide.g., RNA
- SEQ ID NO: 12open reading frame encoding IL-22
- nucleotides and nucleosides of the present disclosurecomprise modified nucleotides or nucleosides.
- modified nucleotides and nucleosidescan be naturally-occurring modified nucleotides and nucleosides or non-naturally occurring modified nucleotides and nucleosides.
- modificationscan include those at the sugar, backbone, or nucleobase portion of the nucleotide and/or nucleoside as are recognized in the art.
- a naturally-occurring modified nucleotide or nucleotide of the disclosureis one as is generally known or recognized in the art.
- Non-limiting examples of such naturally occurring modified nucleotides and nucleotidescan be found, inter alia, in the widely recognized MODOMICS database.
- a non-naturally occurring modified nucleotide or nucleoside of the disclosureis one as is generally known or recognized in the art.
- Non-limiting examples of such non-naturally occurring modified nucleotides and nucleosidescan be found, inter alia, in published US application Nos.
- RNAe.g., mRNA
- at least one RNA (e.g., mRNA) of the present disclosureis not chemically modified and comprises the standard ribonucleotides consisting of adenosine, guanosine, cytosine and uridine.
- nucleotides and nucleosides of the present disclosurecomprise standard nucleoside residues such as those present in transcribed RNA (e.g. A, G, C, or U). In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard deoxyribonucleosides such as those present in DNA (e.g. dA, dG, dC, or dT).
- nucleic acids of the disclosurecan comprise standard nucleotides and nucleosides, naturally-occurring nucleotides and nucleosides, non-naturally-occurring nucleotides and nucleosides, or any combination thereof.
- Nucleic acids of the disclosuree.g., DNA nucleic acids and RNA nucleic acids, such as mRNA nucleic acids
- in some embodimentscomprise various (more than one) different types of standard and/or modified nucleotides and nucleosides.
- a particular region of a nucleic acidcontains one, two or more (optionally different) types of standard and/or modified nucleotides and nucleosides.
- a modified RNA nucleic acide.g., a modified mRNA nucleic acid
- introduced to a cell or organismexhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
- a modified RNA nucleic acid(e.g., a modified mRNA nucleic acid), introduced into a cell or organism, may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
- Nucleic acidse.g., RNA nucleic acids, such as mRNA nucleic acids
- nucleic acide.g., RNA nucleic acids, such as mRNA nucleic acids.
- a “nucleoside”refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as “nucleobase”).
- nucleotiderefers to a nucleoside, including a phosphate group.
- Modified nucleotidesmay by synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides.
- Nucleic acidscan comprise a region or regions of linked nucleosides. Such regions may have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the nucleic acids would comprise regions of nucleotides.
- Modified nucleotide base pairingencompasses not only the standard adenosine- thymine, adenosine-uracil, or guanosine-cytosine base pairs, but also base pairs formed between nucleotides and/or modified nucleotides comprising non-standard or modified bases, wherein the arrangement of hydrogen bond donors and hydrogen bond acceptors permits hydrogen bonding between a non-standard base and a standard base or between two complementary non-standard base structures, such as, for example, in those nucleic acids having at least one chemical modification.
- non-standard base pairingis the base pairing between the modified nucleotide inosine and adenine, cytosine or uracil.
- modified nucleobases in nucleic acidscomprise N1-methyl-pseudouridine (m1 ⁇ ), 1-ethyl- pseudouridine (e1 ⁇ ), 5-methoxy-uridine (mo5U), 5-methyl-cytidine (m5C), and/or pseudouridine ( ⁇ ).
- modified nucleobases in nucleic acidscomprise 5-methoxymethyl uridine, 5-methylthio uridine, 1-methoxymethyl pseudouridine, 5-methyl cytidine, and/or 5-methoxy cytidine.
- the polyribonucleotideincludes a combination of at least two (e.g., 2, 3, 4 or more) of any of the aforementioned modified nucleobases, including but not limited to chemical modifications.
- a RNA nucleic acid of the disclosurecomprises N1-methyl- pseudouridine (m1 ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid.
- a RNA nucleic acid of the disclosurecomprises N1-methyl- pseudouridine (m1 ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid.
- a RNA nucleic acid of the disclosurecomprises pseudouridine ( ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid.
- a RNA nucleic acid of the disclosurecomprises pseudouridine ( ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid.
- a RNA nucleic acid of the disclosurecomprises uridine at one or more or all uridine positions of the nucleic acid.
- nucleic acidse.g., RNA nucleic acids, such as mRNA nucleic acids
- nucleic acidsare uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification.
- a nucleic acidcan be uniformly modified with N1-methyl-pseudouridine, meaning that all uridine residues in the mRNA sequence are replaced with N1-methyl-pseudouridine.
- nucleic acidcan be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above.
- the nucleic acids of the present disclosuremay be partially or fully modified along the entire length of the molecule.
- one or more or all or a given type of nucleotidee.g., purine or pyrimidine, or any one or more or all of A, G, U, C
- nucleotides X in a nucleic acid of the present disclosureare modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C or A+G+C.
- the nucleic acidmay contain from about 1% to about 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, i.e., any one or more of A, G, U or C) or any intervening percentage (e.g., from 1% to 20%, from 1% to 25%, from 1% to 50%, from 1% to 60%, from 1% to 70%, from 1% to 80%, from 1% to 90%, from 1% to 95%, from 10% to 20%, from 10% to 25%, from 10% to 50%, from 10% to 60%, from 10% to 70%, from 10% to 80%, from 10% to 90%, from 10% to 95%, from 10% to 100%, from 20% to 25%, from 20% to 50%, from 20% to 60%, from 20% to 70%, from 20% to 80%, from 20% to 90%, from 20% to 95%, from 20% to 100%, from 50% to 60%, from 50% to 70%, from 50% to 80%, from 50% to 90%, from 50% to 95%, from 50% to 100%, from 70% to
- the nucleic acidsmay contain at a minimum 1% and at maximum 100% modified nucleotides, or any intervening percentage, such as at least 5% modified nucleotides, at least 10% modified nucleotides, at least 25% modified nucleotides, at least 50% modified nucleotides, at least 80% modified nucleotides, or at least 90% modified nucleotides.
- the nucleic acidsmay contain a modified pyrimidine such as a modified uracil or cytosine.
- At least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the uracil in the nucleic acidis replaced with a modified uracil (e.g., a 5-substituted uracil).
- the modified uracilcan be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures).
- At least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the cytosine in the nucleic acidis replaced with a modified cytosine (e.g., a 5-substituted cytosine).
- the modified cytosinecan be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures).
- UTRsUntranslated Regions
- UTRsUntranslated Regions
- UTRsare nucleic acid sections of a polynucleotide before a start codon (5′ UTR) and after a stop codon (3′ UTR) that are not translated.
- a polynucleotidee.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)
- RNAribonucleic acid
- mRNAmessenger RNA
- ORFopen reading frame
- a UTRe.g., 5′ UTR or functional fragment thereof, a 3′ UTR or functional fragment thereof, or a combination thereof.
- a UTRe.g., 5′ UTR or 3′ UTR
- the UTRis homologous to the ORF encoding the IL-22 polypeptide.
- the UTRis heterologous to the ORF encoding the IL-22 polypeptide.
- the polynucleotidecomprises two or more 5′ UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences.
- the polynucleotidecomprises two or more 3′ UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences.
- the 5′ UTR or functional fragment thereof, 3′ UTR or functional fragment thereof, or any combination thereofis sequence optimized.
- the 5′UTR or functional fragment thereof, 3′ UTR or functional fragment thereof, or any combination thereofcomprises at least one chemically modified nucleobase, e.g., N1-methylpseudouracil or 5-methoxyuracil.
- UTRscan have features that provide a regulatory role, e.g., increased or decreased stability, localization and/or translation efficiency.
- a polynucleotide comprising a UTRcan be administered to a cell, tissue, or organism, and one or more regulatory features can be measured using routine methods.
- a functional fragment of a 5′ UTR or 3′ UTRcomprises one or more regulatory features of a full length 5′ or 3′ UTR, respectively.
- Natural 5′UTRsbear features that play roles in translation initiation. They harbor signatures like Kozak sequences that are commonly known to be involved in the process by which the ribosome initiates translation of many genes. Kozak sequences have the consensus CCR(A/G)CCAUGG (SEQ ID NO: 214), where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another ‘G’.5′ UTRs also have been known to form secondary structures that are involved in elongation factor binding. By engineering the features typically found in abundantly expressed genes of specific target organs, one can enhance the stability and protein production of a polynucleotide.
- liver-expressed mRNAsuch as albumin, serum amyloid A, Apolipoprotein A/B/E, transferrin, alpha fetoprotein, erythropoietin, or Factor VIII, can enhance expression of polynucleotides in hepatic cell lines or liver.
- 5′UTR from other tissue-specific mRNA to improve expression in that tissueis possible for muscle (e.g., MyoD, Myosin, Myoglobin, Myogenin, Herculin), for endothelial cells (e.g., Tie-1, CD36), for myeloid cells (e.g., C/EBP, AML1, G-CSF, GM-CSF, CD11b, MSR, Fr-1, i- NOS), for leukocytes (e.g., CD45, CD18), for adipose tissue (e.g., CD36, GLUT4, ACRP30, adiponectin) and for lung epithelial cells (e.g., SP-A/B/C/D).
- musclee.g., MyoD, Myosin, Myoglobin, Myogenin, Herculin
- endothelial cellse.g., Tie-1, CD36
- myeloid cellse.g., C/E
- UTRsare selected from a family of transcripts whose proteins share a common function, structure, feature or property.
- an encoded polypeptidecan belong to a family of proteins (i.e., that share at least one function, structure, feature, localization, origin, or expression pattern), which are expressed in a particular cell, tissue or at some time during development.
- the UTRs from any of the genes or mRNAcan be swapped for any other UTR of the same or different family of proteins to create a new polynucleotide.
- the 5′ UTR and the 3′ UTRcan be heterologous.
- the 5′ UTRcan be derived from a different species than the 3′ UTR.
- the 3′ UTRcan be derived from a different species than the 5′ UTR.
- Co-owned International Patent Application No. PCT/US2014/021522(Publ. No. WO/2014/164253, incorporated herein by reference in its entirety) provides a listing of exemplary UTRs that can be utilized in the polynucleotide of the present invention as flanking regions to an ORF.
- Additional exemplary UTRs of the applicationinclude, but are not limited to, one or more 5′UTR and/or 3′UTR derived from the nucleic acid sequence of: a globin, such as an ⁇ - or ⁇ -globin (e.g., a Xenopus, mouse, rabbit, or human globin); a strong Kozak translational initiation signal; a CYBA (e.g., human cytochrome b-245 ⁇ polypeptide); an albumin (e.g., human albumin7); a HSD17B4 (hydroxysteroid (17- ⁇ ) dehydrogenase); a virus (e.g., a tobacco etch virus (TEV), a Venezuelan equine encephalitis virus (VEEV), a Dengue virus, a cytomegalovirus (CMV) (e.g., CMV immediate early 1 (IE1)), a hepatitis virus (e.g., hepatitis B virus), a Sindbis virus
- the 5′ UTRis selected from the group consisting of a ⁇ -globin 5′ UTR; a 5′UTR containing a strong Kozak translational initiation signal; a cytochrome b- 245 ⁇ polypeptide (CYBA) 5′ UTR; a hydroxysteroid (17- ⁇ ) dehydrogenase (HSD17B4) 5′ UTR; a Tobacco etch virus (TEV) 5′ UTR; a Vietnamese etch virus (TEV) 5′ UTR; a decielen equine encephalitis virus (TEEV) 5′ UTR; a 5′ proximal open reading frame of rubella virus (RV) RNA encoding nonstructural proteins; a Dengue virus (DEN) 5′ UTR; a heat shock protein 70 (Hsp70) 5′ UTR; a eIF4G 5′ UTR; a GLUT15′ UTR; functional fragments thereof and any combination thereof.
- CYBAcytochrome b-
- the 3′ UTRis selected from the group consisting of a ⁇ -globin 3′ UTR; a CYBA 3′ UTR; an albumin 3′ UTR; a growth hormone (GH) 3′ UTR; a VEEV 3′ UTR; a hepatitis B virus (HBV) 3′ UTR; ⁇ -globin 3′UTR; a DEN 3′ UTR; a PAV barley yellow dwarf virus (BYDV-PAV) 3′ UTR; an elongation factor 1 ⁇ 1 (EEF1A1) 3′ UTR; a manganese superoxide dismutase (MnSOD) 3′ UTR; a ⁇ subunit of mitochondrial H(+)-ATP synthase ( ⁇ -mRNA) 3′ UTR; a GLUT13′ UTR; a MEF2A 3′ UTR; a ⁇ -F1-ATPase 3′ UTR; functional fragments thereof and combinations thereof.
- Wild-type UTRs derived from any gene or mRNAcan be incorporated into the polynucleotides of this disclosure.
- a UTRcan be altered relative to a wild type or native UTR to produce a variant UTR, e.g., by changing the orientation or location of the UTR relative to the ORF; or by inclusion of additional nucleotides, deletion of nucleotides, swapping or transposition of nucleotides.
- variants of 5′ or 3′ UTRscan be utilized, for example, mutants of wild type UTRs, or variants wherein one or more nucleotides are added to or removed from a terminus of the UTR.
- one or more synthetic UTRscan be used in combination with one or more non-synthetic UTRs. See, e.g., Mandal and Rossi, Nat. Protoc.20138(3):568-82, the contents of which are incorporated herein by reference in their entirety. UTRs or portions thereof can be placed in the same orientation as in the transcript from which they were selected or can be altered in orientation or location. Hence, a 5′ and/or 3′ UTR can be inverted, shortened, lengthened, or combined with one or more other 5′ UTRs or 3′ UTRs.
- the polynucleotidecomprises multiple UTRs, e.g., a double, a triple or a quadruple 5′ UTR or 3′ UTR.
- a double UTRcomprises two copies of the same UTR either in series or substantially in series.
- a double beta-globin 3′UTRcan be used (see US2010/0129877, the contents of which are incorporated herein by reference in its entirety).
- the polynucleotides of this disclosurecan comprise combinations of features.
- the ORFcan be flanked by a 5′UTR that comprises a strong Kozak translational initiation signal and/or a 3′UTR comprising an oligo(dT) sequence for templated addition of a poly-A tail.
- a 5′UTRcan comprise a first polynucleotide fragment and a second polynucleotide fragment from the same and/or different UTRs (see, e.g., US2010/0293625, herein incorporated by reference in its entirety).
- Other non-UTR sequencescan be used as regions or subregions within the polynucleotides of this disclosure.
- introns or portions of intron sequencescan be incorporated into the polynucleotides of this disclosure.
- the polynucleotide of this disclosurecomprises an internal ribosome entry site (IRES) instead of or in addition to a UTR (see, e.g., Yakubov et al., Biochem. Biophys. Res. Commun.2010394(1):189-193, the contents of which are incorporated herein by reference in their entirety).
- IRESinternal ribosome entry site
- the polynucleotidecomprises an IRES instead of a 5′ UTR sequence.
- the polynucleotidecomprises an ORF and a viral capsid sequence.
- the polynucleotidecomprises a synthetic 5′ UTR in combination with a non-synthetic 3′ UTR.
- the UTRcan also include at least one translation enhancer polynucleotide, translation enhancer element, or translational enhancer elements (collectively, "TEE," which refers to nucleic acid sequences that increase the amount of polypeptide or protein produced from a polynucleotide.
- TEEtranslation enhancer polynucleotide, translation enhancer element, or translational enhancer elements
- the TEEcan be located between the transcription promoter and the start codon.
- the 5′ UTRcomprises a TEE.
- a TEEis a conserved element in a UTR that can promote translational activity of a nucleic acid such as, but not limited to, cap-dependent or cap-independent translation.
- a.5′ UTR sequences 5′ UTR sequencesare important for ribosome recruitment to the mRNA and have been reported to play a role in translation (Hinnebusch A, et al., (2016) Science, 352:6292: 1413-6).
- a polynucleotidee.g., mRNA, comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1-4) or an IL-22-containing fusion protein (e.g., SEQ ID NO: 20-38), which polynucleotide has a 5′ UTR that confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself.
- an IL-22 polypeptidee.g., SEQ ID NO:1-4
- an IL-22-containing fusion proteine.g., SEQ ID NO: 20-38
- a polynucleotide disclosed hereincomprises: (a) a 5′-UTR (e.g., as provided in Table 4 or a variant or fragment thereof); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as described herein), and LNP compositions comprising the same.
- the polynucleotidecomprises a 5′-UTR comprising a sequence provided in Table 4 or a variant or fragment thereof (e.g., a functional variant or fragment thereof).
- the polynucleotide having a 5′ UTR sequence provided in Table 4 or a variant or fragment thereofhas an increase in the half-life of the polynucleotide, e.g., about 1.5-20-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more.
- the increase in half lifeis about 1.5-fold or more.
- the increase in half lifeis about 2-fold or more.
- the increase in half lifeis about 3-fold or more.
- the increase in half lifeis about 4-fold or more.
- the increase in half lifeis about 5-fold or more.
- the polynucleotide having a 5′ UTR sequence provided in Table 4 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the 5′UTRresults in about 1.5-20-fold increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increase in level and/or activityis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more.
- the increase in level and/or activityis about 1.5-fold or more. In an embodiment, the increase in level and/or activity is about 2-fold or more. In an embodiment, the increase in level and/or activity is about 3-fold or more. In an embodiment, the increase in level and/or activity is about 4-fold or more. In an embodiment, the increase in level and/or activity is about 5-fold or more. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 5′ UTR, has a different 5′ UTR, or does not have a 5′ UTR described in Table 4 or a variant or fragment thereof.
- the increase in half-life of the polynucleotideis measured according to an assay that measures the half-life of a polynucleotide.
- the increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotideis measured according to an assay that measures the level and/or activity of a polypeptide.
- the 5′ UTRcomprises a sequence provided in Table 4 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 5′ UTR sequence provided in Table 4, or a variant or a fragment thereof.
- the 5′ UTRcomprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 50, SEQ ID NO: 51, SEQ ID NO: 52, SEQ ID NO: 53, SEQ ID NO: 54, SEQ ID NO: 55, SEQ ID NO: 56, SEQ ID NO: 57 or SEQ ID NO: 58.
- the 5′ UTRcomprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 50.
- the 5′ UTRcomprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 51. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 52. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 53.
- the 5′ UTRcomprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 54. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 55. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 56.
- the 5′ UTRcomprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 57. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 58. In an embodiment, the 5′ UTR comprises the sequence of SEQ ID NO:58. In an embodiment, the 5′ UTR consists of the sequence of SEQ ID NO:58. In an embodiment, a 5′ UTR sequence provided in Table 4 has a first nucleotide which is an A.
- a 5′ UTR sequence provided in Table 4has a first nucleotide which is a G.
- the 5′ UTRcomprises a variant of SEQ ID NO: 50.
- the variant of SEQ ID NO: 50comprises a nucleic acid sequence of Formula A: G G A A A U C G C A A A A (N 2 ) X (N 3 ) X C U (N 4 ) X (N 5 ) X C G C G U U A G A U U U U C U U U U A G U U U U U U C U N 6 N 7 C A A C U A G C A A G C A A G C A A G C A A G C A A G C A A G C A A G C A A G C A A G C A A G C A A A G C U U U U U G U U C U C (N 8 C C)x (SEQ ID NO: 59), wherein: (N 2 ) x is a uracil and x is
- N 2 xis a uracil and x is 0. In an embodiment (N 2 ) x is a uracil and x is 1. In an embodiment (N 2 ) x is a uracil and x is 2. In an embodiment (N 2 ) x is a uracil and x is 3. In an embodiment, (N 2 ) x is a uracil and x is 4. In an embodiment (N 2 ) x is a uracil and x is 5. In an embodiment, (N 3 ) x is a guanine and x is 0. In an embodiment, (N 3 ) x is a guanine and x is 1.
- (N 4 ) xis a cytosine and x is 0. In an embodiment, (N 4 ) x is a cytosine and x is 1. In an embodiment (N 5 ) x is a uracil and x is 0. In an embodiment (N 5 ) x is a uracil and x is 1. In an embodiment (N 5 ) x is a uracil and x is 2. In an embodiment (N 5 ) x is a uracil and x is 3. In an embodiment, (N 5 ) x is a uracil and x is 4. In an embodiment (N 5 ) x is a uracil and x is 5.
- N6is a uracil. In an embodiment, N6 is a cytosine. In an embodiment, N7 is a uracil. In an embodiment, N7 is a guanine. In an embodiment, N8 is an adenine and x is 0. In an embodiment, N8 is an adenine and x is 1. In an embodiment, N8 is a guanine and x is 0. In an embodiment, N8 is a guanine and x is 1. In an embodiment, the 5′ UTR comprises a variant of SEQ ID NO: 50.
- the variant of SEQ ID NO: 50comprises a sequence with at least 50%, 60%, 70%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 50% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 60% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 70% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 80% identity to SEQ ID NO: 50.
- the variant of SEQ ID NO: 50comprises a sequence with at least 90% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 95% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 96% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 97% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 98% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 99% identity to SEQ ID NO: 50.
- the variant of SEQ ID NO: 50comprises a uridine content of at least 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, or 80%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 10%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 20%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 30%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 40%.
- the variant of SEQ ID NO: 50comprises a uridine content of at least 50%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 60%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 70%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 80%. In an embodiment, the variant of SEQ ID NO: 50 comprises at least 2, 3, 4, 5, 6 or 7 consecutive uridines (e.g., a polyuridine tract).
- the polyuridine tract in the variant of SEQ ID NO: 50comprises at least 1-7, 2-7, 3-7, 4-7, 5-7, 6-7, 1-6, 1-5, 1-4, 1-3, 1- 2, 2-6, or 3-5 consecutive uridines. In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises 4 consecutive uridines. In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises 5 consecutive uridines. In an embodiment, the variant of SEQ ID NO: 50 comprises 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 3 polyuridine tracts.
- the variant of SEQ ID NO: 50comprises 4 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 5 polyuridine tracts. In an embodiment, one or more of the polyuridine tracts are adjacent to a different polyuridine tract. In an embodiment, each of, e.g., all, the polyuridine tracts are adjacent to each other, e.g., all of the polyuridine tracts are contiguous. In an embodiment, one or more of the polyuridine tracts are separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides.
- each of, e.g., all of, the polyuridine tractsare separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides.
- a first polyuridine tract and a second polyuridine tractare adjacent to each other.
- a subsequent, e.g., third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth, polyuridine tractis separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides from the first polyuridine tract, the second polyuridine tract, or any one of the subsequent polyuridine tracts.
- a first polyuridine tractis separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides from a subsequent polyuridine tract, e.g., a second, third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth polyuridine tract.
- one or more of the subsequent polyuridine tractsare adjacent to a different polyuridine tract.
- the 5′ UTRcomprises a Kozak sequence, e.g., a GCCRCC nucleotide sequence (SEQ ID NO: 79) wherein R is an adenine or guanine.
- the Kozak sequenceis disposed at the 3′ end of the 5′UTR sequence.
- the polynucleotidee.g., mRNA
- the LNP compositioncomprises: (i) an ionizable lipid, e.g., an amino lipid; (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-lipid.
- the LNP compositions of the disclosureare used in a method of treating metabolic disease in a subject.
- an LNP compositioncomprising a polynucleotide disclosed herein encoding an IL-22 polypeptide, e.g., as described herein, can be administered with an additional agent, e.g., as described herein.
- an additional agente.g., as described herein.
- 3′ UTR sequences 3′UTR sequenceshave been shown to influence translation, half-life, and subcellular localization of mRNAs (Mayr C., Cold Spring Harb Persp Biol 2019 Oct 1;11(10):a034728).
- a polynucleotidee.g., mRNA, comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1), which polynucleotide has a 3′ UTR that confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself.
- an IL-22 polypeptidee.g., SEQ ID NO:1
- a polynucleotide disclosed hereincomprises: (a) a 5′-UTR (e.g., as described herein); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as provided in Table 5 or a variant or fragment thereof), and LNP compositions comprising the same.
- the polynucleotidecomprises a 3′-UTR comprising a sequence provided in Table 5 or a variant or fragment thereof.
- the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more.
- the increase in half-lifeis about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereofresults in a polynucleotide with a mean half-life score of greater than 10.
- the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of Table 5 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in Table 5 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in Table 5, or a fragment thereof.
- the 3′ UTRcomprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 100, SEQ ID NO: 101, SEQ ID NO: 102, SEQ ID NO: 103, SEQ ID NO: 104, SEQ ID NO: 105, SEQ ID NO: 106, SEQ ID NO: 107, SEQ ID NO: 108, SEQ ID NO: 109, SEQ ID NO: 110, SEQ ID NO: 111, SEQ ID NO: 112, SEQ ID NO: 113, SEQ ID NO: 114, SEQ ID NO:115, SEQ ID NO:116, SEQ ID NO:117, SEQ ID NO:118, SEQ ID NO:119, SEQ ID NO:120, SEQ ID NO:121, SEQ ID NO:122, SEQ ID NO:123, SEQ ID NO:124, SEQ ID NO:125, SEQ ID NO:126, SEQ ID NO:127, SEQ ID NO:12
- the 3′ UTRcomprises the sequence of SEQ ID NO: 100, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 100.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 101, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 101.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 102, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 102.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 103, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 103.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 104, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 104.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 105, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 105.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 106, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 106.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 107, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 107.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 108, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 108.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 109, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 109.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 110, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 110.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 111, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 111.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 112, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 112.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 113, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 113.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 114, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 114.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 115, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 115.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 116, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 116.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 117, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 117.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 118, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 118.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 119, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 119.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 120, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 120.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 121, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 121.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 122, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 122.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 123, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 123.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 124, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 124.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 125, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 125.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 126, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 126.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 127, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 127.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 128, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 128.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 129, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 129.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 130, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 130.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 131, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 131.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 132, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 132.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 133, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 133.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 134, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 134.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 135, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 135.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 136, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 136.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 139, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 139.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 140, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 140.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 141, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 141.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 142, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 142.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 143, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 143.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 144, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 144.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 145, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 145.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 146, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 146.
- the 3′ UTRcomprises the sequence of SEQ ID NO: 147, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 147.
- Table 53′ UTR sequences
- the 3′ UTRcomprises a micro RNA (miRNA) binding site, e.g., as described herein, which binds to a miR present in a human cell.
- the 3′ UTRcomprises a miRNA binding site of SEQ ID NO: 212, SEQ ID NO: 174, SEQ ID NO: 152 or a combination thereof.
- the 3′ UTRcomprises a plurality of miRNA binding sites, e.g., 2, 3, 4, 5, 6, 7 or 8 miRNA binding sites.
- the plurality of miRNA binding sitescomprises the same or different miRNA binding sites.
- miR122 bsCAAACACCAUUGUCACACUCCA (SEQ ID NO: 212)
- miR-142-3p bsUCCAUAAAGUAGGAAACACUACA (SEQ ID NO: 174)
- miR-126 bsCGCAUUAUUACUCACGGUACGA (SEQ ID NO: 152)
- a polynucleotideencoding a polypeptide, wherein the polynucleotide comprises: (a) a 5′-UTR, e.g., as described herein; (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as described herein).
- an LNP compositioncomprising a polynucleotide comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1) and comprising a 3′ UTR disclosed herein comprises: (i) an ionizable lipid, e.g., an amino lipid; (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-lipid.
- the LNP compositions of the disclosureare used in a method of treating metabolic disease in a subject.
- an LNP compositioncomprising a polynucleotide disclosed herein encoding an IL-22 polypeptide, e.g., as described herein, can be administered with an additional agent, e.g., as described herein.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUC UGAGUGGGCGGC (SEQ ID NO:139) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:139.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:139 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:139 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:139 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:139 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:139.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGUCUAAGCUGGAGCCUCCUGAGAGACCUGUGUGAACUAUUGAGAAGAUCG GAACAGCUCCUUACUCUGAGGAAGUUGGUACCCCCGUGGUCUUUGAAUAAAG UCUGAGUGGGCGGC (SEQ ID NO:140) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:140.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:140 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:140 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:140 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:140 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:140.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGCAAACACCAUUGUCACACUCCAGCCUCGGUGGCCUAGCU UCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCCAGCCCCUCC UCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUCCAGUGGUCU UUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:141) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:141.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:141 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:141 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:141 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:141 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:141.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGUCCAUAAAGUAGGAAACACUACAGCUGGAGCCUCGGUG GCCUAGCUUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCC AGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUCU GAGUGGGCGGC (SEQ ID NO:142) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:142.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:142 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:142 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:142 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:142 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:142.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUC CAGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:143) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:143.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:143 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:143 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:143 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:143 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:143.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGCAAACACCAUUGUCACACUCCAGCCUCGGUGGCCUAGC UUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCAGCCCCUC CUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUCCAGUGGUCU UUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:144) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:144.
- a variant or fragment thereofe.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:144.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:144 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:144 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:144 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:144 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:144.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGUCCAUAAAGUAGGAAACACUACAGCCUCGGUGGCCUAG CUUCUUGCCCCUUGGGCCUCCAUAAAGUAGGAAACACUACAUCCCCCCAGCCC CUCCUCCCCUUCCUGCACCCGUACCCCCCGCAUUAUUACUCACGGUACGAGUG GUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:145) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:145.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:145 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:145 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:145 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:145 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:145.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGUCCAUAAAGUAGGAAACACUACAGCUGGAGCCUCGGUG GCCUAGCUUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCC AGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUCU GAGUGGGCGGC (SEQ ID NO:146) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:146.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:146 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:146 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:146 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:146 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:146.
- the polynucleotidecomprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUC CAGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:147) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:147.
- the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:147 or a variant or fragment thereofresults in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-lifeis about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-lifeis about 1.5-fold or more.
- the increase in half-lifeis about 2-fold or more.
- the increase in half-lifeis about 3-fold or more.
- the increase in half-lifeis about 4-fold or more.
- the increase in half-lifeis about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:147 or a variant or fragment thereofresults in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increaseis compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:147 or a variant or fragment thereof.
- the polynucleotidecomprises a 3′ UTR sequence provided in SEQ ID NO:147 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:147. 13.
- MicroRNA (miRNA) Binding SitesPolynucleotides of this disclosure can include regulatory elements, for example, microRNA (miRNA) binding sites, transcription factor binding sites, structured mRNA sequences and/or motifs, artificial binding sites engineered to act as pseudo-receptors for endogenous nucleic acid binding molecules, and combinations thereof. In some embodiments, polynucleotides including such regulatory elements are referred to as including “sensor sequences”.
- a polynucleotidee.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)
- RNAribonucleic acid
- mRNAmessenger RNA
- ORFopen reading frame
- miRNA binding site(s)provides for regulation of polynucleotides of this disclosure, and in turn, of the polypeptides encoded therefrom, based on tissue-specific and/or cell-type specific expression of naturally-occurring miRNAs.
- the present inventionalso provides pharmaceutical compositions and Formulations that comprise any of the polynucleotides described above.
- the composition or Formulationfurther comprises a delivery agent.
- the composition or Formulationcan contain a polynucleotide comprising a sequence optimized nucleic acid sequence disclosed herein which encodes a polypeptide.
- the composition or Formulationcan contain a polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a polynucleotide (e.g., an ORF) having significant sequence identity to a sequence optimized nucleic acid sequence disclosed herein which encodes a polypeptide.
- the polynucleotidefurther comprises a miRNA binding site, e.g., a miRNA binding site that binds
- a miRNAe.g., a natural-occurring miRNA
- a miRNA sequencecomprises a “seed” region, i.e., a sequence in the region of positions 2-8 of the mature miRNA.
- a miRNA seedcan comprise positions 2-8 or 2-7 of the mature miRNA.
- microRNAsderive enzymatically from regions of RNA transcripts that fold back on themselves to form short hairpin structures often termed a pre-miRNA (precursor-miRNA).
- a pre-miRNAtypically has a two-nucleotide overhang at its 3′ end, and has 3′ hydroxyl and 5′ phosphate groups.
- This precursor-mRNAis processed in the nucleus and subsequently transported to the cytoplasm where it is further processed by DICER (a RNase III enzyme), to form a mature microRNA of approximately 22 nucleotides.
- DICERa RNase III enzyme
- the mature microRNAis then incorporated into a ribonuclear particle to form the RNA-induced silencing complex, RISC, which mediates gene silencing.
- a miR referred to by number hereincan refer to either of the two mature microRNAs originating from opposite arms of the same pre- miRNA (e.g., either the 3p or 5p microRNA). All miRs referred to herein are intended to include both the 3p and 5p arms/sequences, unless particularly specified by the 3p or 5p designation.
- microRNA binding siterefers to a sequence within a polynucleotide, e.g., within a DNA or within an RNA transcript, including in the 5′UTR and/or 3′UTR, that has sufficient complementarity to all or a region of a miRNA to interact with, associate with or bind to the miRNA.
- a polynucleotide of this disclosurecomprising an ORF encoding a polypeptide of interest and further comprises one or more miRNA binding site(s).
- a 5′ UTR and/or 3′ UTR of the polynucleotidecomprises the one or more miRNA binding site(s).
- a miRNA binding site having sufficient complementarity to a miRNArefers to a degree of complementarity sufficient to facilitate miRNA-mediated regulation of a polynucleotide, e.g., miRNA-mediated translational repression or degradation of the polynucleotide.
- a miRNA binding site having sufficient complementarity to the miRNArefers to a degree of complementarity sufficient to facilitate miRNA-mediated degradation of the polynucleotide, e.g., miRNA-guided RNA- induced silencing complex (RISC)-mediated cleavage of mRNA.
- the miRNA binding sitecan have complementarity to, for example, a 19-25 nucleotide long miRNA sequence, to a 19-23 nucleotide long miRNA sequence, or to a 22 nucleotide long miRNA sequence.
- a miRNA binding sitecan be complementary to only a portion of a miRNA, e.g., to a portion less than 1, 2, 3, or 4 nucleotides of the full length of a naturally-occurring miRNA sequence, or to a portion less than 1, 2, 3, or 4 nucleotides shorter than a naturally-occurring miRNA sequence.
- Full or complete complementaritye.g., full complementarity or complete complementarity over all or a significant portion of the length of a naturally-occurring miRNA
- a miRNA binding siteincludes a sequence that has complementarity (e.g., partial or complete complementarity) with an miRNA seed sequence.
- the miRNA binding siteincludes a sequence that has complete complementarity with a miRNA seed sequence. In some embodiments, a miRNA binding site includes a sequence that has complementarity (e.g., partial or complete complementarity) with an miRNA sequence. In some embodiments, the miRNA binding site includes a sequence that has complete complementarity with a miRNA sequence. In other embodiments, the sequence is not completely complementary. In some embodiments, a miRNA binding site has complete complementarity with a miRNA sequence but for 1, 2, or 3 nucleotide substitutions, terminal additions, and/or truncations. In some embodiments, the miRNA binding site is the same length as the corresponding miRNA.
- the miRNA binding siteis one, two, three, four, five, six, seven, eight, nine, ten, eleven or twelve nucleotide(s) shorter than the corresponding miRNA at the 5′ terminus, the 3′ terminus, or both.
- the microRNA binding siteis two nucleotides shorter than the corresponding microRNA at the 5′ terminus, the 3′ terminus, or both.
- the miRNA binding sites that are shorter than the corresponding miRNAsare still capable of degrading the mRNA incorporating one or more of the miRNA binding sites or preventing the mRNA from translation.
- the miRNA binding sitebinds the corresponding mature miRNA that is part of an active RISC containing Dicer.
- binding of the miRNA binding site to the corresponding miRNA in RISCdegrades the mRNA containing the miRNA binding site or prevents the mRNA from being translated.
- the miRNA binding sitehas sufficient complementarity to miRNA so that a RISC complex comprising the miRNA cleaves the polynucleotide comprising the miRNA binding site.
- the miRNA binding sitehas imperfect complementarity so that a RISC complex comprising the miRNA induces instability in the polynucleotide comprising the miRNA binding site.
- the miRNA binding sitehas imperfect complementarity so that a RISC complex comprising the miRNA represses transcription of the polynucleotide comprising the miRNA binding site.
- the miRNA binding sitehas one, two, three, four, five, six, seven, eight, nine, ten, eleven or twelve mismatch(es) from the corresponding miRNA.
- the miRNA binding sitehas at least about ten, at least about eleven, at least about twelve, at least about thirteen, at least about fourteen, at least about fifteen, at least about sixteen, at least about seventeen, at least about eighteen, at least about nineteen, at least about twenty, or at least about twenty-one contiguous nucleotides complementary to at least about ten, at least about eleven, at least about twelve, at least about thirteen, at least about fourteen, at least about fifteen, at least about sixteen, at least about seventeen, at least about eighteen, at least about nineteen, at least about twenty, or at least about twenty-one, respectively, contiguous nucleotides of the corresponding miRNA.
- the polynucleotideBy engineering one or more miRNA binding sites into a polynucleotide of this disclosure, the polynucleotide can be targeted for degradation or reduced translation, provided the miRNA in question is available. This can reduce off-target effects upon delivery of the polynucleotide. For example, if a polynucleotide of this disclosure is not intended to be delivered to a tissue or cell but ends up is said tissue or cell, then a miRNA abundant in the tissue or cell can inhibit the expression of the gene of interest if one or multiple binding sites of the miRNA are engineered into the 5′ UTR and/or 3′ UTR of the polynucleotide.
- incorporation of one or more miRNA binding sites into an mRNA of the disclosuremay reduce the hazard of off-target effects upon nucleic acid molecule delivery and/or enable tissue-specific regulation of expression of a polypeptide encoded by the mRNA.
- incorporation of one or more miRNA binding sites into an mRNA of the disclosurecan modulate immune responses upon nucleic acid delivery in vivo.
- incorporation of one or more miRNA binding sites into an mRNA of the disclosurecan modulate accelerated blood clearance (ABC) of lipid-comprising compounds and compositions described herein.
- miRNA binding sitescan be removed from polynucleotide sequences in which they naturally occur to increase protein expression in specific tissues.
- a binding site for a specific miRNAcan be removed from a polynucleotide to improve protein expression in tissues or cells containing the miRNA.
- Regulation of expression in multiple tissuescan be accomplished through introduction or removal of one or more miRNA binding sites, e.g., one or more distinct miRNA binding sites.
- the decision whether to remove or insert a miRNA binding sitecan be made based on miRNA expression patterns and/or their profilings in tissues and/or cells in development and/or disease.
- tissues where miRNA are known to regulate mRNA, and thereby protein expressioninclude, but are not limited to, liver (miR-122), muscle (miR-133, miR-206, miR- 208), endothelial cells (miR-17-92, miR-126), myeloid cells (miR-142-3p, miR-142-5p, miR- 16, miR-21, miR-223, miR-24, miR-27), adipose tissue (let-7, miR-30c), heart (miR-1d, miR- 149), kidney (miR-192, miR-194, miR-204), and lung epithelial cells (let-7, miR-133, miR- 126).
- livermiR-122
- musclemiR-133, miR-206, miR- 208
- endothelial cellsmiR-17-92, miR-126
- myeloid cellsmiR-142-3p, miR-142-5p, miR- 16, miR-21, miR
- miRNAsare known to be differentially expressed in immune cells (also called hematopoietic cells), such as antigen presenting cells (APCs) (e.g., dendritic cells and macrophages), macrophages, monocytes, B lymphocytes, T lymphocytes, granulocytes, natural killer cells, etc.
- APCsantigen presenting cells
- Immune cell specific miRNAsare involved in immunogenicity, autoimmunity, the immune-response to infection, inflammation, as well as unwanted immune response after gene therapy and tissue/organ transplantation. Immune cells specific miRNAs also regulate many aspects of development, proliferation, differentiation and apoptosis of hematopoietic cells (immune cells).
- miR-142 and miR-146are exclusively expressed in immune cells, particularly abundant in myeloid dendritic cells. It has been demonstrated that the immune response to a polynucleotide can be shut-off by adding miR- 142 binding sites to the 3′-UTR of the polynucleotide, enabling more stable gene transfer in tissues and cells.
- miR-142efficiently degrades exogenous polynucleotides in antigen presenting cells and suppresses cytotoxic elimination of transduced cells (e.g., Annoni A et al., blood, 2009, 114, 5152-5161; Brown BD, et al., Nat med.2006, 12(5), 585-591; Brown BD, et al., blood, 2007, 110(13): 4144-4152, each of which is incorporated herein by reference in its entirety).
- An antigen-mediated immune responsecan refer to an immune response triggered by foreign antigens, which, when entering an organism, are processed by the antigen presenting cells and displayed on the surface of the antigen presenting cells.
- T cellscan recognize the presented antigen and induce a cytotoxic elimination of cells that express the antigen.
- Introducing one or more (e.g., one, two, or three) miR-142 binding sites into the 5′ UTR and/or 3′UTR of a polynucleotide of this disclosurecan selectively repress gene expression in antigen presenting cells through miR-142 mediated degradation, limiting antigen presentation in antigen presenting cells (e.g., dendritic cells) and thereby preventing antigen-mediated immune response after the delivery of the polynucleotide.
- the polynucleotideis then stably expressed in target tissues or cells without triggering cytotoxic elimination.
- polynucleotides of this disclosurecontain two or more (e.g., two, three, four or more) miR bindings sites from: (i) the group consisting of miR-142, miR-144, miR-150, miR-155 and miR-223 (which are expressed in many hematopoietic cells); or (ii) the group consisting of miR-142, miR150, miR-16 and miR-223 (which are expressed in B cells); or the group consisting of miR-223, miR-451, miR-26a, miR-16 (which are expressed in progenitor hematopoietic cells).
- miR-142, miR-144, miR-150, miR-155 and miR-223which are expressed in many hematopoietic cells
- miR-142, miR150, miR-16 and miR-223which are expressed in B cells
- miR-223, miR-451, miR-26a, miR-16which are expressed in progenitor hema
- miR-142 and miR-126may also be beneficial to combine various miRs such that multiple cell types of interest are targeted at the same time (e.g., miR-142 and miR-126 to target many cells of the hematopoietic lineage and endothelial cells).
- polynucleotides of this disclosurecomprise two or more (e.g., two, three, four or more) miRNA bindings sites, wherein: (i) at least one of the miRs targets cells of the hematopoietic lineage (e.g., miR-142, miR-144, miR-150, miR-155 or miR-223) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or (ii) at least one of the miRs targets B cells (e.g., miR-142, miR150, miR-16 or miR-223) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or (iii) at least one of the miRs targets progenitor hematopoietic cells (e.g., miR
- polynucleotides of the present inventioncan comprise one or more miRNA binding sequences that bind to one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro-inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells).
- miRNA binding sequencesthat bind to one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro-inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells).
- incorporation into an mRNA of one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro- inflammatory cytokines and/or chemokinesreduces or inhibits immune cell activation (e.g., B cell activation, as measured by frequency of activated B cells) and/or cytokine production (e.g., production of IL-6, IFN- ⁇ and/or TNF ⁇ ).
- incorporation into an mRNA of one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro- inflammatory cytokines and/or chemokinescan reduce or inhibit an anti-drug antibody (ADA) response against a protein of interest encoded by the mRNA.
- ADAanti-drug antibody
- polynucleotides of this disclosurecan comprise one or more miR binding sequences that bind to one or more miRNAs expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro-inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells).
- incorporation into an mRNA of one or more miR binding sitesreduces or inhibits accelerated blood clearance (ABC) of the lipid-comprising compound or composition for use in delivering the mRNA.
- incorporation of one or more miR binding sites into an mRNAreduces serum levels of anti-PEG anti-IgM (e.g, reduces or inhibits the acute production of IgMs that recognize polyethylene glycol (PEG) by B cells) and/or reduces or inhibits proliferation and/or activation of plasmacytoid dendritic cells following administration of a lipid-comprising compound or composition comprising the mRNA.
- serum levels of anti-PEG anti-IgMe.g, reduces or inhibits the acute production of IgMs that recognize polyethylene glycol (PEG) by B cells
- PEGpolyethylene glycol
- miR sequencesmay correspond to any known microRNA expressed in immune cells, including but not limited to those taught in US Publication US2005/0261218 and US Publication US2005/0059005, the contents of which are incorporated herein by reference in their entirety.
- Non-limiting examples of miRs expressed in immune cellsinclude those expressed in spleen cells, myeloid cells, dendritic cells, plasmacytoid dendritic cells, B cells, T cells and/or macrophages.
- miR-142-3p, miR-142-5p, miR-16, miR-21, miR-223, miR-24 and miR-27are expressed in myeloid cells
- miR-155is expressed in dendritic cells
- miR-146is upregulated in macrophages upon TLR stimulation
- miR-126is expressed in plasmacytoid dendritic cells.
- the miR(s)is expressed abundantly or preferentially in immune cells.
- miR-142miR-142-3p and/or miR-142-5p
- miR-126miR-126- 3p and/or miR-126-5p
- miR-146miR-146-3p and/or miR-146-5p
- miR-155miR-155- 3p and/or miR155-5p
- the polynucleotide of this disclosurecomprises three copies of the same miRNA binding site.
- use of three copies of the same miR binding sitecan exhibit beneficial properties as compared to use of a single miRNA binding site.
- the polynucleotide of this disclosurecomprises two or more (e.g., two, three, four) copies of at least two different miR binding sites expressed in immune cells.
- the polynucleotide of this disclosurecomprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-142-3p.
- the polynucleotide of this disclosurecomprises binding sites for miR-142-3p and miR-155 (miR-155-3p or miR-155- 5p), miR-142-3p and miR-146 (miR-146-3 or miR-146-5p), or miR-142-3p and miR-126 (miR-126-3p or miR-126-5p).
- the polynucleotide of this disclosurecomprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-126-3p.
- the polynucleotide of this disclosurecomprises binding sites for miR-126-3p and miR-155 (miR-155-3p or miR-155- 5p), miR-126-3p and miR-146 (miR-146-3p or miR-146-5p), or miR-126-3p and miR-142 (miR-142-3p or miR-142-5p).
- the polynucleotide of this disclosurecomprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-142-5p.
- the polynucleotide of this disclosurecomprises binding sites for miR-142-5p and miR-155 (miR-155-3p or miR-155- 5p), miR-142-5p and miR-146 (miR-146-3 or miR-146-5p), or miR-142-5p and miR-126 (miR-126-3p or miR-126-5p).
- the polynucleotide of this disclosurecomprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-155-5p.
- the polynucleotide of this disclosurecomprises binding sites for miR-155-5p and miR-142 (miR-142-3p or miR-142- 5p), miR-155-5p and miR-146 (miR-146-3 or miR-146-5p), or miR-155-5p and miR-126 (miR-126-3p or miR-126-5p).
- a polynucleotide of this disclosurecomprises a miRNA binding site, wherein the miRNA binding site comprises one or more nucleotide sequences selected from Table 6, including one or more copies of any one or more of the miRNA binding site sequences.
- a polynucleotide of this disclosurefurther comprises at least one, two, three, four, five, six, seven, eight, nine, ten, or more of the same or different miRNA binding sites selected from Table 6, including any combination thereof.
- the miRNA binding sitebinds to miR-142 or is complementary to miR-142.
- the miR-142comprises SEQ ID NO:172.
- the miRNA binding sitebinds to miR-142-3p or miR-142-5p.
- the miR-142-3p binding sitecomprises SEQ ID NO:174.
- the miR-142-5p binding sitecomprises SEQ ID NO:210.
- the miRNA binding sitecomprises a nucleotide sequence at least 80%, at least 85%, at least 90%, at least 95%, or 100% identical to SEQ ID NO:174 or SEQ ID NO:210. In some embodiments, the miRNA binding site binds to miR-126 or is complementary to miR-126. In some embodiments, the miR-126 comprises SEQ ID NO: 150. In some embodiments, the miRNA binding site binds to miR-126-3p or miR-126-5p. In some embodiments, the miR-126-3p binding site comprises SEQ ID NO: 152. In some embodiments, the miR-126-5p binding site comprises SEQ ID NO: 154.
- the miRNA binding sitecomprises a nucleotide sequence at least 80%, at least 85%, at least 90%, at least 95%, or 100% identical to SEQ ID NO: 152 or SEQ ID NO: 154.
- the 3′ UTRcomprises two miRNA binding sites, wherein a first miRNA binding site binds to miR-142 and a second miRNA binding site binds to miR-126.
- a miRNA binding siteis inserted in the polynucleotide of this disclosure in any position of the polynucleotide (e.g., the 5′ UTR and/or 3′ UTR).
- the 5′ UTRcomprises a miRNA binding site.
- the 3′ UTRcomprises a miRNA binding site.
- the 5′ UTR and the 3′ UTRcomprise a miRNA binding site.
- the insertion site in the polynucleotidecan be anywhere in the polynucleotide as long as the insertion of the miRNA binding site in the polynucleotide does not interfere with the translation of a functional polypeptide in the absence of the corresponding miRNA; and in the presence of the miRNA, the insertion of the miRNA binding site in the polynucleotide and the binding of the miRNA binding site to the corresponding miRNA are capable of degrading the polynucleotide or preventing the translation of the polynucleotide.
- a miRNA binding siteis inserted in at least about 30 nucleotides downstream from the stop codon of an ORF in a polynucleotide of this disclosure comprising the ORF. In some embodiments, a miRNA binding site is inserted in at least about 10 nucleotides, at least about 15 nucleotides, at least about 20 nucleotides, at least about 25 nucleotides, at least about 30 nucleotides, at least about 35 nucleotides, at least about 40 nucleotides, at least about 45 nucleotides, at least about 50 nucleotides, at least about 55 nucleotides, at least about 60 nucleotides, at least about 65 nucleotides, at least about 70 nucleotides, at least about 75 nucleotides, at least about 80 nucleotides, at least about 85 nucleotides, at least about 90 nucleotides, at least about 95 nucleotides, or at least about 100 nucle
- a miRNA binding siteis inserted in about 10 nucleotides to about 100 nucleotides, about 20 nucleotides to about 90 nucleotides, about 30 nucleotides to about 80 nucleotides, about 40 nucleotides to about 70 nucleotides, about 50 nucleotides to about 60 nucleotides, about 45 nucleotides to about 65 nucleotides downstream from the stop codon of an ORF in a polynucleotide of this disclosure.
- a miRNA binding siteis inserted within the 3′ UTR immediately following the stop codon of the coding region within the polynucleotide of this disclosure, e.g., mRNA.
- a miRNA binding siteis inserted immediately following the final stop codon. In some embodiments, a miRNA binding site is inserted further downstream of the stop codon, in which case there are 3′ UTR bases between the stop codon and the miR binding site(s). In some embodiments, one or more miRNA binding sites can be positioned within the 5′ UTR at one or more possible insertion sites.
- a codon optimized open reading frame encoding a polypeptide of interestcomprises a stop codon and the at least one microRNA binding site is located within the 3′ UTR 1-100 nucleotides after the stop codon.
- the codon optimized open reading frame encoding the polypeptide of interestcomprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR 30-50 nucleotides after the stop codon.
- the codon optimized open reading frame encoding the polypeptide of interestcomprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR at least 50 nucleotides after the stop codon.
- the codon optimized open reading frame encoding the polypeptide of interestcomprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR immediately after the stop codon, or within the 3′ UTR 15-20 nucleotides after the stop codon or within the 3′ UTR 70-80 nucleotides after the stop codon.
- the 3′ UTRcomprises more than one miRNA binding site (e.g., 2-4 miRNA binding sites), wherein there can be a spacer region (e.g., of 10-100, 20-70 or 30-50 nucleotides in length) between each miRNA binding site.
- the 3′ UTRcomprises a spacer region between the end of the miRNA binding site(s) and the poly A tail nucleotides.
- a spacer region of 10-100, 20-70 or 30-50 nucleotides in lengthcan be situated between the end of the miRNA binding site(s) and the beginning of the poly A tail.
- a codon optimized open reading frame encoding a polypeptide of interestcomprises a start codon and the at least one microRNA binding site is located within the 5′ UTR 1-100 nucleotides before (upstream of) the start codon.
- the codon optimized open reading frame encoding the polypeptide of interestcomprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR 10-50 nucleotides before (upstream of) the start codon.
- the codon optimized open reading frame encoding the polypeptide of interestcomprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR at least 25 nucleotides before (upstream of) the start codon.
- the codon optimized open reading frame encoding the polypeptide of interestcomprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR immediately before the start codon, or within the 5′ UTR 15-20 nucleotides before the start codon or within the 5′ UTR 70-80 nucleotides before the start codon.
- the 5′ UTRcomprises more than one miRNA binding site (e.g., 2-4 miRNA binding sites), wherein there can be a spacer region (e.g., of 10-100, 20-70 or 30-50 nucleotides in length) between each miRNA binding site.
- the 3′ UTRcomprises more than one stop codon, wherein at least one miRNA binding site is positioned downstream of the stop codons.
- a 3′ UTRcan comprise 1, 2 or 3 stop codons.
- triple stop codonsinclude: UGAUAAUAG (SEQ ID NO:182), UGAUAGUAA (SEQ ID NO:183), UAAUGAUAG (SEQ ID NO:184), UGAUAAUAA (SEQ ID NO:185), UGAUAGUAG (SEQ ID NO:186), UAAUGAUGA (SEQ ID NO:187), UAAUAGUAG (SEQ ID NO:188), UGAUGAUGA (SEQ ID NO:179), UAAUAAUAA (SEQ ID NO:180), and UAGUAGUAG (SEQ ID NO:181).
- 1, 2, 3 or 4 miRNA binding sitese.g., miR-142-3p binding sites
- miRNA binding sitescan be positioned immediately adjacent to the stop codon(s) or at any number of nucleotides downstream of the final stop codon.
- these binding sitescan be positioned directly next to each other in the construct (i.e., one after the other) or, alternatively, spacer nucleotides can be positioned between each binding site.
- the 3′ UTRcomprises three stop codons with a single miR-142- 3p binding site located downstream of the 3rd stop codon.
- the polynucleotide of this disclosurecomprises a 5′ UTR, a codon optimized open reading frame encoding a polypeptide of interest, a 3′ UTR comprising the at least one miRNA binding site for a miR expressed in immune cells, and a 3′ tailing region of linked nucleosides.
- the 3′ UTRcomprises 1-4, at least two, one, two, three or four miRNA binding sites for miRs expressed in immune cells, preferably abundantly or preferentially expressed in immune cells.
- the at least one miRNA expressed in immune cellsis a miR-142- 3p microRNA binding site.
- the miR-142-3p microRNA binding sitecomprises the sequence shown in SEQ ID NO: 174.
- the at least one miRNA expressed in immune cellsis a miR-126 microRNA binding site.
- the miR-126 binding siteis a miR-126-3p binding site.
- the miR-126-3p microRNA binding sitecomprises the sequence shown in SEQ ID NO: 152.
- Non-limiting exemplary sequences for miRs to which a microRNA binding site(s) of the disclosure can bindinclude the following: miR-142-3p (SEQ ID NO: 173), miR-142-5p (SEQ ID NO: 175), miR-146-3p (SEQ ID NO: 155), miR-146-5p (SEQ ID NO: 156), miR- 155-3p (SEQ ID NO: 157), miR-155-5p (SEQ ID NO: 158), miR-126-3p (SEQ ID NO: 151), miR-126-5p (SEQ ID NO: 153), miR-16-3p (SEQ ID NO: 159), miR-16-5p (SEQ ID NO: 160), miR-21-3p (SEQ ID NO: 161), miR-21-5p (SEQ ID NO: 162), miR-223-3p (SEQ ID NO: 163), miR-223-5p (SEQ ID NO: 164), miR-24-3p (SEQ ID NO: 165), miR-24-5p
- miR sequences expressed in immune cellsare known and available in the art, for example at the University of Manchester’s microRNA database, miRBase. Sites that bind any of the aforementioned miRs can be designed based on Watson-Crick complementarity to the miR, typically 100% complementarity to the miR, and inserted into an mRNA construct of the disclosure as described herein.
- a polynucleotide of the present invention(e.g., and mRNA, e.g., the 3′ UTR thereof) can comprise at least one miRNA bindingsite to thereby reduce or inhibit accelerated blood clearance, for example by reducing or inhibiting production of IgMs, e.g., against PEG, by B cells and/or reducing or inhibiting proliferation and/or activation of pDCs, and can comprise at least one miRNA bindingsite for modulating tissue expression of an encoded protein of interest.
- miRNA gene regulationcan be influenced by the sequence surrounding the miRNA such as, but not limited to, the species of the surrounding sequence, the type of sequence (e.g., heterologous, homologous, exogenous, endogenous, or artificial), regulatory elements in the surrounding sequence and/or structural elements in the surrounding sequence.
- the miRNAcan be influenced by the 5′UTR and/or 3′UTR.
- a non- human 3′UTRcan increase the regulatory effect of the miRNA sequence on the expression of a polypeptide of interest compared to a human 3′ UTR of the same sequence type.
- other regulatory elements and/or structural elements of the 5′ UTRcan influence miRNA mediated gene regulation.
- a regulatory element and/or structural elementis a structured IRES (Internal Ribosome Entry Site) in the 5′ UTR, which is necessary for the binding of translational elongation factors to initiate protein translation. EIF4A2 binding to this secondarily structured element in the 5′-UTR is necessary for miRNA mediated gene expression (Meijer HA et al., Science, 2013, 340, 82-85, herein incorporated by reference in its entirety).
- the polynucleotides of this disclosurecan further include this structured 5′ UTR in order to enhance microRNA mediated gene regulation. At least one miRNA binding site can be engineered into the 3′ UTR of a polynucleotide of this disclosure.
- At least two, at least three, at least four, at least five, at least six, at least seven, at least eight, at least nine, at least ten, or more miRNA binding sitescan be engineered into a 3′ UTR of a polynucleotide of this disclosure.
- 1 to 10, 1 to 9, 1 to 8, 1 to 7, 1 to 6, 1 to 5, 1 to 4, 1 to 3, 2, or 1 miRNA binding sitescan be engineered into the 3′UTR of a polynucleotide of this disclosure.
- miRNA binding sites incorporated into a polynucleotide of this disclosurecan be the same or can be different miRNA sites.
- a combination of different miRNA binding sites incorporated into a polynucleotide of this disclosurecan include combinations in which more than one copy of any of the different miRNA sites are incorporated.
- miRNA binding sites incorporated into a polynucleotide of this disclosurecan target the same or different tissues in the body.
- tissue-, cell-type-, or disease-specific miRNA binding sites in the 3′-UTR of a polynucleotide of this disclosurethe degree of expression in specific cell types (e.g., myeloid cells, endothelial cells, etc.) can be reduced.
- a miRNA binding sitecan be engineered near the 5′ terminus of the 3′UTR, about halfway between the 5′ terminus and 3′ terminus of the 3′UTR and/or near the 3′ terminus of the 3′ UTR in a polynucleotide of this disclosure.
- a miRNA binding sitecan be engineered near the 5′ terminus of the 3′UTR and about halfway between the 5′ terminus and 3′ terminus of the 3′UTR.
- a miRNA binding sitecan be engineered near the 3′ terminus of the 3′UTR and about halfway between the 5′ terminus and 3′ terminus of the 3′ UTR.
- a miRNA binding sitecan be engineered near the 5′ terminus of the 3′ UTR and near the 3′ terminus of the 3′ UTR.
- a 3′UTRcan comprise 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 miRNA binding sites.
- the miRNA binding sitescan be complementary to a miRNA, miRNA seed sequence, and/or miRNA sequences flanking the seed sequence.
- the expression of a polynucleotide of this disclosurecan be controlled by incorporating at least one sensor sequence in the polynucleotide and Formulating the polynucleotide for administration.
- a polynucleotide of this disclosurecan be targeted to a tissue or cell by incorporating a miRNA binding site and Formulating the polynucleotide in a lipid nanoparticle comprising an ionizable amino lipid, including any of the lipids described herein.
- a polynucleotide of this disclosurecan be engineered for more targeted expression in specific tissues, cell types, or biological conditions based on the expression patterns of miRNAs in the different tissues, cell types, or biological conditions. Through introduction of tissue-specific miRNA binding sites, a polynucleotide of this disclosure can be designed for optimal protein expression in a tissue or cell, or in the context of a biological condition.
- a polynucleotide of this disclosurecan be designed to incorporate miRNA binding sites that either have 100% identity to known miRNA seed sequences or have less than 100% identity to miRNA seed sequences.
- a polynucleotide of this disclosurecan be designed to incorporate miRNA binding sites that have at least: 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to known miRNA seed sequences.
- the miRNA seed sequencecan be partially mutated to decrease miRNA binding affinity and as such result in reduced downmodulation of the polynucleotide.
- a miRNA sequencecan be incorporated into the loop of a stem loop.
- a miRNA seed sequencecan be incorporated in the loop of a stem loop and a miRNA binding site can be incorporated into the 5′ or 3′ stem of the stem loop.
- the miRNA sequence in the 5′ UTRcan be used to stabilize a polynucleotide of this disclosure described herein.
- a miRNA sequence in the 5′ UTR of a polynucleotide of this disclosurecan be used to decrease the accessibility of the site of translation initiation such as, but not limited to a start codon. See, e.g., Matsuda et al., PLoS One.201011(5):e15057; incorporated herein by reference in its entirety, which used antisense locked nucleic acid (LNA) oligonucleotides and exon-junction complexes (EJCs) around a start codon (-4 to +37 where the A of the AUG codons is +1) in order to decrease the accessibility to the first start codon (AUG).
- LNAantisense locked nucleic acid
- EJCsexon-junction complexes
- a polynucleotide of this disclosurecan comprise a miRNA sequence, instead of the LNA or EJC sequence described by Matsuda et al, near the site of translation initiation in order to decrease the accessibility to the site of translation initiation.
- the site of translation initiationcan be prior to, after or within the miRNA sequence.
- the site of translation initiationcan be located within a miRNA sequence such as a seed sequence or binding site.
- a polynucleotide of this disclosurecan include at least one miRNA in order to dampen the antigen presentation by antigen presenting cells.
- the miRNAcan be the complete miRNA sequence, the miRNA seed sequence, the miRNA sequence without the seed, or a combination thereof.
- a miRNA incorporated into a polynucleotide of this disclosurecan be specific to the hematopoietic system.
- a miRNA incorporated into a polynucleotide of this disclosure to dampen antigen presentationis miR-142-3p.
- a polynucleotide of this disclosurecan include at least one miRNA in order to dampen expression of the encoded polypeptide in a tissue or cell of interest.
- a polynucleotide of this disclosurecan include at least one miR-142-3p binding site, miR-142-3p seed sequence, miR-142-3p binding site without the seed, miR-142-5p binding site, miR-142-5p seed sequence, miR-142-5p binding site without the seed, miR-146 binding site, miR-146 seed sequence and/or miR-146 binding site without the seed sequence.
- a polynucleotide of this disclosurecan comprise at least one miRNA binding site in the 3′UTR in order to selectively degrade mRNA therapeutics in the immune cells to subdue unwanted immunogenic reactions caused by therapeutic delivery.
- the miRNA binding sitecan make a polynucleotide of this disclosure more unstable in antigen presenting cells.
- these miRNAsinclude miR-142-5p, miR-142-3p, miR-146a-5p, and miR-146-3p.
- a polynucleotide of this disclosurecomprises at least one miRNA sequence in a region of the polynucleotide that can interact with a RNA binding protein.
- the polynucleotide of this disclosure(e.g., a RNA, e.g., an mRNA) comprising (i) a sequence-optimized nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, variant thereof, or a fusion protein comprising the same) and (ii) a miRNA binding site (e.g., a miRNA binding site that binds to miR-142) and/or a miRNA binding site that binds to miR-126. 14.
- a sequence-optimized nucleotide sequencee.g., an ORF
- an IL-22 polypeptidee.g., the wild-type sequence, functional fragment, variant thereof, or a fusion protein comprising the same
- a miRNA binding sitee.g., a miRNA binding site that binds to miR-142
- miRNA binding sitee.g.,
- Regions having a 5′ CapThe disclosure also includes a polynucleotide that comprises both a 5′ Cap and a polynucleotide of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide to be expressed).
- the 5′ cap structure of a natural mRNAis involved in nuclear export, increasing mRNA stability and binds the mRNA Cap Binding Protein (CBP), which is responsible for mRNA stability in the cell and translation competency through the association of CBP with poly(A) binding protein to form the mature cyclic mRNA species.
- CBPmRNA Cap Binding Protein
- the capfurther assists the removal of 5′ proximal introns during mRNA splicing.
- Endogenous mRNA moleculescan be 5′-end capped generating a 5′-ppp-5′- triphosphate linkage between a terminal guanosine cap residue and the 5′-terminal transcribed sense nucleotide of the mRNA molecule.
- This 5′-guanylate capcan then be methylated to generate an N7-methyl-guanylate residue.
- the ribose sugars of the terminal and/or anteterminal transcribed nucleotides of the 5′ end of the mRNAcan optionally also be 2′-O- methylated.5′-decapping through hydrolysis and cleavage of the guanylate cap structure can target a nucleic acid molecule, such as an mRNA molecule, for degradation.
- the polynucleotides of the present inventionincorporate a cap moiety.
- polynucleotides of the present inventioncomprise a non- hydrolyzable cap structure preventing decapping and thus increasing mRNA half-life. Because cap structure hydrolysis requires cleavage of 5′-ppp-5′ phosphorodiester linkages, modified nucleotides can be used during the capping reaction.
- Vaccinia Capping Enzymefrom New England Biolabs (Ipswich, MA) can be used with ⁇ -thio- guanosine nucleotides according to the manufacturer's instructions to create a phosphorothioate linkage in the 5′-ppp-5′ cap.
- Additional modified guanosine nucleotidescan be used such as ⁇ -methyl-phosphonate and seleno-phosphate nucleotides.
- Additional modificationsinclude, but are not limited to, 2′-O-methylation of the ribose sugars of 5′-terminal and/or 5′-anteterminal nucleotides of the polynucleotide (as mentioned above) on the 2′-hydroxyl group of the sugar ring.
- Multiple distinct 5′-cap structurescan be used to generate the 5′-cap of a nucleic acid molecule, such as a polynucleotide that functions as an mRNA molecule.
- Cap analogswhich herein are also referred to as synthetic cap analogs, chemical caps, chemical cap analogs, or structural or functional cap analogs, differ from natural (i.e., endogenous, wild-type or physiological) 5′- caps in their chemical structure, while retaining cap function.
- Cap analogscan be chemically (i.e., non-enzymatically) or enzymatically synthesized and/or linked to the polynucleotides of this disclosure.
- the Anti-Reverse Cap Analog (ARCA) capcontains two guanines linked by a 5′-5′-triphosphate group, wherein one guanine contains an N7 methyl group as well as a 3′-O-methyl group (i.e., N7,3′-O-dimethyl-guanosine-5′-triphosphate-5′-guanosine (m 7 G- 3′mppp-G; which can equivalently be designated 3′ O-Me-m 7 G(5′)ppp(5′)G).
- mCAPwhich is similar to ARCA but has a 2′-O-methyl group on guanosine (i.e., N7,2′-O-dimethyl-guanosine-5′-triphosphate-5′-guanosine, m 7 Gm- ppp-G).
- the capis m 7 G-ppp-Gm-A (i.e., N7,guanosine-5′-triphosphate-2′-O- dimethyl-guanosine-adenosine).
- the capis a dinucleotide cap analog.
- the dinucleotide cap analogcan be modified at different phosphate positions with a boranophosphate group or a phosphoroselenoate group such as the dinucleotide cap analogs described in U.S. Patent No. US 8,519,110, the contents of which are herein incorporated by reference in its entirety.
- the capis a cap analog is a N7-(4-chlorophenoxyethyl) substituted dinucleotide form of a cap analog known in the art and/or described herein.
- Non- limiting examples of a N7-(4-chlorophenoxyethyl) substituted dinucleotide form of a cap analoginclude a N7-(4-chlorophenoxyethyl)-G(5′)ppp(5′)G and a N7-(4- chlorophenoxyethyl)-m 3′-O G(5′)ppp(5′)G cap analog (See, e.g., the various cap analogs and the methods of synthesizing cap analogs described in Kore et al.
- a cap analog of the present inventionis a 4- chloro/bromophenoxyethyl analog.
- Polynucleotides of this disclosurecan also be capped post-manufacture (whether IVT or chemical synthesis), using enzymes, in order to generate more authentic 5′-cap structures.
- the phrase "more authentic"refers to a feature that closely mirrors or mimics, either structurally or functionally, an endogenous or wild type feature.
- a "more authentic" featureis better representative of an endogenous, wild-type, natural or physiological cellular function and/or structure as compared to synthetic features or analogs, etc., of the prior art, or which outperforms the corresponding endogenous, wild-type, natural or physiological feature in one or more respects.
- Non-limiting examples of more authentic 5′cap structures of the present inventionare those that, among other things, have enhanced binding of cap binding proteins, increased half-life, reduced susceptibility to 5′ endonucleases and/or reduced 5′decapping, as compared to synthetic 5′cap structures known in the art (or to a wild-type, natural or physiological 5′cap structure).
- recombinant Vaccinia Virus Capping Enzyme and recombinant 2′-O-methyltransferase enzymecan create a canonical 5′-5′-triphosphate linkage between the 5′-terminal nucleotide of a polynucleotide and a guanine cap nucleotide wherein the cap guanine contains an N7 methylation and the 5′- terminal nucleotide of the mRNA contains a 2′-O-methyl.
- Cap1 structureis termed the Cap1 structure.
- Cap structuresinclude, but are not limited to, 7mG(5′)ppp(5′)N1pN2p (cap 0), 7mG(5′)ppp(5′)N1mpNp (cap 1), and 7mG(5′)- ppp(5′)N1mpN2mp (cap 2).
- Cap structuresinclude, but are not limited to, 7mG(5′)ppp(5′)N1pN2p (cap 0), 7mG(5′)ppp(5′)N1mpNp (cap 1), and 7mG(5′)- ppp(5′)N1mpN2mp (cap 2).
- capping chimeric polynucleotides post-manufacturecan be more efficient as nearly 100% of the chimeric polynucleotides can be capped.
- 5′ terminal capscan include endogenous caps or cap analogs.
- a 5′ terminal capcan comprise a guanine analog.
- Useful guanine analogsinclude, but are not limited to, inosine, N1-methyl-guanosine, 2′fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino-guanosine, LNA- guanosine, and 2-azido-guanosine.
- capsincluding those that can be used in co- transcriptional capping methods for ribonucleic acid (RNA) synthesis, using RNA polymerase, e.g., wild type RNA polymerase or variants thereof, e.g., such as those variants described herein.
- RNA polymerasee.g., wild type RNA polymerase or variants thereof, e.g., such as those variants described herein.
- capscan be added when RNA is produced in a “one- pot” reaction, without the need for a separate capping reaction.
- the methodsin some embodiments, comprise reacting a polynucleotide template with an RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript.
- capincludes the inverted G nucleotide and can comprise one or more additional nucleotides 3’ of the inverted G nucleotide, e.g., 1, 2, 3, or more nucleotides 3’ of the inverted G nucleotide and 5’ to the 5’ UTR, e.g., a 5’ UTR described herein.
- Exemplary capscomprise a sequence of GG, GA, or GGA, wherein the underlined, italicized G is an in inverted G nucleotide followed by a 5’-5’-triphosphate group.
- a capcomprises a compound of formula (C-I)
- ring B 1is a modified or unmodified Guanine; ring B 2 and ring B 3 each independently is a nucleobase or a modified nucleobase;
- X 2is O, S(O) p , NR 24 or CR 25 R 26 in which p is 0, 1, or 2;
- Y 0is O or CR 6 R 7 ;
- Y1is O, S(O) n , CR 6 R 7 , or NR 8 , in which n is 0, 1 , or 2; each --- is a single bond or absent, wherein when each --- is a single bond, Yi is O, S(O) n , CR6R7, or NR8; and when each --- is absent, Y1 is void;
- Y 2is (OP(O)R 4 ) m in which m is 0, 1, or 2, or -O-(CR 40 R 41 )u-Q 0 -(CR 42 R 43 )
- a cap analogmay include any of the cap analogs described in international publication WO 2017/066797, published on 20 April 2017, incorporated by reference herein in its entirety.
- the B 2 middle positioncan be a non-ribose molecule, such as arabinose.
- R 2is ethyl-based.
- a capcomprises the following structure: In other embodiments, a cap comprises the following structure:
- a capcomprises the following structure: In still other embodiments, a cap comprises the following structure: In some embodiments, R is an alkyl (e.g., C 1 -C 6 alkyl). In some embodiments, R is a methyl group (e.g., C 1 alkyl). In some embodiments, R is an ethyl group (e.g., C 2 alkyl). In some embodiments, a cap comprises a sequence selected from the following sequences: GAA, GAC, GAG, GAU, GCA, GCC, GCG, GCU, GGA , GGC, GGG, GGU, GUA, GUC, GUG, and GUU. In some embodiments, a cap comprises GAA.
- a capcomprises GAC. In some embodiments, a cap comprises GAG. In some embodiments, a cap comprises GAU. In some embodiments, a cap comprises GCA. In some embodiments, a cap comprises GCC. In some embodiments, a cap comprises GCG. In some embodiments, a cap comprises GCU. In some embodiments, a cap comprises GGA. In some embodiments, a cap comprises GGC. In some embodiments, a cap comprises GGG. In some embodiments, a cap comprises GGU. In some embodiments, a cap comprises GUA. In some embodiments, a cap comprises GUC. In some embodiments, a cap comprises GUG. In some embodiments, a cap comprises GUU.
- a capcomprises a sequence selected from the following sequences: m 7 GpppApA, m 7 GpppApC, m 7 GpppApG, m 7 GpppApU, m 7 GpppCpA, m 7 GpppCpC, m 7 GpppCpG, m 7 GpppCpU, m 7 GpppGpA, m 7 GpppGpC, m 7 GpppGpG, m 7 GpppGpU, m 7 GpppUpA, m 7 GpppUpC, m 7 GpppUpG, and m 7 GpppUpU.
- a capcomprises m 7 GpppApA. In some embodiments, a cap comprises m 7 GpppApC. In some embodiments, a cap comprises m 7 GpppApG. In some embodiments, a cap comprises m 7 GpppApU. In some embodiments, a cap comprises m 7 GpppCpA. In some embodiments, a cap comprises m 7 GpppCpC. In some embodiments, a cap comprises m 7 GpppCpG. In some embodiments, a cap comprises m 7 GpppCpU. In some embodiments, a cap comprises m 7 GpppGpA. In some embodiments, a cap comprises m 7 GpppGpC.
- a capcomprises m 7 GpppGpG. In some embodiments, a cap comprises m 7 GpppGpU. In some embodiments, a cap comprises m 7 GpppUpA. In some embodiments, a cap comprises m 7 GpppUpC. In some embodiments, a cap comprises m 7 GpppUpG. In some embodiments, a cap comprises m 7 GpppUpU.
- a capin some embodiments, comprises a sequence selected from the following sequences: m 7 G 3′OMe pppApA, m 7 G 3′OMe pppApC, m 7 G 3′OMe pppApG, m 7 G 3′OMe pppApU, m 7 G 3′OMe pppCpA, m 7 G 3′OMe pppCpC, m 7 G 3′OMe pppCpG, m 7 G 3′OMe pppCpU, m 7 G 3′OMe pppGpA, m 7 G 3′OMe pppGpC, m 7 G 3′OMe pppGpG, m 7 G 3′OMe pppGpU, m 7 G 3′OMe pppUpA, m 7 G 3′OMe pppUpC, m 7 G 3′OMe pppUpG, and m 7 G 3′OMe pppUpU
- a capcomprises m 7 G 3 ′ OMe pppApA. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppApC. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppApG. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppApU. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppCpA. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppCpC. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppCpG.
- a capcomprises m 7 G 3′OMe pppCpU. In some embodiments, a cap comprises m 7 G 3′OMe pppGpA. In some embodiments, a cap comprises m 7 G 3′OMe pppGpC. In some embodiments, a cap comprises m 7 G 3′OMe pppGpG. In some embodiments, a cap comprises m 7 G 3′OMe pppGpU. In some embodiments, a cap comprises m 7 G 3′OMe pppUpA. In some embodiments, a cap comprises m 7 G 3′OMe pppUpC. In some embodiments, a cap comprises m 7 G 3′OMe pppUpG.
- a capcomprises m 7 G 3′OMe pppUpU.
- a capin other embodiments, comprises a sequence selected from the following sequences: m 7 G 3′OMe pppA 2′OMe pA, m 7 G 3′OMe pppA 2′OMe pC, m 7 G 3′OMe pppA 2′OMe pG, m 7 G 3′OMe pppA 2′OMe pU, m 7 G 3′OMe pppC 2′OMe pA, m 7 G 3′OMe pppC 2′OMe pC, m 7 G 3′OMe pppC 2′OMe pG, m 7 G 3′OMe pppC 2′OMe pU, m 7 G 3′OMe pppG 2′OMe pA, m 7 G 3′OMe pppG 2′OMe pC, m 7 G 3′OMe pppG 2′OMe
- a capcomprises m 7 G 3′OMe pppA 2′OMe pA. In some embodiments, a cap comprises m 7 G 3′OMe pppA 2′OMe pC. In some embodiments, a cap comprises m 7 G 3′OMe pppA 2′OMe pG. In some embodiments, a cap comprises m 7 G 3′OMe pppA 2′OMe pU. In some embodiments, a cap comprises m 7 G 3′OMe pppC 2′OMe pA. In some embodiments, a cap comprises m 7 G3′OMepppC2′OMepC.
- a capcomprises m 7 G 3 ′ OMe pppC 2 ′ OMe pG. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppC 2 ′ OMe pU. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppG 2 ′ OMe pA. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppG 2 ′ OMe pC. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppG 2 ′ OMe pG.
- a capcomprises m 7 G 3 ′ OMe pppG 2 ′ OMe pU. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppU 2 ′ OMe pA. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppU 2 ′ OMe pC. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppU 2 ′ OMe pG. In some embodiments, a cap comprises m 7 G 3 ′ OMe pppU 2 ′ OMe pU.
- a capin still other embodiments, comprises a sequence selected from the following sequences: m 7 GpppA 2 ′ OMe pA, m 7 GpppA 2 ′ OMe pC, m 7 GpppA 2 ′ OMe pG, m 7 GpppA 2 ′ OMe pU, m 7 GpppC 2 ′ OMe pA, m 7 GpppC 2 ′ OMe pC, m 7 GpppC 2 ′ OMe pG, m 7 GpppC 2 ′ OMe pU, m 7 GpppG 2 ′ OMe pA, m 7 GpppG 2 ′ OMe pC, m 7 GpppG 2 ′ OMe pG, m 7 GpppG 2 ′ OMe pC, m 7 GpppG 2 ′ OMe pG, m 7 GpppG 2 ′ OMe
- a capcomprises m 7 GpppA 2 ′ OMe pA. In some embodiments, a cap comprises m 7 GpppA 2 ′ OMe pC. In some embodiments, a cap comprises m 7 GpppA 2 ′ OMe pG. In some embodiments, a cap comprises m 7 GpppA 2 ′ OMe pU. In some embodiments, a cap comprises m 7 GpppC 2′OMe pA. In some embodiments, a cap comprises m 7 GpppC 2′OMe pC. In some embodiments, a cap comprises m 7 GpppC 2′OMe pG.
- a trinucleotide capcomprises m 7 GpppC 2′OMe pU. In some embodiments, a cap comprises m 7 GpppG 2′OMe pA. In some embodiments, a cap comprises m 7 GpppG 2′OMe pC. In some embodiments, a cap comprises m 7 GpppG 2′OMe pG. In some embodiments, a cap comprises m 7 GpppG 2′OMe pU. In some embodiments, a cap comprises m 7 GpppU 2′OMe pA. In some embodiments, a cap comprises m 7 GpppU 2′OMe pC.
- a capcomprises m 7 GpppU 2′OMe pG. In some embodiments, a cap comprises m 7 GpppU 2′OMe pU. In some embodiments, a cap comprises m 7 Gpppm 6 A 2’Ome pG. In some embodiments, a cap comprises m 7 Gpppe 6 A 2’Ome pG. In some embodiments, a cap comprises GAG. In some embodiments, a cap comprises GCG. In some embodiments, a cap comprises GUG. In some embodiments, a cap comprises GGG.
- a capcomprises any one of the following structures:
- the capcomprises m7 GpppN 1 N 2 N 3 , where N 1 , N 2 , and N 3 are optional (i.e., can be absent or one or more can be present) and are independently a natural, a modified, or an unnatural nucleoside base.
- m7 Gis further methylated, e.g., at the 3’ position.
- the m7 Gcomprises an O-methyl at the 3’ position.
- N 1 , N 2 , and N 3if present, optionally, are independently an adenine, a uracil, a guanidine, a thymine, or a cytosine.
- one or more (or all) of N 1 , N 2 , and N 3if present, are methylated, e.g., at the 2’ position.
- one or more (or all) of N 1 , N 2 , and N 3, if presenthave an O-methyl at the 2’ position.
- the capcomprises the following structure: wherein B 1 , B 2 , and B 3 are independently a natural, a modified, or an unnatural nucleoside based; and R 1 , R 2 , R 3 , and R 4 are independently OH or O-methyl.
- R 3is O-methyl and R 4 is OH.
- R 3 and R 4are O-methyl.
- R 4is O-methyl.
- R 1is OH, R 2 is OH, R 3 is O- methyl, and R 4 is OH.
- R 1is OH, R 2 is OH, R 3 is O-methyl, and R 4 is O-methyl.
- R 1 and R 2is O-methyl, R 3 is O-methyl, and R 4 is OH. In some embodiments, at least one of R 1 and R 2 is O-methyl, R 3 is O-methyl, and R 4 is O-methyl.
- B 1 , B 3 , and B 3are natural nucleoside bases. In some embodiments, at least one of B 1 , B 2 , and B 3 is a modified or unnatural base. In some embodiments, at least one of B 1 , B 2 , and B 3 is N6-methyladenine. In some embodiments, B 1 is adenine, cytosine, thymine, or uracil.
- B 1is adenine
- B 2is uracil
- B 3is adenine
- R 1 and R 2are OH
- R 3 and R 4are O-methyl
- B 1is adenine
- B 2is uracil
- B 3is adenine
- the capcomprises a sequence selected from the following sequences: GAAA, GACA, GAGA, GAUA, GCAA, GCCA, GCGA, GCUA, GGAA, GGCA, GGGA, GGUA, GUCA, and GUUA.
- the capcomprises a sequence selected from the following sequences: GAAG, GACG, GAGG, GAUG, GCAG, GCCG, GCGG, GCUG, GGAG, GGCG, GGGG, GGUG, GUCG, GUGG, and GUUG.
- the capcomprises a sequence selected from the following sequences: GAAU, GACU, GAGU, GAUU, GCAU, GCCU, GCGU, GCUU, GGAU, GGCU, GGGU, GGUU, GUAU, GUCU, GUGU, and GUUU.
- the capcomprises a sequence selected from the following sequences: GAAC, GACC, GAGC, GAUC, GCAC, GCCC, GCGC, GCUC, GGAC, GGCC, GGGC, GGUC, GUAC, GUCC, GUGC, and GUUC.
- a capin some embodiments, comprises a sequence selected from the following sequences: m 7 G 3 ′ OMe pppApApN, m 7 G 3 ′ OMe pppApCpN, m 7 G 3 ′ OMe pppApGpN, m 7 G 3 ′ OMe pppApUpN, m 7 G 3 ′ OMe pppCpApN, m 7 G 3 ′ OMe pppCpCpN, m 7 G 3 ′ OMe pppCpGpN, m 7 G 3 ′ OMe pppCpUpN, m 7 G 3 ′ OMe pppGpApN, m 7 G 3 ′ OMe pppGpCpN, m 7 G 3 ′ OMe pppGpCpN, m 7 G 3 ′ OMe pppGpApN, m 7 G 3 ′ OMe
- a capin other embodiments, comprises a sequence selected from the following sequences: m 7 G 3 ′ OMe pppA 2 ′ OMe pApN, m 7 G 3 ′ OMe pppA 2 ′ OMe pCpN, m 7 G 3 ′ OMe pppA 2 ′ OMe pGpN, m 7 G 3 ′ OMe pppA 2 ′ OMe pUpN, m 7 G 3 ′ OMe pppC 2 ′ OMe pApN, m 7 G 3 ′ OMe pppC 2 ′ OMe pCpN, m 7 G 3 ′ OMe pppC 2 ′ OMe pGpN, m 7 G 3 ′ OMe pppC 2 ′ OMe pGpN, m 7 G 3 ′ OMe pppC 2 ′ OMe pUpN, m 7 G
- a capin still other embodiments, comprises a sequence selected from the following sequences: m 7 GpppA 2′OMe pApN, m 7 GpppA 2′OMe pCpN, m 7 GpppA 2′OMe pGpN, m 7 GpppA 2′OMe pUpN, m 7 GpppC 2′OMe pApN, m 7 GpppC 2′OMe pCpN, m 7 GpppC 2′OMe pGpN, m 7 GpppC 2 ′ OMe pUpN, m 7 GpppG 2 ′ OMe pApN, m 7 GpppG 2 ′ OMe pCpN, m 7 GpppG 2 ′ OMe pCpN, m 7 GpppG 2 ′ OMe pGpN, m 7 GpppG 2 ′ OMe pCpN, m 7 G
- a capin other embodiments, comprises a sequence selected from the following sequences: m 7 G 3′OMe pppA 2′OMe pA 2′OMe pN, m 7 G 3′OMe pppA 2′OMe pC 2′OMe pN, m 7 G3′OMepppA2′OMepG2′OMepN, m 7 G3′OMepppA2′OMepU2′OMepN, m 7 G3′OMepppC2′OMepA2′OMepN, m 7 G 3′OMe pppC 2′OMe pC 2′OMe pN, m 7 G 3′OMe pppC 2′OMe pG 2′OMe pN, m 7 G 3′OMe pppC 2′OMe pU 2′OMe pN, m 7 G 3 ′OMe pppG 2′OMe pA 2 ′ OMe
- a capin still other embodiments, comprises a sequence selected from the following sequences: m 7 GpppA 2′OMe pA 2′OMe pN, m 7 GpppA 2′OMe pC 2′OMe pN, m 7 GpppA 2′OMe pG 2′OMe pN, m 7 GpppA 2′OMe pU 2′OMe pN, m 7 GpppC 2′OMe pA 2′OMe pN, m 7 GpppC 2′OMe pC 2′OMe pN, m 7 GpppC 2′OMe pG 2′OMe pN, m 7 GpppC 2′OMe pU 2′OMe pN, m 7 GpppG 2′OMe pA 2′OMe pN, m 7 GpppG 2′OMe pC 2′OMe pN, m 7 GpppG 2′OMe pA 2
- a capcomprises GGAG. In some embodiments, a cap comprises the following structure: 15. Poly-A Tails
- the polynucleotides of the present disclosuree.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22- containing fusion protein
- terminal groups on the poly-A tailcan be incorporated for stabilization.
- a poly- A tailcomprises des-3′ hydroxyl tails.
- a long chain of adenine nucleotidescan be added to a polynucleotide such as an mRNA molecule in order to increase stability.
- a polynucleotidesuch as an mRNA molecule
- the 3′ end of the transcriptcan be cleaved to free a 3′ hydroxyl.
- poly-A polymeraseadds a chain of adenine nucleotides to the RNA.
- polyadenylationadds a poly-A tail that can be between, for example, approximately 80 to approximately 250 residues long, including approximately 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240 or 250 residues long.
- the poly-A tailis 100 nucleotides in length (SEQ ID NO:195).
- PolyA tailscan also be added after the construct is exported from the nucleus.
- terminal groups on the poly A tailcan be incorporated for stabilization.
- Polynucleotides of the present inventioncan include des-3′ hydroxyl tails.
- the polynucleotides of the present inventioncan be designed to encode transcripts with alternative polyA tail structures including histone mRNA. According to Norbury, "Terminal uridylation has also been detected on human replication-dependent histone mRNAs. The turnover of these mRNAs is thought to be important for the prevention of potentially toxic histone accumulation following the completion or inhibition of chromosomal DNA replication.
- mRNAsare distinguished by their lack of a 3 ⁇ poly(A) tail, the function of which is instead assumed by a stable stem–loop structure and its cognate stem–loop binding protein (SLBP); the latter carries out the same functions as those of PABP on polyadenylated mRNAs" (Norbury, "Cytoplasmic RNA: a case of the tail wagging the dog," Nature Reviews Molecular Cell Biology; AOP, published online 29 August 2013; doi:10.1038/nrm3645) the contents of which are incorporated herein by reference in its entirety.
- Unique poly-A tail lengthsprovide certain advantages to the polynucleotides of the present invention.
- the length of a poly-A tailwhen present, is greater than 30 nucleotides in length.
- the poly-A tailis greater than 35 nucleotides in length (e.g., at least or greater than about 35, 40, 45, 50, 55, 60, 70, 80, 90, 100, 120, 140, 160, 180, 200, 250, 300, 350, 400, 450, 500, 600, 700, 800, 900, 1,000, 1,100, 1,200, 1,300, 1,400, 1,500, 1,600, 1,700, 1,800, 1,900, 2,000, 2,500, and 3,000 nucleotides).
- the polynucleotide or region thereofincludes from about 30 to about 3,000 nucleotides (e.g., from 30 to 50, from 30 to 100, from 30 to 250, from 30 to 500, from 30 to 750, from 30 to 1,000, from 30 to 1,500, from 30 to 2,000, from 30 to 2,500, from 50 to 100, from 50 to 250, from 50 to 500, from 50 to 750, from 50 to 1,000, from 50 to 1,500, from 50 to 2,000, from 50 to 2,500, from 50 to 3,000, from 100 to 500, from 100 to 750, from 100 to 1,000, from 100 to 1,500, from 100 to 2,000, from 100 to 2,500, from 100 to 3,000, from 500 to 750, from 500 to 1,000, from 500 to 1,500, from 500 to 2,000, from 500 to 2,500, from 500 to 3,000, from 1,000 to 1,500, from 1,000 to 2,000, from 1,000 to 2,500, from 1,000 to 3,000, from 1,500 to 2,000, from 1,500 to 2,500, from 1,500 to 3,000, from from about 30 to
- the poly-A tailis designed relative to the length of the overall polynucleotide or the length of a particular region of the polynucleotide. This design can be based on the length of a coding region, the length of a particular feature or region or based on the length of the ultimate product expressed from the polynucleotides. In this context, the poly-A tail can be 10, 20, 30, 40, 50, 60, 70, 80, 90, or 100% greater in length than the polynucleotide or feature thereof. The poly-A tail can also be designed as a fraction of the polynucleotides to which it belongs.
- the poly-A tailcan be 10, 20, 30, 40, 50, 60, 70, 80, or 90% or more of the total length of the construct, a construct region or the total length of the construct minus the poly-A tail.
- engineered binding sites and conjugation of polynucleotides for Poly-A binding proteincan enhance expression.
- multiple distinct polynucleotidescan be linked together via the PABP (Poly-A binding protein) through the 3′-end using modified nucleotides at the 3′-terminus of the poly-A tail.
- Transfection experimentscan be conducted in relevant cell lines at and protein production can be assayed by ELISA at 12hr, 24hr, 48hr, 72hr and day 7 post- transfection.
- the polynucleotides of the present inventionare designed to include a polyA-G quartet region.
- the G-quartetis a cyclic hydrogen bonded array of four guanine nucleotides that can be formed by G-rich sequences in both DNA and RNA.
- the G-quartetis incorporated at the end of the poly-A tail.
- the resultant polynucleotideis assayed for stability, protein production and other parameters including half-life at various time points. It has been discovered that the polyA-G quartet results in protein production from an mRNA equivalent to at least 75% of that seen using a poly-A tail of 120 nucleotides alone (SEQ ID NO:196).
- the polyA tailcomprises an alternative nucleoside, e.g., inverted thymidine.
- PolyA tails comprising an alternative nucleoside, e.g., inverted thymidinemay be generated as described herein. For instance, mRNA constructs may be modified by ligation to stabilize the poly(A) tail.
- Ligationmay be performed using 0.5-1.5 mg/mL mRNA (5′ Cap1, 3′ A100), 50 mM Tris-HCl pH 7.5, 10 mM MgCl 2 , 1 mM TCEP, 1000 units/mL T4 RNA Ligase 1, 1 mM ATP, 20% w/v polyethylene glycol 8000, and 5:1 molar ratio of modifying oligo to mRNA.
- Modifying oligohas a sequence of 5’-phosphate- AAAAAAAAAAAAAAAAAAAAAA-(inverted deoxythymidine (idT) (SEQ ID NO:209)) (see below). Ligation reactions are mixed and incubated at room temperature ( ⁇ 22°C) for, e.g., 4 hours.
- Stable tail mRNAare purified by, e.g., dT purification, reverse phase purification, hydroxyapatite purification, ultrafiltration into water, and sterile filtration.
- the resulting stable tail-containing mRNAscontain the following structure at the 3’end, starting with the polyA region: A 100 -UCUAGAAAAAAAAAAAAAAAAAA-inverted deoxythymidine (SEQ ID NO:211).
- Modifying oligo to stabilize tail(5’-phosphate-AAAAAAAAAAAAAAAAAA- (inverted deoxythymidine)(SEQ ID NO:209)):
- the polyA tailcomprises A100-UCUAG-A20-inverted deoxy- thymidine (SEQ ID NO:211).
- the polyA tailconsists of A100-UCUAG- A20-inverted deoxy-thymidine (SEQ ID NO:211).
- Start codon regionThis disclosure also includes a polynucleotide that comprises both a start codon region and the polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein).
- the polynucleotides of the present inventioncan have regions that are analogous to or function like a start codon region.
- the translation of a polynucleotidecan initiate on a codon that is not the start codon AUG.
- Translation of the polynucleotidecan initiate on an alternative start codon such as, but not limited to, ACG, AGG, AAG, CTG/CUG, GTG/GUG, ATA/AUA, ATT/AUU, TTG/UUG (see Touriol et al. Biology of the Cell 95 (2003) 169-178 and Matsuda and Mauro PLoS ONE, 20105:11; the contents of each of which are herein incorporated by reference in its entirety).
- the translation of a polynucleotidebegins on the alternative start codon ACG.
- polynucleotide translationbegins on the alternative start codon CTG or CUG.
- the translation of a polynucleotidebegins on the alternative start codon GTG or GUG.
- Nucleotides flanking a codon that initiates translationsuch as, but not limited to, a start codon or an alternative start codon, are known to affect the translation efficiency, the length and/or the structure of the polynucleotide. (See, e.g., Matsuda and Mauro PLoS ONE, 20105:11; the contents of which are herein incorporated by reference in its entirety).
- Masking any of the nucleotides flanking a codon that initiates translationcan be used to alter the position of translation initiation, translation efficiency, length and/or structure of a polynucleotide.
- a masking agentcan be used near the start codon or alternative start codon in order to mask or hide the codon to reduce the probability of translation initiation at the masked start codon or alternative start codon.
- masking agentsinclude antisense locked nucleic acids (LNA) polynucleotides and exon- junction complexes (EJCs) (See, e.g., Matsuda and Mauro describing masking agents LNA polynucleotides and EJCs (PLoS ONE, 20105:11); the contents of which are herein incorporated by reference in its entirety).
- a masking agentcan be used to mask a start codon of a polynucleotide in order to increase the likelihood that translation will initiate on an alternative start codon.
- a masking agentcan be used to mask a first start codon or alternative start codon in order to increase the chance that translation will initiate on a start codon or alternative start codon downstream to the masked start codon or alternative start codon.
- a start codon or alternative start codoncan be located within a perfect complement for a miRNA binding site. The perfect complement of a miRNA binding site can help control the translation, length and/or structure of the polynucleotide similar to a masking agent.
- the start codon or alternative start codoncan be located in the middle of a perfect complement for a miRNA binding site.
- the start codon or alternative start codoncan be located after the first nucleotide, second nucleotide, third nucleotide, fourth nucleotide, fifth nucleotide, sixth nucleotide, seventh nucleotide, eighth nucleotide, ninth nucleotide, tenth nucleotide, eleventh nucleotide, twelfth nucleotide, thirteenth nucleotide, fourteenth nucleotide, fifteenth nucleotide, sixteenth nucleotide, seventeenth nucleotide, eighteenth nucleotide, nineteenth nucleotide, twentieth nucleotide or twenty-first nucleotide.
- the start codon of a polynucleotidecan be removed from the polynucleotide sequence in order to have the translation of the polynucleotide begin on a codon that is not the start codon.
- Translation of the polynucleotidecan begin on the codon following the removed start codon or on a downstream start codon or an alternative start codon.
- the start codon ATG or AUGis removed as the first 3 nucleotides of the polynucleotide sequence in order to have translation initiate on a downstream start codon or alternative start codon.
- the polynucleotide sequence where the start codon was removedcan further comprise at least one masking agent for the downstream start codon and/or alternative start codons in order to control or attempt to control the initiation of translation, the length of the polynucleotide and/or the structure of the polynucleotide.
- Stop Codon RegionThis disclosure also includes a polynucleotide that comprises both a stop codon region and the polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein).
- the polynucleotides of the present inventioncan include at least two stop codons before the 3′ untranslated region (UTR).
- the stop codoncan be selected from TGA, TAA and TAG in the case of DNA, or from UGA, UAA and UAG in the case of RNA.
- the polynucleotides of the present inventioninclude the stop codon TGA in the case or DNA, or the stop codon UGA in the case of RNA, and one additional stop codon.
- the addition stop codoncan be TAA or UAA.
- the polynucleotides of the present inventioninclude three consecutive stop codons, four stop codons, or more. 18.
- An Identification and Ratio Determination (IDR) sequenceis a sequence of a biological molecule (e.g., nucleic acid or protein) that, when combined with the sequence of a target biological molecule, serves to identify the target biological molecule.
- an IDR sequenceis a heterologous sequence that is incorporated within or appended to a sequence of a target biological molecule and can be used as a reference to identify the target molecule.
- a nucleic acidcomprises (i) a target sequence of interest (e.g., a coding sequence encoding a therapeutic and/or antigenic peptide or protein); and (ii) a unique IDR sequence.
- a target sequence of intereste.g., a coding sequence encoding a therapeutic and/or antigenic peptide or protein
- a unique IDR sequencee.g., an RNA species (e.g., RNA having a given coding sequence) may comprise an IDR sequence that differs from the IDR sequence of other RNA species (e.g., RNA(s) having different coding sequence(s)).
- Each IDR sequencethus identifies a particular RNA species, and so the abundance of IDR sequences may be measured to determine the abundance of each RNA species in a composition.
- RNA speciesUse of distinct IDR sequences to identify RNA species allows for analysis of multivalent RNA compositions (e.g., containing multiple RNA species) containing RNA species with similar coding sequences and/or lengths, which could otherwise be difficult to distinguish using PCR- or chromatography-based analysis of full-length RNAs.
- Each RNA species in a multivalent RNA compositionmay comprise an IDR sequence that is not a sequence isomer of an IDR sequence of another RNA species in a multivalent RNA composition (e.g., the IDR sequence does not have the same number of adenosine nucleotides, the same number of cytosine nucleotides, the same number of guanine nucleotides, and the same number of uracil nucleotides, as another IDR sequence in the composition, even if those sequences have different sequences).
- Having identical nucleotide compositionscauses sequence isomers to have the same mass, presenting a challenge to distinguishing sequence isomers using mass-based identification methods (e.g., mass spectrometry).
- Each RNA species in a multivalent RNA compositionmay comprise an IDR sequence having a mass that differs from the mass of IDR sequences of each other RNA species in a multivalent RNA composition.
- the mass of each IDR sequencemay differ from the mass of other IDR sequences by at least 9 Da, at least 25 Da, at least 25 Da, or at least 50 Da.
- Use of IDR sequences with distinct massesallows RNA fragments comprising different IDR sequences to be distinguished using mass-based analysis methods (e.g., mass spectrometry), which do not require reverse transcription, amplification, or sequencing of RNAs.
- Each RNA species in an RNA compositionmay comprises an IDR sequence with a different length.
- each IDR sequencemay have a length independently selected from 0 to 25 nucleotides.
- the length of a nucleic acidinfluences the rate at which the nucleic acid traverses a chromatography column, and so the use of IDR sequences of different lengths on different RNA species allows RNA fragments having different IDR sequences to be distinguished using chromatography-based methods (e.g., LC-UV).
- IDR sequencesmay be chosen such that no IDR sequence comprises a start codon, ‘AUG’. Lack of a start codon in an IDR sequence prevents undesired translation of nucleotide sequences within and/or downstream from the IDR sequence.
- IDR sequencesmay be chosen such that no IDR sequence comprises a recognition site for a restriction enzyme.
- no IDR sequencecomprises a recognition site for XbaI, ‘UCUAG’.
- Lack of a recognition site for a restriction enzymee.g., XbaI recognition site ‘UCUAG’) allows the restriction enzyme to be used in generating and modifying a DNA template for in vitro transcription, without affecting the IDR sequence or sequence of the transcribed RNA. 19.
- any of the polynucleotides disclosed hereincan comprise one, two, three, or all of the following elements: (a) a 5’-UTR, e.g., as described herein; (b) a coding region comprising a stop element (e.g., as described herein); (c) a 3’-UTR (e.g., as described herein) and; optionally (d) a 3’ stabilizing region, e.g., as described herein. Also disclosed herein are LNP compositions comprising the same.
- a polynucleotide of the disclosurecomprises (a) a 5’ UTR described in Table 4 or a variant or fragment thereof and (b) a coding region comprising a stop element provided herein.
- the polynucleotidefurther comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein.
- the polynucleotidefurther comprises a 3’ stabilizing region, e.g., as described herein.
- a polynucleotide of the disclosurecomprises (a) a 5’ UTR described in Table 4 or a variant or fragment thereof and (c) a 3’ UTR described in Table 5 or a variant or fragment thereof.
- the polynucleotidefurther comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein.
- the polynucleotidefurther comprises a 3’ stabilizing region, e.g., as described herein.
- a polynucleotide of the disclosurecomprises (c) a 3’ UTR described in Table 5 or a variant or fragment thereof and (b) a coding region comprising a stop element provided herein.
- the polynucleotidecomprises a sequence provided in Table 7.
- the polynucleotidefurther comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein.
- the polynucleotidefurther comprises a 3’ stabilizing region, e.g., as described herein.
- a polynucleotide of the disclosurecomprises (a) a 5’ UTR described in Table 4 or a variant or fragment thereof; (b) a coding region comprising a stop element provided herein; and (c) a 3’ UTR described in Table 5 or a variant or fragment thereof.
- the polynucleotidefurther comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein.
- the polynucleotidefurther comprises a 3’ stabilizing region, e.g., as described herein. Table 7: Exemplary 3’ UTR and stop element sequences
- a polynucleotide of the present disclosurecomprises from 5′ to 3′ end: (i) a 5′ cap such as provided above; (ii) a 5′ UTR, such as the sequences provided above; (iii) an ORF encoding an IL-22 polypeptide, wherein the ORF has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of any of SEQ ID NO:300-303; (iv) at least one stop codon; (v) a 3′ UTR, such as the sequences provided above; and (vi) a poly-A tail provided above.
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding a serum albumin polypeptide, comprises from 5′ to 3′ end: (i) a 5′ cap such as provided above; (ii) a 5′ UTR, such as the sequences provided above; (iii) an ORF encoding a human serum albumin polypeptide, wherein the ORF has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of any of SEQ ID NO:304-307; (iv) at least one stop codon; (v) a 3′ UTR, such as the sequences provided above; and (vi) a poly-A tail provided above.
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein, comprises from 5′ to 3′ end: (i) a 5′ cap such as provided above; (ii) a 5′ UTR, such as the sequences provided above; (iii) an ORF encoding a human IL-22 polypeptide or an IL-22-containing fusion protein, wherein the ORF has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of any one of SEQ ID NO:310-328; (iv) at least one stop codon; (v) a 3′ UTR, such as the sequences provided above; and (vi) a poly-A tail provided above.
- a 5′ capsuch as provided above
- a 5′ UTRsuch as the sequences provided above
- an ORFen
- the polynucleotidefurther comprises a miRNA binding site, e.g., a miRNA binding site that binds to miRNA-142.
- the 5′ UTRcomprises the miRNA binding site.
- the 3′ UTRcomprises the miRNA binding site.
- a polynucleotide of the present disclosurecomprises a nucleotide sequence encoding a polypeptide sequence at least 70%, at least 80%, at least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96% , at least 97%, at least 98%, at least 99%, or 100% identical to the protein sequence of a wild type human IL-22 (SEQ ID NO:1 or SEQ ID NO:3).
- a polynucleotide of the present disclosurecomprises a nucleotide sequence encoding a polypeptide sequence at least 70%, at least 80%, at least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96% , at least 97%, at least 98%, at least 99%, or 100% identical to the protein sequence of a human serum albumin having the amino acid sequence of SEQ ID NO:5.
- a polynucleotide of the present disclosurecomprises a nucleotide sequence encoding a polypeptide sequence at least 70%, at least 80%, at least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96% , at least 97%, at least 98%, at least 99%, or 100% identical to the protein sequence of a wild type mouse IL-22 (SEQ ID NO:2 or SEQ ID NO:4).
- a polynucleotide of the present disclosurecomprises a nucleotide sequence encoding a polypeptide sequence at least 70%, at least 80%, at least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96% , at least 97%, at least 98%, at least 99%, or 100% identical to the protein sequence of a mouse serum albumin having the amino acid sequence of SEQ ID NO:6 or SEQ ID NO:8.
- a polynucleotide of the present disclosurecomprises a nucleotide sequence encoding a polypeptide sequence at least 70%, at least 80%, at least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96% , at least 97%, at least 98%, at least 99%, or 100% identical to the protein sequence of a human IL-22 polypeptide having the amino acid sequence of SEQ ID NO:3.
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding a polypeptide, comprises (1) a 5′ cap such as provided above, for example, m 7 Gp-ppGm-A, (2) a 5′ UTR, (3) a nucleotide sequence ORF of any of SEQ ID NO:300-303, (3) a stop codon, (4) a 3′UTR, and (5) a poly-A tail provided above, for example, a poly-A tail of SEQ ID NO:195 or A100- UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding a polypeptide, comprises (1) a 5′ cap such as provided above, for example, m 7 Gp-ppGm-A, (2) a 5′ UTR, (3) a nucleotide sequence ORF of any of SEQ ID NO:304-307, (3) a stop codon, (4) a 3′UTR, and (5) a poly-A tail provided above, for example, a poly-A tail of SEQ ID NO:195 or A100- UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding a polypeptide, comprises (1) a 5′ cap such as provided above, for example, C1, (2) a 5′ UTR, (3) a nucleotide sequence ORF of any of SEQ ID NO:300-303, (3) a stop codon, (4) a 3′UTR, and (5) a poly-A tail provided above, for example, a poly-A tail of SEQ ID NO:195 or A100-UCUAG-A20- inverted deoxy-thymidine (SEQ ID NO:211).
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding a polypeptide, comprises (1) a 5′ cap such as provided above, for example, C1, (2) a 5′ UTR, (3) a nucleotide sequence ORF of any of SEQ ID NO:304-307, (3) a stop codon, (4) a 3′UTR, and (5) a poly-A tail provided above, for example, a poly-A tail of SEQ ID NO:195 or A100-UCUAG-A20- inverted deoxy-thymidine (SEQ ID NO:211).
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding a polypeptide, comprises (1) a 5′ cap such as provided above, for example, C1 or m 7 Gp-ppGm-A, (2) a 5′ UTR, (3) a nucleotide sequence ORF selected from SEQ ID NO:310-328, (3) a stop codon, (4) a 3′UTR, and (5) a poly-A tail provided above, for example, a poly-A tail of SEQ ID NO:195 or A100- UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
- SEQ ID NO:350consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO: 310, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:351consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 56, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:311, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:352consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:312, and 3′ UTR of SEQ ID NO: 139.
- SEQ ID NO:353consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:313, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:354consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 56, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:314, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:355consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 56, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:315, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:356consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 56, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:316, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:357consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 56, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:317, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:358consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO:50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:318, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:359consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:319, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:360consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:320, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:361consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:321, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:362consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:322, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:363consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:323, and 3′ UTR of SEQ ID NO: 125.
- SEQ ID NO:364consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:324, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:365consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:325, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:366consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:326, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:367consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:327, and 3′ UTR of SEQ ID NO: 108.
- SEQ ID NO:368consists from 5′ to 3′ end: 5′ UTR of SEQ ID NO: 50, serum albumin-IL-22 nucleotide ORF of SEQ ID NO:328, and 3′ UTR of SEQ ID NO: 108.
- all uracils thereinare replaced by N1-methylpseudouracil.
- all uracils thereinare replaced by N1-methylpseudouracil.
- in a construct with SEQ ID NO:351all uracils therein are replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:352, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:352, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:353, all uracils therein are replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:354, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:354, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:355, all uracils therein are replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:356, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:356 all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:357, all uracils therein are replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:358, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:358, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:359, all uracils therein are replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:362, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:362, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:363, all uracils therein are replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:364, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:364, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:365, all uracils therein are replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:366, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:366, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:367, all uracils therein are replaced by N1 methylpseudouracil.
- all uracils thereinare replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:368, all uracils therein are replaced by N1 methylpseudouracil. In certain embodiments, in a construct with SEQ ID NO:368, all uracils therein are replaced by N1 methylpseudouracil.
- a polynucleotide of the present disclosurefor example a polynucleotide comprising an mRNA nucleotide sequence encoding an IL-22 polypeptide, comprises (1) a 5′ cap such as provided above, for example, m 7 Gp-ppGm-A or C1, (2) a nucleotide sequence selected from SEQ ID NO:350-368, and (3) a poly-A tail provided above, for example, a poly A tail of ⁇ 100 residues, e.g., SEQ ID NO:195 or A100-UCUAG- A20-inverted deoxy-thymidine (SEQ ID NO:211).
- RNA constructsincluding ORFs encoding human IL-22, human serum albumin, or a fusion protein comprising serum albumin and IL-22 polypeptides 21.
- Methods of Making PolynucleotidesThe present disclosure also provides methods for making a polynucleotide of this disclosure (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide) or a complement thereof.
- a polynucleotidee.g., a RNA, e.g., an mRNA
- IVTTin vitro transcription
- a polynucleotidee.g., a RNA, e.g., an mRNA
- a polynucleotidecan be constructed by chemical synthesis using an oligonucleotide synthesizer.
- a polynucleotidee.g., a RNA, e.g., an mRNA
- encoding an IL-22 polypeptideis made by using a host cell.
- a polynucleotidee.g., a RNA, e.g., an mRNA
- encoding an IL-22 polypeptideis made by one or more combination of the IVT, chemical synthesis, host cell expression, or any other methods known in the art.
- Naturally occurring nucleosides, non-naturally occurring nucleosides, or combinations thereof,can totally or partially naturally replace occurring nucleosides present in the candidate nucleotide sequence and can be incorporated into a sequence-optimized nucleotide sequence (e.g., a RNA, e.g., an mRNA) encoding an IL-22 polypeptide.
- the resultant polynucleotidescan then be examined for their ability to produce protein and/or produce a therapeutic outcome.
- a polynucleotide disclosed hereincan be constructed using in vitro transcription.
- a polynucleotide (e.g., an mRNA) disclosed hereincan be constructed by chemical synthesis using an oligonucleotide synthesizer.
- a polynucleotide (e.g., an mRNA) disclosed hereinis made by using a host cell.
- a polynucleotide (e.g., an mRNA) disclosed hereinis made by one or more combination of the IVT, chemical synthesis, host cell expression, or any other methods known in the art.
- Naturally occurring nucleosides, non-naturally occurring nucleosides, or combinations thereofcan totally or partially naturally replace occurring nucleosides present in the candidate nucleotide sequence and can be incorporated into a sequence-optimized nucleotide sequence (e.g., an mRNA) encoding an IL-22 polypeptide.
- RNA transcripte.g., mRNA transcript
- a RNA polymerasee.g., a T7 RNA polymerase or a T7 RNA polymerase variant
- the present disclosureprovides methods of performing an IVT (in vitro transcription) reaction, comprising contacting a DNA template with the RNA polymerase (e.g., a T7 RNA polymerase, such as a T7 RNA polymerase variant) in the presence of nucleoside triphosphates and buffer under conditions that result in the production of RNA transcripts.
- RNA polymerasee.g., a T7 RNA polymerase, such as a T7 RNA polymerase variant
- capping methodse.g., co- transcriptional capping methods or other methods known in the art.
- a capping methodcomprises reacting a polynucleotide template with a T7 RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript.
- IVT conditionstypically require a purified linear DNA template containing a promoter, nucleoside triphosphates, a buffer system that includes dithiothreitol (DTT) and magnesium ions, and a RNA polymerase.
- DTTdithiothreitol
- RNA polymerasea buffer system that includes dithiothreitol
- Typical IVT reactionsare performed by incubating a DNA template with a RNA polymerase and nucleoside triphosphates, including GTP, ATP, CTP, and UTP (or nucleotide analogs) in a transcription buffer.
- a RNA transcript having a 5 ⁇ terminal guanosine triphosphateis produced from this reaction.
- a deoxyribonucleic acidis simply a nucleic acid template for RNA polymerase.
- a DNA templatemay include a polynucleotide encoding an IL-22 polypeptide.
- a DNA templatein some embodiments, includes a RNA polymerase promoter (e.g., a T7 RNA polymerase promoter) located 5' from and operably linked to polynucleotide encoding an IL-22 polypeptide.
- a DNA templatemay also include a nucleotide sequence encoding a polyadenylation (polyA) tail located at the 3' end of the gene of interest.
- Polypeptides of interestinclude, but are not limited to, biologics, antibodies, antigens (vaccines), and therapeutic proteins.
- RNA transcriptin some embodiments, is the product of an IVT reaction and, as will be understood by one of ordinary skill in the art, the DNA template for making an RNA molecule is known based on base complementarity.
- a RNA transcriptin some embodiments, is a messenger RNA (mRNA) that includes a nucleotide sequence encoding a polypeptide of interest linked to a polyA tail.
- the mRNAis modified mRNA (mmRNA), which includes at least one modified nucleotide.
- mmRNAmodified mRNA
- a nucleotideincludes a nitrogenous base, a five-carbon sugar (ribose or deoxyribose), and at least one phosphate group.
- Nucleotidesinclude nucleoside monophosphates, nucleoside diphosphates, and nucleoside triphosphates.
- a nucleoside monophosphate (NMP)includes a nucleobase linked to a ribose and a single phosphate;
- a nucleoside diphosphate (NDP)includes a nucleobase linked to a ribose and two phosphates;
- a nucleoside triphosphate (NTP)includes a nucleobase linked to a ribose and three phosphates.
- Nucleotide analogsare compounds that have the general structure of a nucleotide or are structurally similar to a nucleotide.
- Nucleotide analogsinclude an analog of the nucleobase, an analog of the sugar and/or an analog of the phosphate group(s) of a nucleotide.
- a nucleosideincludes a nitrogenous base and a 5-carbon sugar. Thus, a nucleoside plus a phosphate group yields a nucleotide.
- Nucleoside analogsare compounds that have the general structure of a nucleoside or are structurally similar to a nucleoside.
- Nucleoside analogsfor example, include an analog of the nucleobase and/or an analog of the sugar of a nucleoside.
- nucleotideincludes naturally-occurring nucleotides, synthetic nucleotides and modified nucleotides, unless indicated otherwise.
- naturally-occurring nucleotides used for the production of RNAinclude adenosine triphosphate (ATP), guanosine triphosphate (GTP), cytidine triphosphate (CTP), uridine triphosphate (UTP), and 5-methyluridine triphosphate (m 5 UTP).
- ATPadenosine triphosphate
- GTPguanosine triphosphate
- CTPcytidine triphosphate
- UTPuridine triphosphate
- m 5 UTP5-methyluridine triphosphate
- adenosine diphosphateADP
- GDPguanosine diphosphate
- CDPcytidine diphosphate
- UDPuridine diphosphate
- nucleotide analogsinclude, but are not limited to, antiviral nucleotide analogs, phosphate analogs (soluble or immobilized, hydrolyzable or non-hydrolyzable), dinucleotide, trinucleotide, tetranucleotide, e.g., a cap analog, or a precursor/substrate for enzymatic capping (vaccinia or ligase), a nucleotide labeled with a functional group to facilitate ligation/conjugation of cap or 5 ⁇ moiety (IRES), a nucleotide labeled with a 5 ⁇ PO 4 to facilitate ligation of cap or 5 ⁇ moiety, or a nucleotide labeled with
- antiviral nucleotide/nucleoside analogsinclude, but are not limited, to Ganciclovir, Entecavir, Telbivudine, Vidarabine and Cidofovir.
- Modified nucleotidesmay include modified nucleobases.
- RNA transcript(e.g., mRNA transcript) of the present disclosure may include a modified nucleobase selected from pseudouridine ( ⁇ ), 1-methylpseudouridine (m1 ⁇ ), 1- ethylpseudouridine, 2-thiouridine, 4’-thiouridine, 2-thio-1-methyl-1-deaza-pseudouridine, 2- thio-1-methyl-pseudouridine, 2-thio-5-aza-uridine , 2-thio-dihydropseudouridine, 2-thio- dihydrouridine, 2-thio-pseudouridine, 4-methoxy-2-thio-pseudouridine, 4-methoxy- pseudouridine, 4-thio-1-methyl-pseudouridine, 4-thio-pseudouridine, 5-aza-uridine, dihydropseudouridine, 5-methyluridine, 5-methoxyuridine (mo5U) and 2’-O-methyl uridine
- a RNA transcript(e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified nucleobases.
- the nucleoside triphosphates (NTPs) as provided hereinmay comprise unmodified or modified ATP, modified or unmodified UTP, modified or unmodified GTP, and/or modified or unmodified CTP.
- NTPs of an IVT reactioncomprise unmodified ATP.
- NTPs of an IVT reactioncomprise modified ATP.
- NTPs of an IVT reactioncomprise unmodified UTP.
- NTPs of an IVT reactioncomprise modified UTP.
- NTPs of an IVT reactioncomprise unmodified GTP. In some embodiments, NTPs of an IVT reaction comprise modified GTP. In some embodiments, NTPs of an IVT reaction comprise unmodified CTP. In some embodiments, NTPs of an IVT reaction comprise modified CTP.
- concentration of nucleoside triphosphates and cap analog present in an IVT reactionmay vary. In some embodiments, NTPs and cap analog are present in the reaction at equimolar concentrations. In some embodiments, the molar ratio of cap analog (e.g., trinucleotide cap) to nucleoside triphosphates in the reaction is greater than 1:1.
- the molar ratio of cap analog to nucleoside triphosphates in the reactionmay be 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1, 15:1, 20:1, 25:1, 50:1, or 100:1.
- the molar ratio of cap analog (e.g., trinucleotide cap) to nucleoside triphosphates in the reactionis less than 1:1.
- the molar ratio of cap analog (e.g., trinucleotide cap) to nucleoside triphosphates in the reactionmay be 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:15, 1:20, 1:25, 1:50, or 1:100.
- the composition of NTPs in an IVT reactionmay also vary.
- ATPmay be used in excess of GTP, CTP and UTP.
- an IVT reactionmay include 7.5 millimolar GTP, 7.5 millimolar CTP, 7.5 millimolar UTP, and 3.75 millimolar ATP.
- the same IVT reactionmay include 3.75 millimolar cap analog (e.g., trinucleotide cap).
- the molar ratio of G:C:U:A:capis 1:1:1:0.5:0.5. In some embodiments, the molar ratio of G:C:U:A:cap is 1:1:0.5:1:0.5. In some embodiments, the molar ratio of G:C:U:A:cap is 1:0.5:1:1:0.5. In some embodiments, the molar ratio of G:C:U:A:cap is 0.5:1:1:1:0.5.
- a RNA transcript(e.g., mRNA transcript) includes a modified nucleobase selected from pseudouridine ( ⁇ ), 1-methylpseudouridine (m 1 ⁇ ), 5- methoxyuridine (mo 5 U), 5-methylcytidine (m 5 C), ⁇ -thio-guanosine and ⁇ -thio-adenosine.
- a RNA transcript(e.g., mRNA transcript) includes a combination of at least two (e.g., 2, 3, 4 or more) of the foregoing modified nucleobases.
- a RNA transcript(e.g., mRNA transcript) includes pseudouridine ( ⁇ ).
- a RNA transcript(e.g., mRNA transcript) includes 1-methylpseudouridine (m 1 ⁇ ). In some embodiments, a RNA transcript (e.g., mRNA transcript) includes 5-methoxyuridine (mo 5 U). In some embodiments, a RNA transcript (e.g., mRNA transcript) includes 5-methylcytidine (m 5 C). In some embodiments, a RNA transcript (e.g., mRNA transcript) includes ⁇ -thio-guanosine. In some embodiments, a RNA transcript (e.g., mRNA transcript) includes ⁇ -thio-adenosine.
- the polynucleotidee.g., RNA polynucleotide, such as mRNA polynucleotide
- RNA polynucleotidesuch as mRNA polynucleotide
- m 1 ⁇1-methylpseudouridine
- a polynucleotidecan be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as any of those set forth above.
- the polynucleotidee.g., RNA polynucleotide, such as mRNA polynucleotide
- the buffer systemcontains tris.
- the concentration of tris used in an IVT reactionmay be at least 10 mM, at least 20 mM, at least 30 mM, at least 40 mM, at least 50 mM, at least 60 mM, at least 70 mM, at least 80 mM, at least 90 mM, at least 100 mM or at least 110 mM phosphate.
- the concentration of phosphateis 20-60 mM or 10-100 mM.
- the buffer systemcontains dithiothreitol (DTT).
- DTTdithiothreitol
- the concentration of DTT used in an IVT reactionmay be at least 1 mM, at least 5 mM, or at least 50 mM. In some embodiments, the concentration of DTT used in an IVT reaction is 1-50 mM or 5-50 mM. In some embodiments, the concentration of DTT used in an IVT reaction is 5 mM. In some embodiments, the buffer system contains magnesium.
- the molar ratio of NTP to magnesium ions (Mg 2+ ; e.g., MgCl 2 ) present in an IVT reactionis 1:1 to 1:5.
- the molar ratio of NTP to magnesium ionsmay be 1:1, 1:2, 1:3, 1:4 or 1:5.
- the molar ratio of NTP plus cap analog (e.g., trinucleotide cap, such as GAG) to magnesium ions (Mg 2+ ; e.g., MgCl 2 ) present in an IVT reactionis 1:1 to 1:5.
- the molar ratio of NTP+trinucleotide cap (e.g., GAG) to magnesium ionsmay be 1:1, 1:2, 1:3, 1:4 or 1:5.
- the buffer systemcontains Tris-HCl, spermidine (e.g., at a concentration of 1-30 mM), TRITON ® X-100 (polyethylene glycol p-(1,1,3,3- tetramethylbutyl)-phenyl ether) and/or polyethylene glycol (PEG).
- nucleoside triphosphatesNTPs
- a polymerasesuch as T7 RNA polymerase
- the RNA polymerasee.g., T7 RNA polymerase variant
- a reactione.g., an IVT reaction
- the RNA polymerasemay be present in a reaction at a concentration of 0.01 mg/mL, 0.05 mg/ml, 0.1 mg/ml, 0.5 mg/ml or 1.0 mg/ml.
- the polynucleotide of the present disclosureis an IVT polynucleotide.
- the basic components of an mRNA moleculeinclude at least a coding region, a 5′UTR, a 3′UTR, a 5′ cap and a poly-A tail.
- the IVT polynucleotides of the present disclosurecan function as mRNA but are distinguished from wild-type mRNA in their functional and/or structural design features which serve, e.g., to overcome existing problems of effective polypeptide production using nucleic-acid based therapeutics.
- the primary construct of an IVT polynucleotidecomprises a first region of linked nucleotides that is flanked by a first flanking region and a second flaking region. This first region can include, but is not limited to, the encoded IL-22 polypeptide, a serum albumin polypeptide, or an IL-22-containing fusion protein.
- the first flanking regioncan include a sequence of linked nucleosides which function as a 5’ untranslated region (UTR) such as the 5’ UTR of any of the nucleic acids encoding the native 5’ UTR of the polypeptide or a non- native 5’UTR such as, but not limited to, a heterologous 5’ UTR or a synthetic 5’ UTR.
- the IVT encoding an IL-22 polypeptidecan comprise at its 5 terminus a signal sequence region encoding one or more signal sequences.
- the flanking regioncan comprise a region of linked nucleotides comprising one or more complete or incomplete 5′ UTRs sequences.
- the flanking regioncan also comprise a 5′ terminal cap.
- the second flanking regioncan comprise a region of linked nucleotides comprising one or more complete or incomplete 3′ UTRs which can encode the native 3’ UTR of an IL-22 polypeptide, or a non-native 3’ UTR such as, but not limited to, a heterologous 3’ UTR or a synthetic 3’ UTR.
- the flanking regioncan also comprise a 3′ tailing sequence.
- the 3’ tailing sequencecan be, but is not limited to, a polyA tail, a polyA-G quartet and/or a stem loop sequence.
- IVT polynucleotide architectureand methods of making a polynucleotide are disclosed in International PCT application WO 2017/201325, filed on 18 May 2017, the entire contents of which are hereby incorporated by reference.
- Chemical synthesis Standard methodscan be applied to synthesize an isolated polynucleotide sequence encoding an isolated polypeptide of interest, such as a polynucleotide of this disclosure (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide).
- a single DNA or RNA oligomer containing a codon-optimized nucleotide sequence coding for the particular isolated polypeptidecan be synthesized.
- RNAe.g., an mRNA
- a polynucleotide disclosed hereincan be chemically synthesized using chemical synthesis methods and potential nucleobase substitutions known in the art. See, for example, International Publication Nos. WO2014093924, WO2013052523; WO2013039857, WO2012135805, WO2013151671; U.S. Publ. No. US20130115272; or U.S. Pat. Nos.
- compositions and formulationsthat comprise any of the polynucleotides described above.
- the composition or formulationfurther comprises a delivery agent.
- the composition or formulationcan contain a polynucleotide comprising a sequence optimized nucleic acid sequence disclosed herein which encodes an IL-22 polypeptide.
- the composition or formulationcan contain a polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a polynucleotide (e.g., an ORF) having significant sequence identity to a sequence optimized nucleic acid sequence disclosed herein which encodes an IL-22 polypeptide.
- the polynucleotidefurther comprises a miRNA binding site, e.g., a miRNA binding site that binds miR-126, miR-142, miR-144, miR-146, miR-150, miR-155, miR-16, miR-21, miR-223, miR-24, miR- 27 and miR-26a.
- compositions or formulationcan optionally comprise one or more additional active substances, e.g., therapeutically and/or prophylactically active substances.
- Pharmaceutical compositions or formulation of the present disclosurecan be sterile and/or pyrogen-free. General considerations in the formulation and/or manufacture of pharmaceutical agents can be found, for example, in Remington: The Science and Practice of Pharmacy 21 st ed., Lippincott Williams & Wilkins, 2005 (incorporated herein by reference in its entirety).
- compositionsare administered to humans, human patients or subjects.
- the phrase "active ingredient"generally refers to polynucleotides to be delivered as described herein.
- Formulations and pharmaceutical compositions described hereincan be prepared by any method known or hereafter developed in the art of pharmacology. In general, such preparatory methods include the step of associating the active ingredient with an excipient and/or one or more other accessory ingredients, and then, if necessary and/or desirable, dividing, shaping and/or packaging the product into a desired single- or multi-dose unit.
- a pharmaceutical composition or formulation in accordance with the present disclosurecan be prepared, packaged, and/or sold in bulk, as a single unit dose, and/or as a plurality of single unit doses.
- a "unit dose"refers to a discrete amount of the pharmaceutical composition comprising a predetermined amount of the active ingredient.
- the amount of the active ingredientis generally equal to the dosage of the active ingredient that would be administered to a subject and/or a convenient fraction of such a dosage such as, for example, one-half or one-third of such a dosage.
- Relative amounts of the active ingredient, the pharmaceutically acceptable excipient, and/or any additional ingredients in a pharmaceutical composition in accordance with the present disclosurecan vary, depending upon the identity, size, and/or condition of the subject being treated and further depending upon the route by which the composition is to be administered.
- the compositions and formulations described hereincan contain at least one polynucleotide of the present disclosure.
- the composition or formulationcan contain 1, 2, 3, 4 or 5 polynucleotides of the present disclosure.
- compositions or formulations described hereincan comprise more than one type of polynucleotide.
- the composition or formulationcan comprise a polynucleotide in linear and circular form.
- the composition or formulationcan comprise a circular polynucleotide and an in vitro transcribed (IVT) polynucleotide.
- IVTin vitro transcribed
- the composition or formulationcan comprise an IVT polynucleotide, a chimeric polynucleotide and a circular polynucleotide.
- compositions and formulationsare principally directed to pharmaceutical compositions and formulations that are suitable for administration to humans, it will be understood by the skilled artisan that such compositions are generally suitable for administration to any other animal, e.g., to non-human animals, e.g. non-human mammals.
- the present disclosureprovides pharmaceutical formulations that comprise a polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide).
- the polynucleotides described hereincan be Formulated using one or more excipients to: (1) increase stability; (2) increase cell transfection; (3) permit the sustained or delayed release (e.g., from a depot formulation of the polynucleotide); (4) alter the biodistribution (e.g., target the polynucleotide to specific tissues or cell types); (5) increase the translation of encoded protein in vivo; and/or (6) alter the release profile of encoded protein in vivo.
- excipientsto: (1) increase stability; (2) increase cell transfection; (3) permit the sustained or delayed release (e.g., from a depot formulation of the polynucleotide); (4) alter the biodistribution (e.g., target the polynucleotide to specific tissues or cell types); (5) increase the translation of encoded protein in vivo; and/or (6) alter the release profile of encoded protein in vivo.
- the polynucleotide(e.g., a RNA, e.g., a mRNA) disclosed herein is Formulated with a delivery agent comprising LNP-1A, LNP 1-B, LNP-2A, LNP-2B, LNP-3A, or LNP-3B.
- the polynucleotide(e.g., a RNA, e.g., a mRNA) of the present disclosure is Formulated with LNP-1A.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-1B.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-2A.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-2B.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) of the present disclosureis Formulated with LNP-3A.
- the polynucleotide(e.g., a RNA, e.g., a mRNA) of the present disclosure is Formulated with LNP-3B.
- a pharmaceutically acceptable excipientincludes, but are not limited to, any and all solvents, dispersion media, or other liquid vehicles, dispersion or suspension aids, diluents, granulating and/or dispersing agents, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, binders, lubricants or oil, coloring, sweetening or flavoring agents, stabilizers, antioxidants, antimicrobial or antifungal agents, osmolality adjusting agents, pH adjusting agents, buffers, chelants, cyoprotectants, and/or bulking agents, as suited to the particular dosage form desired.
- diluentsinclude, but are not limited to, calcium or sodium carbonate, calcium phosphate, calcium hydrogen phosphate, sodium phosphate, lactose, sucrose, cellulose, microcrystalline cellulose, kaolin, mannitol, sorbitol, etc., and/or combinations thereof.
- Exemplary surface active agents and/or emulsifiersinclude, but are not limited to, natural emulsifiers (e.g., acacia, agar, alginic acid, sodium alginate, tragacanth, chondrux, cholesterol, xanthan, pectin, gelatin, egg yolk, casein, wool fat, cholesterol, wax, and lecithin), sorbitan fatty acid esters (e.g., polyoxyethylene sorbitan monooleate [TWEEN®80], sorbitan monopalmitate [SPAN®40], glyceryl monooleate, polyoxyethylene esters, polyethylene glycol fatty acid esters (e.g., CREMOPHOR®), polyoxyethylene ethers (e.g., polyoxyethylene lauryl ether [BRIJ®30]), PLUORINC®F 68, POLOXAMER®188, etc.
- natural emulsifierse.g., acacia, a
- binding agentsinclude, but are not limited to, starch, gelatin, sugars (e.g., sucrose, glucose, dextrose, dextrin, molasses, lactose, lactitol, mannitol), amino acids (e.g., glycine), natural and synthetic gums (e.g., acacia, sodium alginate), ethylcellulose, hydroxyethylcellulose, hydroxypropyl methylcellulose, etc., and combinations thereof.
- Oxidationis a potential degradation pathway for mRNA, especially for liquid mRNA formulations. In order to prevent oxidation, antioxidants can be added to the formulations.
- antioxidantsinclude, but are not limited to, alpha tocopherol, ascorbic acid, ascorbyl palmitate, benzyl alcohol, butylated hydroxyanisole, m-cresol, methionine, butylated hydroxytoluene, monothioglycerol, sodium or potassium metabisulfite, propionic acid, propyl gallate, sodium ascorbate, etc., and combinations thereof.
- Exemplary chelating agentsinclude, but are not limited to, ethylenediaminetetraacetic acid (EDTA), citric acid monohydrate, disodium edetate, fumaric acid, malic acid, phosphoric acid, sodium edetate, tartaric acid, trisodium edetate, etc., and combinations thereof.
- Exemplary antimicrobial or antifungal agentsinclude, but are not limited to, benzalkonium chloride, benzethonium chloride, methyl paraben, ethyl paraben, propyl paraben, butyl paraben, benzoic acid, hydroxybenzoic acid, potassium or sodium benzoate, potassium or sodium sorbate, sodium propionate, sorbic acid, etc., and combinations thereof.
- Exemplary preservativesinclude, but are not limited to, vitamin A, vitamin C, vitamin E, beta-carotene, citric acid, ascorbic acid, butylated hydroxyanisol, ethylenediamine, sodium lauryl sulfate (SLS), sodium lauryl ether sulfate (SLES), etc., and combinations thereof.
- the pH of polynucleotide solutionsis maintained between pH 5 and pH 8 to improve stability.
- Exemplary buffers to control pHcan include, but are not limited to sodium phosphate, sodium citrate, sodium succinate, histidine (or histidine-HCl), sodium malate, sodium carbonate, etc., and/or combinations thereof.
- Exemplary lubricating agentsinclude, but are not limited to, magnesium stearate, calcium stearate, stearic acid, silica, talc, malt, hydrogenated vegetable oils, polyethylene glycol, sodium benzoate, sodium or magnesium lauryl sulfate, etc., and combinations thereof.
- the pharmaceutical composition or formulation described herecan contain a cryoprotectant to stabilize a polynucleotide described herein during freezing.
- Exemplary cryoprotectantsinclude, but are not limited to mannitol, sucrose, trehalose, lactose, glycerol, dextrose, etc., and combinations thereof.
- the pharmaceutical composition or formulation described herecan contain a bulking agent in lyophilized polynucleotide formulations to yield a "pharmaceutically elegant" cake, stabilize the lyophilized polynucleotides during long term (e.g., 36 month) storage.
- exemplary bulking agents of the present disclosurecan include, but are not limited to sucrose, trehalose, mannitol, glycine, lactose, raffinose, and combinations thereof.
- the pharmaceutical composition or formulationfurther comprises a delivery agent.
- the delivery agent of the present disclosurecan include, without limitation, liposomes, lipid nanoparticles, lipidoids, polymers, lipoplexes, microvesicles, exosomes, peptides, proteins, cells transfected with polynucleotides, hyaluronidase, nanoparticle mimics, nanotubes, conjugates, and combinations thereof.
- compositions or Formulations of the present disclosurecomprise the polynucleotides described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide) that is covalently linked to a carrier or targeting group, or including two encoding regions that together produce a fusion protein (e.g., bearing a targeting group and therapeutic protein or peptide) as a conjugate.
- the conjugatecan be a peptide that extends half life or selectively directs the nanoparticle to neurons in a tissue or organism, or assists in crossing the blood-brain barrier.
- the conjugatesinclude a naturally occurring substance, such as a protein (e.g., human serum albumin (HSA), low-density lipoprotein (LDL), high-density lipoprotein (HDL), or globulin); an carbohydrate (e.g., a dextran, pullulan, chitin, chitosan, inulin, cyclodextrin or hyaluronic acid); or a lipid.
- the ligandcan also be a recombinant or synthetic molecule, such as a synthetic polymer, e.g., a synthetic polyamino acid, an oligonucleotide (e.g., an aptamer).
- polyamino acidsexamples include polyamino acid is a polylysine (PLL), poly L-aspartic acid, poly L-glutamic acid, styrene-maleic acid anhydride copolymer, poly(L-lactide-co- glycolied) copolymer, divinyl ether-maleic anhydride copolymer, N-(2- hydroxypropyl)methacrylamide copolymer (HMPA), polyethylene glycol (PEG), polyvinyl alcohol (PVA), polyurethane, poly(2-ethylacryllic acid), N-isopropylacrylamide polymers, or polyphosphazine.
- PLLpolylysine
- poly L-aspartic acidpoly L-glutamic acid
- styrene-maleic acid anhydride copolymerpoly(L-lactide-co- glycolied) copolymer
- divinyl ether-maleic anhydride copolymerdivinyl ether-maleic an
- polyaminesinclude: polyethylenimine, polylysine (PLL), spermine, spermidine, polyamine, pseudopeptide-polyamine, peptidomimetic polyamine, dendrimer polyamine, arginine, amidine, protamine, cationic lipid, cationic porphyrin, quaternary salt of a polyamine, or an alpha helical peptide.
- the conjugatecan function as a carrier for the polynucleotide disclosed herein.
- the conjugatecan comprise a cationic polymer such as, but not limited to, polyamine, polylysine, polyalkylenimine, and polyethylenimine that can be grafted to with poly(ethylene glycol).
- conjugates and their preparationsare described in U.S. Pat. No.6,586,524 and U.S. Pub. No. US20130211249, each of which herein is incorporated by reference in its entirety.
- the conjugatescan also include targeting groups, e.g., a cell or tissue targeting agent, e.g., a lectin, glycoprotein, lipid or protein, e.g., an antibody, that binds to a specified cell type such as a kidney cell.
- a cell or tissue targeting agente.g., a lectin, glycoprotein, lipid or protein, e.g., an antibody, that binds to a specified cell type such as a kidney cell.
- a targeting groupcan be a thyrotropin, melanotropin, lectin, glycoprotein, surfactant protein A, Mucin carbohydrate, multivalent lactose, multivalent galactose, N-acetyl-galactosamine, N-acetyl-glucosamine multivalent mannose, multivalent fucose, glycosylated polyaminoacids, multivalent galactose, transferrin, bisphosphonate, polyglutamate, polyaspartate, a lipid, cholesterol, a steroid, bile acid, folate, vitamin B12, biotin, an RGD peptide, an RGD peptide mimetic or an aptamer.
- Targeting groupscan be proteins, e.g., glycoproteins, or peptides, e.g., molecules having a specific affinity for a co-ligand, or antibodies e.g., an antibody, that binds to a specified cell type such as an endothelial cell or bone cell.
- Targeting groupscan also include hormones and hormone receptors. They can also include non-peptidic species, such as lipids, lectins, carbohydrates, vitamins, cofactors, multivalent lactose, multivalent galactose, N- acetyl-galactosamine, N-acetyl-glucosamine multivalent mannose, multivalent frucose, or aptamers.
- the ligandcan be, for example, a lipopolysaccharide, or an activator of p38 MAP kinase.
- the targeting groupcan be any ligand that is capable of targeting a specific receptor. Examples include, without limitation, folate, GalNAc, galactose, mannose, mannose-6P, apatamers, integrin receptor ligands, chemokine receptor ligands, transferrin, biotin, serotonin receptor ligands, PSMA, endothelin, GCPII, somatostatin, LDL, and HDL ligands.
- the targeting groupis an aptamer.
- the aptamercan be unmodified or have any combination of modifications disclosed herein.
- the targeting groupcan be a glutathione receptor (GR)-binding conjugate for targeted delivery across the blood-central nervous system barrier as described in, e.g., U.S. Pub. No. US2013021661012 (herein incorporated by reference in its entirety).
- the conjugatecan be a synergistic biomolecule-polymer conjugate, which comprises a long-acting continuous-release system to provide a greater therapeutic efficacy.
- the synergistic biomolecule-polymer conjugatecan be those described in U.S. Pub. No. US20130195799.
- the conjugatecan be an aptamer conjugate as described in Intl. Pat. Pub. No. WO2012040524.
- the conjugatecan be an amine containing polymer conjugate as described in U.S. Pat. No. 8,507,653. Each of the references is herein incorporated by reference in its entirety.
- the polynucleotidescan be conjugated to SMARTT POLYMER TECHNOLOGY® (PHASERX®, Inc. Seattle, WA).
- the polynucleotides described hereinare covalently conjugated to a cell penetrating polypeptide, which can also include a signal sequence or a targeting sequence.
- the conjugatescan be designed to have increased stability, and/or increased cell transfection; and/or altered the biodistribution (e.g., targeted to specific tissues or cell types).
- the polynucleotides described hereincan be conjugated to an agent to enhance delivery.
- the agentcan be a monomer or polymer such as a targeting monomer or a polymer having targeting blocks as described in Intl. Pub. No. WO2011062965.
- the agentcan be a transport agent covalently coupled to a polynucleotide as described in, e.g., U.S. Pat. Nos.6,835.393 and 7,374,778.
- the agentcan be a membrane barrier transport enhancing agent such as those described in U.S. Pat. Nos.7,737,108 and 8,003,129. Each of the references is herein incorporated by reference in its entirety. 23.
- polynucleotides, pharmaceutical compositions and Formulations described aboveare used in the preparation, manufacture and therapeutic use of to treat and/or prevent metabolic diseases, disorders or conditions.
- the polynucleotides, compositions and Formulations of the present disclosureare used to treat and/or prevent metabolic disease.
- the polynucleotides, polypeptides, pharmaceutical compositions, and formulations of the present disclosureare used in a method of treating or delaying the onset and/or progression of metabolic disease in a subject (e.g., a human subject), comprising: administering to the subject an effective amount of any of the polynucleotides, polypeptides, pharmaceutical compositions, and formulations described above.
- the polynucleotides, polypeptides, pharmaceutical compositions, and formulations of the present disclosureare used in a method of increasing serum IL-22 levels in a subject (e.g., a human subject), comprising: administering to the subject an effective amount of any of the the polynucleotides, polypeptides, pharmaceutical compositions, and formulations described above.
- the polynucleotides, polypeptides, pharmaceutical compositions, and formulations of the present disclosureare used in a method of increasing serum IL-22 levels in a subject (e.g., a human subject), comprising: administering to the subject an effective amount of any of the the polynucleotides, polypeptides, pharmaceutical compositions, and formulations described above.
- the polynucleotides, polypeptides, pharmaceutical compositions, and formulations of the present disclosureare used in a method of increasing IL-22 activity in a subject (e.g., a human subject), comprising: administering to the subject an effective amount of any of the the polynucleotides, polypeptides, pharmaceutical compositions, and formulations described above.
- the present disclosurerelates to use of any of the polynucleotides, polypeptides, or pharmaceutical compostions or formulations discussed above in the manufacture of a medicament for treating and/or preventing an IL-22-related disease, treating and/or preventing metabolic disease, treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject.
- the polynucleotides, pharmaceutical compositions and formulations of the present disclosureare used in methods for increasing IL-22 levels in a subject in need thereof, e.g., a subject with metabolic disease.
- one aspect of the present disclosureprovides a method of alleviating the signs and symptoms of metabolic disease in a subject comprising the administration of a composition or formulation comprising a polynucleotide encoding IL-22 or an IL-22-polypeptide-containing fusion protein to that subject (e.g, an mRNA encoding an IL-22 polypeptide).
- a composition or formulationcomprising a polynucleotide encoding IL-22 or an IL-22-polypeptide-containing fusion protein to that subject (e.g, an mRNA encoding an IL-22 polypeptide).
- the administration of an effective amount of a polynucleotide, pharmaceutical composition or Formulation of this disclosureincreases body weight of a human subject.
- the administration of the polynucleotide, pharmaceutical composition or Formulation of this disclosureresults in an increase in body weight within a short period of time (e.g., within about 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 5 days, 7 days, 14 days, 24 days, 48 days, or 60 days) after administration of the polynucleotide, pharmaceutical composition or Formulation of this disclosure.
- a short period of timee.g., within about 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 5 days, 7 days, 14 days, 24 days, 48 days, or 60 days
- the administration of an effective amount of a polynucleotide, pharmaceutical composition or Formulation of this disclosuremaintains body weight of a human subject.
- Replacement therapyis a potential treatment for metabolic disease.
- the polynucleotides, e.g., mRNA, disclosed hereincomprise one or more sequences encoding an IL-22 polypeptide that is suitable for use in gene replacement therapy for metabolic disease.
- the present disclosuretreats a lack of IL-22 or IL-22 activity, or decreased or abnornal IL-22 activity in a subject by providing a polynucleotide, e.g., mRNA, that encodes an IL-22 polypeptide to the subject.
- the polynucleotideis sequence-optimized.
- the polynucleotide(e.g., an mRNA) comprises a nucleic acid sequence (e.g., an ORF) encoding an IL-22 polypeptide, wherein the nucleic acid is sequence-optimized, e.g., by modifying its G/C, uridine, or thymidine content, and/or the polynucleotide comprises at least one chemically modified nucleoside.
- the polynucleotidecomprises a miRNA binding site, e.g., a miRNA binding site that binds miRNA-142.
- the administration of a composition or Formulation comprising polynucleotide, pharmaceutical composition or Formulation of the present disclosure to a subjectresults in an increase in serum IL-22 levels to a level at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 100%, at least 500%, or at least 1000% higher than the level observed prior to the administration of the composition or Formulation.
- the administration of a composition or Formulation comprising an mRNA encoding an IL-22 polypeptide to a subjectresults in an increase of IL-22 signaling activity in the serum of subject to a level at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 100%, at least 500%, or at least 1000% or more of the activity level expected in a normal subject, e.g., a human not suffering from metabolic disease.
- the administration of the polynucleotide, pharmaceutical composition or Formulation of the present disclosureresults in expression of IL-22 protein in the serum of a subject that persists for a period of time sufficient to allow significant activation of downstream signaling pathways to occur.
- the polynucleotides, pharmaceutical compositions, or formulations of the present disclosurecan be repeatedly administered such that IL-22 protein is expressed at a therapeutic level for a period of time sufficient to have a beneficial biological effect as described herein.
- the expression of the encoded polypeptideis increased.
- the polynucleotideincreases IL-22 expression levels in serum when introduced, e.g., by at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or to 100% with respect to the IL-22 expression level before the polynucleotide is introduced.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectresults in a reduction in body weight of the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, or at least about 50%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectis sufficient to reduce body weight of the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, or at least about 50%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectresults in a reduction in total cholesterol levels (e.g., total serum cholesterol) in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- total cholesterol levelse.g., total serum cholesterol
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectis sufficient to reduce total cholesterol levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectresults in a reduction in total HDL cholesterol levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectis sufficient to reduce total HDL cholesterol levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectresults in a reduction in total LDL cholesterol levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectis sufficient to reduce total LDL cholesterol levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectresults in a reduction in total triglyceride levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectis sufficient to reduce total triglyceride levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectresults in a reduction in liver steatosis in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectis sufficient to reduce liver steatosis in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectresults in a reduction in blood glucose levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the administration of a composition or formulation comprising a polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subjectis sufficient to reduce blood glucose levels in the subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
- the method or usecomprises administering a polynucleotide, e.g., mRNA, comprising a nucleotide sequence having sequence similarity to a polynucleotide of SEQ ID NO:300-303, wherein the polynucleotide encodes an IL-22 polypeptide.
- the method or usecomprises administering a polynucleotide, e.g., mRNA, comprising a nucleotide sequence having sequence similarity to a polynucleotide of SEQ ID NO:304-307, wherein the polynucleotide encodes a serum albumin polypeptide.
- the method or usecomprises administering a polynucleotide, e.g., mRNA, comprising a nucleotide sequence having sequence similarity to a polynucleotide of any of SEQ ID NO:310-328, wherein the polynucleotide encodes an IL-22 polypeptide-containing fusion protein.
- the method or usecomprises administering a polynucleotide, e.g., mRNA, comprising a nucleotide sequence having sequence similarity to a polynucleotide of any of SEQ ID NO:350-368, wherein the polynucleotide encodes an IL-22 polypeptide-containing fusion protein.
- aspects of the present disclosurerelate to transplantation of cells containing polynucleotides to a mammalian subject.
- Administration of cells to mammalian subjectsis known to those of ordinary skill in the art, and includes, but is not limited to, local implantation (e.g., topical or subcutaneous administration), organ delivery or systemic injection (e.g., intravenous injection or inhalation), and the Formulation of cells in pharmaceutically acceptable carriers.
- the present disclosurealso provides methods to increase IL-22 activity in a subject in need thereof, e.g., a subject with metabolic disease, comprising administering to the subject a therapeutically effective amount of a composition or Formulation comprising mRNA encoding an IL-22 polypeptide disclosed herein, e.g., a human IL-22 polypeptide, a mutant thereof, or a fusion protein comprising a human IL-22.
- the IL-22 activity measured after administration to a subject in need thereof, e.g., a subject with metabolic diseaseis at least the normal IL-22 activity level observed in healthy human subjects.
- the IL-22 activity measured after administrationis at higher than the IL-22 activity level observed in metabolic disease patients, e.g., untreated metabolic disease patients.
- the increase in IL-22 activity in a subject in need thereof, e.g., a subject with metabolic disease, after administering to the subject a therapeutically effective amount of a composition or Formulation comprising mRNA encoding an IL-22 polypeptide disclosed hereinis at least about 5, at least about 10, at least about 15, at least about 20, at least about 25, at least about 30, at least about 35, at least about 40, at least about 45, at least about 50, at least about 55, at least about 60, at least about 65, at least about 70, at least about 75, at least about 80, at least about 85, at least about 90, at least about 95, at least about 100, or greater than 100 percent of the normal IL-22 activity level observed in healthy human subjects.
- the increase in IL-22 activity above the IL-22 activity level observed in metabolic disease patients after administering to the subject a composition or Formulation comprising an mRNA encoding an IL-22 polypeptide disclosed hereinis maintained for at least 1 day, at least 2 days, at least 3 days, at least 4 days, at least 5 days, at least 6 days, at least 7 days, at least 8 days, at least 9 days, at least 10 days, at least 12 days, at least 14 days, at least 21 days, or at least 28 days.
- the present disclosurealso provides a method to treat, prevent, or ameliorate the symptoms of metabolic disease (e.g., high blood pressure, obesity, insulin resistance, etc.) in a metabolic disease patient comprising administering to the subject a therapeutically effective amount of a composition or Formulation comprising mRNA encoding an IL-22 polypeptide disclosed herein.
- a therapeutically effective amount of a composition or Formulation comprising mRNA encoding an IL-22 polypeptide disclosed hereinresults in reducing the symptoms of metabolic disease.
- the polynucleotidese.g., mRNA
- pharmaceutical compositions and Formulations used in the methods of this disclosurecomprise a uracil- modified sequence encoding an IL-22 polypeptide disclosed herein and a miRNA binding site disclosed herein, e.g., a miRNA binding site that binds to miR-142 and/or a miRNA binding site that binds to miR-126.
- the uracil-modified sequence encoding an IL-22 polypeptidecomprises at least one chemically modified nucleobase, e.g., N1-methylpseudouracil or 5-methoxyuracil.
- At least 95% of a type of nucleobase (e.g., uracil) in a uracil-modified sequence encoding an IL-22 polypeptide of this disclosureare modified nucleobases.
- at least 95% of uracil in a uracil- modified sequence encoding an IL-22 polypeptideis 1-N-methylpseudouridine or 5- methoxyuridine.
- the polynucleotide(e.g., a RNA, e.g., a mRNA) disclosed herein is Formulated with a delivery agent comprising, e.g., LNP-1A, LNP-1B, LNP-2A, LNP-2B, LNP-3A, or LNP-3B.
- a delivery agentcomprising, e.g., LNP-1A, LNP-1B, LNP-2A, LNP-2B, LNP-3A, or LNP-3B.
- the polynucleotide(e.g., a RNA, e.g., a mRNA) disclosed herein is Formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., Compound II or Compound B; or a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound I or Compound VI, or any combination thereof.
- a delivery agentcomprising, e.g., a compound having the Formula (I), e.g., Compound II or Compound B; or a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound I or Compound VI, or any combination thereof.
- the delivery agentcomprises an ionizable amino lipid (e.g., Compound II, VI, or B), a helper lipid (e.g., DSPC), a sterol (e.g., Cholesterol), and a PEG lipid (e.g., Compound I or PEG-DMG), e.g., with a mole ratio in the range of about (i) 40-50 mol% ionizable amino lipid (e.g., Compound II, VI, or B), optionally 45-50 mol% ionizable amino lipid, for example, 45-46 mol%, 46-47 mol%, 47-48 mol%, 48-49 mol%, or 49-50 mol% for example about 45 mol%, 45.5 mol%, 46 mol%, 46.5 mol%, 47 mol%, 47.5 mol%, 48 mol%, 48.5 mol%, 49 mol%, or 49.5 mol%; (ii) 30-45
- the delivery agentcomprises Compound B, Cholesterol, DSPC, and Compound I with a mole ratio of 47:39:11:3.
- an encoded proteine.g., signaling protein
- a subjecte.g., from a preclinical test subject (rodent, primate, etc.) or from a clinical subject (human).
- the therapeutic effectiveness of a drug or a treatment of the instant inventioncan be characterized or determined by measuring the level of activity of an encoded protein (e.g., signaling protein) in a sample or in samples taken from a subject (e.g., from a preclinical test subject (rodent, primate, etc.) or from a clinical subject (human).
- an encoded proteine.g., signaling protein
- the therapeutic effectiveness of a drug or a treatment of the instant inventioncan be characterized or determined by measuring the level of an appropriate biomarker in sample(s) taken from a subject.
- IL-22 Protein Expression LevelsCertain aspects of this disclosure feature measurement, determination and/or monitoring of the expression level or levels of IL-22 protein in a subject, for example, in an animal (e.g., rodents, primates, and the like) or in a human subject. Animals include normal, healthy or wild type animals, as well as animal models for use in understanding metabolic disease and treatments thereof.
- Exemplary animal modelsinclude rodent models, for example, IL-22 deficient mice also referred to as IL-22 mice.
- IL-22 protein expression levelscan be measured or determined by any art-recognized method for determining protein levels in biological samples, e.g., from blood samples or a needle biopsy.
- levelor "level of a protein” as used herein, preferably means the weight, mass or concentration of the protein within a sample or a subject. It will be understood by the skilled artisan that in certain embodiments the sample may be subjected, e.g., to any of the following: purification, precipitation, separation, e.g.
- centrifugation and/or HPLCand subsequently subjected to determining the level of the protein, e.g., using mass and/or spectrometric analysis.
- enzyme-linked immunosorbent assayELISA
- protein purification, separation and LC-MScan be used as a means for determining the level of a protein according to this disclosure.
- an mRNA therapy of this disclosureresults in increased IL-22 protein expression levels in the tissue (e.g., serum) of the subject (e.g., 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold increase and/or increased to at least 50%, at least 60%, at least 70%, at least 75%, 80%, at least 85%, at least 90%, at least 95%, or at least 100% of normal levels) for at least 6 hours, at least 12 hours, at least 24 hours, at least 36 hours, at least 48 hours, at least 60 hours, at least 72 hours, at least 84 hours, at least 96 hours, at least 108 hours, at least 122 hours after administration of a single dose of the mRNA therapy.
- IL-22 Protein and Signaling Activity Levelfeature measurement, determination, and/or monitoring of the activity level(s) (i.e., signaling activity level(s)) of IL-22 protein in a subject, for example, in an animal (e.g., rodent, primate, and the like) or in a human subject.
- Activity levelscan be measured or determined by any art-recognized method for determining signaling activity levels in biological samples.
- the term "activity level” or “signaling activity level” as used herein,preferably means the activity of the signaling protein per volume, mass or weight of sample or total protein within a sample.
- the "activity level” or “signaling activity level”is described in terms of units per milliliter of fluid (e.g., bodily fluid, e.g., serum, plasma, urine and the like) or is described in terms of units per weight of tissue or per weight of protein (e.g., total protein) within a sample.
- Units (“U”) of enzyme activitycan be described in terms of weight or mass of substrate hydrolyzed per unit time.
- an mRNA therapy of this disclosurefeatures a pharmaceutical composition comprising a dose of mRNA effective to result in at least 5 U/mg, at least 10 U/mg, at least 20 U/mg, at least 30 U/mg, at least 40 U/mg, at least 50 U/mg, at least 60 U/mg, at least 70 U/mg, at least 80 U/mg, at least 90 U/mg, at least 100 U/mg, or at least 150 U/mg of IL-22 activity in tissue (e.g., liver) between 6 and 12 hours, or between 12 and 24, between 24 and 48, or between 48 and 72 hours post administration (e.g., at 48 or at 72 hours post administration).
- tissuee.g., liver
- an mRNA therapy of this disclosureresults in increased IL-22 activity levels in the serum of the subject (e.g., 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold increase and/or increased to at least 50%, at least 60%, at least 70%, at least 75%, 80%, at least 85%, at least 90%, at least 95%, or at least 100% of normal levels) for at least 6 hours, at least 12 hours, at least 24 hours, or at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30 or more days after administration of a single dose of the mRNA therapy.
- IL-22 activity levels in the serum of the subjecte.g., 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold increase and/or increased
- an mRNA therapy of this disclosurefeatures a pharmaceutical composition comprising a single intravenous dose of mRNA that results in the above-described levels of activity.
- an mRNA therapy of this disclosurefeatures a pharmaceutical composition which can be administered in multiple single unit intravenous doses of mRNA that maintain the above-described levels of activity.
- IL-22as a Biomarker
- aspects of this disclosurefeature determining the level (or levels) of a biomarker determined in a sample as compared to a level (e.g., a reference level) of the same or another biomarker in another sample, e.g., from the same patient, from another patient, from a control and/or from the same or different time points, and/or a physiologic level, and/or an elevated level, and/or a supraphysiologic level, and/or a level of a control.
- physiologic levels of biomarkersfor example, levels in normal or wild type animals, normal or healthy subjects, and the like, in particular, the level or levels characteristic of subjects who are healthy and/or normal functioning.
- the phrase “elevated level”means amounts greater than normally found in a normal or wild type preclinical animal or in a normal or healthy subject, e.g. a human subject.
- the term “supraphysiologic”means amounts greater than normally found in a normal or wild type preclinical animal or in a normal or healthy subject, e.g. a human subject, optionally producing a significantly enhanced physiologic response.
- the term “comparing” or “compared to”preferably means the mathematical comparison of the two or more values, e.g., of the levels of the biomarker(s).
- Comparing or comparison tocan be in the context, for example, of comparing to a control value, e.g., as compared to a reference blood, serum, plasma, and/or tissue level, in said subject prior to administration (e.g., in a person suffering from metabolic disease) or in a normal or healthy subject.
- Comparing or comparison tocan also be in the context, for example, of comparing to a control value, e.g., as compared to a reference blood, serum, plasma and/or tissue level in said subject prior to administration (e.g., in a person suffering from metabolic disease) or in a normal or healthy subject.
- a “control”is preferably a sample from a subject wherein the metabolic disease status of said subject is known.
- a controlis a sample of a healthy patient.
- the controlis a sample from at least one subject having a known metabolic disease status, for example, a severe, mild, or healthy metabolic disease status, e.g. a control patient.
- controlis a sample from a subject not being treated for metabolic disease.
- controlis a sample from a single subject or a pool of samples from different subjects and/or samples taken from the subject(s) at different time points.
- levelor “level of a biomarker” as used herein, preferably means the mass, weight or concentration of a biomarker of this disclosure within a sample or a subject. It will be understood by the skilled artisan that in certain embodiments the sample may be subjected to, e.g., one or more of the following: substance purification, precipitation, separation, e.g. centrifugation and/or HPLC and subsequently subjected to determining the level of the biomarker, e.g.
- LC-MScan be used as a means for determining the level of a biomarker according to this disclosure.
- determining the level of a biomarker as used hereincan mean methods which include quantifying an amount of at least one substance in a sample from a subject, for example, in a bodily fluid from the subject (e.g., serum, plasma, urine, lymph, etc.) or in a tissue of the subject.
- reference levelas used herein can refer to levels (e.g., of a biomarker) in a subject prior to administration of an mRNA therapy of this disclosure (e.g., in a person suffering from metabolic disease) or in a normal or healthy subject.
- normal subjector “healthy subject” refers to a subject not suffering from symptoms associated with metabolic disease.
- a sample from a healthy subjectis used as a control sample, or the known or standardized value for the level of biomarker from samples of healthy or normal subjects is used as a control.
- comparing the level of the biomarker in a sample from a subject in need of treatment for metabolic disease or in a subject being treated for metabolic disease to a control level of the biomarkercomprises comparing the level of the biomarker in the sample from the subject (in need of treatment or being treated for metabolic disease) to a baseline or reference level, wherein if a level of the biomarker in the sample from the subject (in need of treatment or being treated for metabolic disease) is elevated, increased or higher compared to the baseline or reference level, this is indicative that the subject is suffering from metabolic disease and/or is in need of treatment; and/or wherein if a level of the biomarker in the sample from the subject (in need of treatment or being treated for metabolic disease) is decreased or lower compared to the baseline level this is indicative that the subject is not suffering from, is successfully being treated for metabolic disease, or is not in need of treatment for metabolic disease.
- the stronger the reductione.g., at least 2-fold, at least 3- fold, at least 4-fold, at least 5-fold, at least 6-fold, at least 7-fold, at least 8-fold, at least 10- fold, at least 20-fold, at least-30 fold, at least 40-fold, at least 50-fold reduction and/or at least 10%, at least 20%, at least 30% at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 100% reduction) of the level of a biomarker, within a certain time period, e.g., within 6 hours, within 12 hours, 24 hours, 36 hours, 48 hours, 60 hours, or 72 hours, and/or for a certain duration of time, e.g., 48 hours, 72 hours, 96 hours, 120 hours, 144 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, 18 months, 24 months
- a specified time periode.g., post-administration, for example, of a single intravenous dose
- Exemplary time periodsinclude 12, 24, 48, 72, 96, 120 or 144 hours post administration, in particular 24, 48, 72 or 96 hours post administration.
- a sustained reduction in substrate levelsis particularly indicative of mRNA therapeutic dosing and/or administration regimens successful for treatment of metabolic disease. Such sustained reduction can be referred to herein as “duration” of effect.
- a bodily fluide.g., plasma, serum, urine, e.g., urinary sediment
- tissue(s) in a subjectwithin 1, 2, 3, 4, 5, 6, 7, 8 or more days following administration is indicative of a successful therapeutic approach.
- sustained reduction in substrate (e.g., biomarker) levels in one or more samplesis preferred.
- substratee.g., biomarker
- a single dose of an mRNA therapy of this disclosureis about 0.2 to about 0.8 mgs/kg (mpk), about 0.3 to about 0.7 mpk, about 0.4 to about 0.8 mpk, or about 0.5 mpk.
- a single dose of an mRNA therapy of this disclosureis less than 1.5 mpk, less than 1.25 mpk, less than 1 mpk, or less than 0.75 mpk. 24.
- Compositions and Formulations for UseCertain aspects of this disclosure are directed to compositions or Formulations comprising any of the polynucleotides disclosed above.
- the composition or Formulationcomprises: (i) a polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a sequence-optimized nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, or variant thereof), wherein the polynucleotide comprises at least one chemically modified nucleobase, e.g., N1-methylpseudouracil or 5-methoxyuracil (e.g., wherein at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, at least about 99%, or 100% of the uracils are N1-methylpseudouracils or 5- methoxyuracils), and wherein the polynucleotide further comprises a miRNA, e
- the delivery agentis a lipid nanoparticle comprising Compound II, Compound VI, a salt or a stereoisomer thereof, or any combination thereof.
- the delivery agentcomprises an ionizable amino lipid (e.g., Compound II, VI, or B), a helper lipid (e.g., DSPC), a sterol (e.g., Cholesterol), and a PEG lipid (e.g., Compound I or PEG-DMG), e.g., with a mole ratio in the range of about (i) 40-50 mol% ionizable amino lipid (e.g., Compound II, VI, or B), optionally 45-50 mol% ionizable amino lipid, for example, 45-46 mol%, 46-47 mol%, 47-48 mol%, 48-49 mol%, or 49-50 mol% for example about 45 mol%, 45.5 mol%, 46 mol%, 46.5 mol
- the delivery agentcomprises Compound B, Cholesterol, DSPC, and Compound I with a mole ratio of 47:39:11:3.
- the uracil or thymine content of the ORF relative to the theoretical minimum uracil or thymine content of a nucleotide sequence encoding the IL-22 polypeptideis between about 100% and about 150%.
- the polynucleotides, compositions or Formulations aboveare used to treat and/or prevent IL-22-related diseases, disorders or conditions, e.g., metabolic disease. 25.
- compositionscan be administered by any route that results in a therapeutically effective outcome, such as intravenous (into a vein) administration. These also include, but are not limited to subcutaneous (SC) (under the skin), intravenous (IV) bolus, intravenous drip, and intramuscular (IM) (into a muscle).
- SCsubcutaneous
- IVintravenous
- IMintramuscular
- compositionscan be administered in a way that allows them cross the blood-brain barrier, vascular barrier, or other epithelial barrier.
- a Formulation for a route of administrationcan include at least one inactive ingredient.
- compositionscan be administered intravenously.
- compositionscan be administered subcutaneously.
- compositionscan be administered intramuscularly. 26. Definitions In order that the present disclosure can be more readily understood, certain terms are first defined. As used in this application, except as otherwise expressly provided herein, each of the following terms shall have the meaning set forth below. Additional definitions are set forth throughout the application. This disclosure includes embodiments in which exactly one member of the group is present in, employed in, or otherwise relevant to a given product or process. This disclosure includes embodiments in which more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process. In this specification and the appended claims, the singular forms “a”, “an” and “the” include plural referents unless the context clearly dictates otherwise.
- nucleotidesare referred to by their commonly accepted single-letter codes. Unless otherwise indicated, nucleic acids are written left to right in 5′ to 3′ orientation. Nucleobases are referred to herein by their commonly known one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Accordingly, A represents adenine, C represents cytosine, G represents guanine, T represents thymine, U represents uracil.
- Amino acidsare referred to herein by either their commonly known three letter symbols or by the one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Unless otherwise indicated, amino acid sequences are written left to right in amino to carboxy orientation. About: The term "about” as used in connection with a numerical value throughout the specification and the claims denotes an interval of accuracy, familiar and acceptable to a person skilled in the art, such interval of accuracy is ⁇ 10 %. Where ranges are given, endpoints are included.
- administered in combinationmeans that two or more agents are administered to a subject at the same time or within an interval such that there can be an overlap of an effect of each agent on the patient. In some embodiments, they are administered within about 60, 30, 15, 10, 5, or 1 minute of one another.
- amino acid substitutionrefers to replacing an amino acid residue present in a parent or reference sequence (e.g., a wild type IL-22 sequence) with another amino acid residue.
- An amino acidcan be substituted in a parent or reference sequence (e.g., a wild type IL-22 polypeptide sequence), for example, via chemical peptide synthesis or through recombinant methods known in the art. Accordingly, a reference to a "substitution at position X" refers to the substitution of an amino acid present at position X with an alternative amino acid residue.
- substitution patternscan be described according to the schema AnY, wherein A is the single letter code corresponding to the amino acid naturally or originally present at position n, and Y is the substituting amino acid residue.
- substitution patternscan be described according to the schema An(YZ), wherein A is the single letter code corresponding to the amino acid residue substituting the amino acid naturally or originally present at position X, and Y and Z are alternative substituting amino acid residue.
- substitutionsare conducted at the nucleic acid level, i.e., substituting an amino acid residue with an alternative amino acid residue is conducted by substituting the codon encoding the first amino acid with a codon encoding the second amino acid.
- animalrefers to any member of the animal kingdom. In some embodiments, “animal” refers to humans at any stage of development. In some embodiments, “animal” refers to non-human animals at any stage of development. In certain embodiments, the non-human animal is a mammal (e.g., a rodent, a mouse, a rat, a rabbit, a monkey, a dog, a cat, a sheep, cattle, a primate, or a pig). In some embodiments, animals include, but are not limited to, mammals, birds, reptiles, amphibians, fish, and worms.
- mammalsinclude, but are not limited to, mammals, birds, reptiles, amphibians, fish, and worms.
- the animalis a transgenic animal, genetically-engineered animal, or a clone.
- the term “approximately,” as applied to one or more values of interest,refers to a value that is similar to a stated reference value.
- the term “approximately”refers to a range of values that fall within 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or less in either direction (greater than or less than) of the stated reference value unless otherwise stated or otherwise evident from the context (except where such number would exceed 100% of a possible value).
- the term “associated with”means that the symptom, measurement, characteristic, or status in question is linked to the diagnosis, development, presence, or progression of that disease.
- associationcan, but need not, be causatively linked to the disease.
- symptoms, sequelae, or any effects causing a decrease in the quality of life of a patient of metabolic diseaseare considered associated with metabolic disease and in some embodiments of the present invention can be treated, ameliorated, or prevented by administering the polynucleotides of the present invention to a subject in need thereof.
- associationWhen used with respect to two or more moieties, the terms “associated with,” “conjugated,” “linked,” “attached,” and “tethered,” when used with respect to two or more moieties, means that the moieties are physically associated or connected with one another, either directly or via one or more additional moieties that serves as a linking agent, to form a structure that is sufficiently stable so that the moieties remain physically associated under the conditions in which the structure is used, e.g., physiological conditions.
- An “association”need not be strictly through direct covalent chemical bonding. It can also suggest ionic or hydrogen bonding or a hybridization based connectivity sufficiently stable such that the "associated" entities remain physically associated.
- BiocompatibleAs used herein, the term “biocompatible” means compatible with living cells, tissues, organs or systems posing little to no risk of injury, toxicity or rejection by the immune system.
- BiodegradableAs used herein, the term “biodegradable” means capable of being broken down into innocuous products by the action of living things.
- Biologically activeAs used herein, the phrase “biologically active” refers to a characteristic of any substance that has activity in a biological system and/or organism. For instance, a substance that, when administered to an organism, has a biological effect on that organism, is considered to be biologically active.
- a polynucleotide of the present inventioncan be considered biologically active if even a portion of the polynucleotide is biologically active or mimics an activity considered biologically relevant.
- ChimeraAs used herein, "chimera” is an entity having two or more incongruous or heterogeneous parts or regions.
- a chimeric moleculecan comprise a first part comprising an IL-22 polypeptide, and a second part (e.g., genetically fused to the first part) comprising a second therapeutic protein (e.g., a protein with a distinct enzymatic activity, an antigen binding moiety, or a moiety capable of extending the plasma half life of IL-22, for example, an Fc region of an antibody).
- Sequence Optimizationrefers to a process or series of processes by which nucleobases in a reference nucleic acid sequence are replaced with alternative nucleobases, resulting in a nucleic acid sequence with improved properties, e.g., improved protein expression or decreased immunogenicity.
- the goal in sequence optimizationis to produce a synonymous nucleotide sequence than encodes the same polypeptide sequence encoded by the reference nucleotide sequence.
- Codon substitutionThe terms "codon substitution” or "codon replacement” in the context of sequence optimization refer to replacing a codon present in a reference nucleic acid sequence with another codon. A codon can be substituted in a reference nucleic acid sequence, for example, via chemical peptide synthesis or through recombinant methods known in the art.
- references to a "substitution” or “replacement” at a certain location in a nucleic acid sequence (e.g., an mRNA) or within a certain region or subsequence of a nucleic acid sequence (e.g., an mRNA)refer to the substitution of a codon at such location or region with an alternative codon.
- the terms "coding region” and “region encoding” and grammatical variants thereof,refer to an Open Reading Frame (ORF) in a polynucleotide that upon expression yields a polypeptide or protein.
- ORFOpen Reading Frame
- stereoisomermeans any geometric isomer (e.g., cis- and trans- isomer), enantiomer, or diastereomer of a compound.
- the present disclosureencompasses any and all stereoisomers of the compounds described herein, including stereomerically pure forms (e.g., geometrically pure, enantiomerically pure, or diastereomerically pure) and enantiomeric and stereoisomeric mixtures, e.g., racemates. Enantiomeric and stereomeric mixtures of compounds and means of resolving them into their component enantiomers or stereoisomers are well-known.
- isotopesrefers to atoms having the same atomic number but different mass numbers resulting from a different number of neutrons in the nuclei.
- isotopes of hydrogeninclude tritium and deuterium.
- a compound, salt, or complex of the present disclosurecan be prepared in combination with solvent or water molecules to form solvates and hydrates by routine methods.
- Contactingmeans establishing a physical connection between two or more entities. For example, contacting a mammalian cell with a nanoparticle composition means that the mammalian cell and a nanoparticle are made to share a physical connection. Methods of contacting cells with external entities both in vivo and ex vivo are well known in the biological arts.
- contacting a nanoparticle composition and a mammalian cell disposed within a mammalcan be performed by varied routes of administration (e.g., intravenous, intramuscular, intradermal, and subcutaneous) and can involve varied amounts of nanoparticle compositions.
- routes of administratione.g., intravenous, intramuscular, intradermal, and subcutaneous
- nanoparticle compositionse.g., more than one mammalian cell can be contacted by a nanoparticle composition.
- Conservative amino acid substitutionis one in which the amino acid residue in a protein sequence is replaced with an amino acid residue having a similar side chain.
- Families of amino acid residues having similar side chainshave been defined in the art, including basic side chains (e.g., lysine, arginine, or histidine), acidic side chains (e.g., aspartic acid or glutamic acid), uncharged polar side chains (e.g., glycine, asparagine, glutamine, serine, threonine, tyrosine, or cysteine), nonpolar side chains (e.g., alanine, valine, leucine, isoleucine, proline, phenylalanine, methionine, or tryptophan), beta- branched side chains (e.g., threonine, valine, isoleucine) and aromatic side chains (e.g., tyrosine, phenylalanine, tryptophan, or histidine).
- basic side chainse.g., lysine, arginine, or histidine
- acidic side chainse.g
- amino acid substitutionis considered to be conservative.
- a string of amino acidscan be conservatively replaced with a structurally similar string that differs in order and/or composition of side chain family members.
- Non-conservative amino acid substitutionsinclude those in which (i) a residue having an electropositive side chain (e.g., Arg, His or Lys) is substituted for, or by, an electronegative residue (e.g., Glu or Asp), (ii) a hydrophilic residue (e.g., Ser or Thr) is substituted for, or by, a hydrophobic residue (e.g., Ala, Leu, Ile, Phe or Val), (iii) a cysteine or proline is substituted for, or by, any other residue, or (iv) a residue having a bulky hydrophobic or aromatic side chain (e.g., Val, His, Ile or Trp) is substituted for, or by, one having a smaller side chain (e.g., Ala or Ser) or no side chain (e.g., Gly).
- a residue having an electropositive side chaine.g., Arg, His or Lys
- an electronegative residuee.g.,
- amino acid substitutionscan be readily identified by workers of ordinary skill.
- a substitutioncan be taken from any one of D- alanine, glycine, beta-alanine, L-cysteine and D-cysteine.
- a replacementcan be any one of D-lysine, arginine, D-arginine, homo-arginine, methionine, D-methionine, ornithine, or D- ornithine.
- substitutions in functionally important regions that can be expected to induce changes in the properties of isolated polypeptidesare those in which (i) a polar residue, e.g., serine or threonine, is substituted for (or by) a hydrophobic residue, e.g., leucine, isoleucine, phenylalanine, or alanine; (ii) a cysteine residue is substituted for (or by) any other residue; (iii) a residue having an electropositive side chain, e.g., lysine, arginine or histidine, is substituted for (or by) a residue having an electronegative side chain, e.g., glutamic acid or aspartic acid; or (iv) a residue having a bulky side chain, e.g., phenylalanine, is substituted for (or by) one not having such a side chain, e.g., glycine.
- a polar residuee.g
- non-conservative substitutionscan accordingly have little or no effect on biological properties.
- conservedrefers to nucleotides or amino acid residues of a polynucleotide sequence or polypeptide sequence, respectively, that are those that occur unaltered in the same position of two or more sequences being compared. Nucleotides or amino acids that are relatively conserved are those that are conserved amongst more related sequences than nucleotides or amino acids appearing elsewhere in the sequences.
- two or more sequencesare said to be "completely conserved” if they are 100% identical to one another. In some embodiments, two or more sequences are said to be "highly conserved” if they are at least 70% identical, at least 80% identical, at least 90% identical, or at least 95% identical to one another. In some embodiments, two or more sequences are said to be "highly conserved” if they are about 70% identical, about 80% identical, about 90% identical, about 95%, about 98%, or about 99% identical to one another.
- two or more sequencesare said to be "conserved” if they are at least 30% identical, at least 40% identical, at least 50% identical, at least 60% identical, at least 70% identical, at least 80% identical, at least 90% identical, or at least 95% identical to one another. In some embodiments, two or more sequences are said to be "conserved” if they are about 30% identical, about 40% identical, about 50% identical, about 60% identical, about 70% identical, about 80% identical, about 90% identical, about 95% identical, about 98% identical, or about 99% identical to one another. Conservation of sequence can apply to the entire length of an polynucleotide or polypeptide or can apply to a portion, region or feature thereof.
- Controlled Releaserefers to a pharmaceutical composition or compound release profile that conforms to a particular pattern of release to effect a therapeutic outcome.
- Cyclic or CyclizedAs used herein, the term “cyclic” refers to the presence of a continuous loop. Cyclic molecules need not be circular, only joined to form an unbroken chain of subunits. Cyclic molecules such as the engineered RNA or mRNA of the present invention can be single units or multimers or comprise one or more components of a complex or higher order structure.
- DeliveringAs used herein, the term “delivering” means providing an entity to a destination.
- delivering a polynucleotide to a subjectcan involve administering a nanoparticle composition including the polynucleotide to the subject (e.g., by an intravenous, intramuscular, intradermal, or subcutaneous route).
- Administration of a nanoparticle composition to a mammal or mammalian cellcan involve contacting one or more cells with the nanoparticle composition.
- Delivery Agentrefers to any substance that facilitates, at least in part, the in vivo, in vitro, or ex vivo delivery of a polynucleotide to targeted cells.
- domainrefers to a motif of a polypeptide having one or more identifiable structural or functional characteristics or properties (e.g., binding capacity, serving as a site for protein-protein interactions).
- Dosing regimenAs used herein, a “dosing regimen” or a “dosing regimen” is a schedule of administration or physician determined regimen of treatment, prophylaxis, or palliative care.
- Effective AmountAs used herein, the term “effective amount” of an agent is that amount sufficient to effect beneficial or desired results, for example, clinical results, and, as such, an "effective amount” depends upon the context in which it is being applied.
- an effective amount of an agentis, for example, an amount of mRNA expressing sufficient IL-22 to ameliorate, reduce, eliminate, or prevent the symptoms associated with the IL-22 deficiency, as compared to the severity of the symptom observed without administration of the agent.
- the term "effective amount”can be used interchangeably with "effective dose,” “therapeutically effective amount,” or “therapeutically effective dose.”
- EncapsulateAs used herein, the term “encapsulate” means to enclose, surround or encase.
- Encapsulation efficiencyrefers to the amount of a polynucleotide that becomes part of a nanoparticle composition, relative to the initial total amount of polynucleotide used in the preparation of a nanoparticle composition. For example, if 97 mg of polynucleotide are encapsulated in a nanoparticle composition out of a total 100 mg of polynucleotide initially provided to the composition, the encapsulation efficiency can be given as 97%. As used herein, “encapsulation” can refer to complete, substantial, or partial enclosure, confinement, surrounding, or encasement.
- enhanced deliverymeans delivery of more (e.g., at least 1.5 fold more, at least 2-fold more, at least 3-fold more, at least 4-fold more, at least 5-fold more, at least 6-fold more, at least 7-fold more, at least 8-fold more, at least 9-fold more, at least 10-fold more) of a polynucleotide by a nanoparticle to a target tissue of interest (e.g., mammalian liver) compared to the level of delivery of a polynucleotide by a control nanoparticle to a target tissue of interest (e.g., MC3, KC2, or DLinDMA).
- a target tissue of intereste.g., mammalian liver
- the level of delivery of a nanoparticle to a particular tissuecan be measured by comparing the amount of protein produced in a tissue to the weight of said tissue, comparing the amount of polynucleotide in a tissue to the weight of said tissue, comparing the amount of protein produced in a tissue to the amount of total protein in said tissue, or comparing the amount of polynucleotide in a tissue to the amount of total polynucleotide in said tissue.
- a surrogatesuch as an animal model (e.g., a rat model).
- expression of a nucleic acid sequencerefers to one or more of the following events: (1) production of an mRNA template from a DNA sequence (e.g., by transcription); (2) processing of an mRNA transcript (e.g., by splicing, editing, 5′ cap formation, and/or 3′ end processing); (3) translation of an mRNA into a polypeptide or protein; and (4) post-translational modification of a polypeptide or protein.
- a "Formulation"includes at least a polynucleotide and one or more of a carrier, an excipient, and a delivery agent. Fragment: A "fragment,” as used herein, refers to a portion.
- fragments of proteinscan comprise polypeptides obtained by digesting full-length protein isolated from cultured cells.
- a fragmentis a subsequences of a full length protein (e.g., IL-22) wherein N-terminal, and/or C-terminal, and/or internal subsequences have been deleted.
- the fragments of a protein of the present inventionare functional fragments.
- FunctionalAs used herein, a "functional" biological molecule is a biological molecule in a form in which it exhibits a property and/or activity by which it is characterized.
- a functional fragment of a polynucleotide of the present inventionis a polynucleotide capable of expressing a functional IL-22 fragment.
- a functional fragment of IL-22refers to a fragment of wild type IL-22 (i.e., a fragment of any of its naturally occurring isoforms), or a mutant or variant thereof, wherein the fragment retains a least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95% of the biological activity of the corresponding full length protein.
- Metabolic DiseaseAs use herein the terms “metabolic disease” or “metabolic disorder” refer to diseases or disorders, respectively, affecting metabolism. As non-limiting examples, metabolic disease includes and is associated with high blood pressure, high blood sugar, high cholesterol, obesity, and diabetes. Numerous clinical variants of metabolic disease are known in the art.
- the terms “IL-22 signaling activity” and “IL-22 activity”are used interchangeably in the present disclosure and refer to the ability of IL-22 to mediates cellular responses by binding to the IL-22 receptor 1 (IL-22R1), inducing conformational changes that enable binding of the IL-22/IL-22R1 complex by the IL-10 receptor ⁇ (IL-10R ⁇ ).
- Cellular responsesinclude but are not limited to the downstream activation of the extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK).
- ERKextracellular signal-regulated kinases
- JNKc-Jun N-terminal kinase
- MAPKmitogen-activated protein kinase
- IL-22promotes the expression of downstream innate immune mediators, mitogenic modulators, and antiapoptotic modulators that promote cell survival and proliferation.
- helper lipidrefers to a compound or molecule that includes a lipidic moiety (for insertion into a lipid layer, e.g., lipid bilayer) and a polar moiety (for interaction with physiologic solution at the surface of the lipid layer).
- the helper lipidis a phospholipid.
- a function of the helper lipidis to “complement” the amino lipid and increase the fusogenicity of the bilayer and/or to help facilitate endosomal escape, e.g., of nucleic acid delivered to cells.
- Helper lipidsare also believed to be a key structural component to the surface of the LNP.
- homologyrefers to the overall relatedness between polymeric molecules, e.g. between nucleic acid molecules (e.g. DNA molecules and/or RNA molecules) and/or between polypeptide molecules. Generally, the term “homology” implies an evolutionary relationship between two molecules. Thus, two molecules that are homologous will have a common evolutionary ancestor. In the context of the present invention, the term homology encompasses both to identity and similarity.
- polymeric moleculesare considered to be "homologous" to one another if at least 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% of the monomers in the molecule are identical (exactly the same monomer) or are similar (conservative substitutions).
- the term “homologous”necessarily refers to a comparison between at least two sequences (polynucleotide or polypeptide sequences).
- Identityrefers to the overall monomer conservation between polymeric molecules, e.g., between polynucleotide molecules (e.g.
- DNA molecules and/or RNA moleculesDNA molecules and/or RNA molecules) and/or between polypeptide molecules.
- Calculation of the percent identity of two polynucleotide sequencescan be performed by aligning the two sequences for optimal comparison purposes (e.g., gaps can be introduced in one or both of a first and a second nucleic acid sequences for optimal alignment and non- identical sequences can be disregarded for comparison purposes).
- the length of a sequence aligned for comparison purposesis at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, or 100% of the length of the reference sequence.
- the nucleotides at corresponding nucleotide positionsare then compared.
- the percent identity between the two sequencesis a function of the number of identical positions shared by the sequences, taking into account the number of gaps, and the length of each gap, which needs to be introduced for optimal alignment of the two sequences.
- the comparison of sequences and determination of percent identity between two sequencescan be accomplished using a mathematical algorithm. When comparing DNA and RNA, thymine (T) and uracil (U) can be considered equivalent. Suitable software programs are available from various sources, and for alignment of both protein and nucleotide sequences.
- Bl2seqperforms a comparison between two sequences using either the BLASTN or BLASTP algorithm.
- BLASTNis used to compare nucleic acid sequences
- BLASTPis used to compare amino acid sequences.
- Other suitable programsare, e.g., Needle, Stretcher, Water, or Matcher, part of the EMBOSS suite of bioinformatics programs and also available from the European Bioinformatics Institute (EBI) at www.ebi.ac.uk/Tools/psa.
- Sequence alignmentscan be conducted using methods known in the art such as MAFFT, Clustal (ClustalW, Clustal X or Clustal Omega), MUSCLE, etc.
- Different regions within a single polynucleotide or polypeptide target sequence that aligns with a polynucleotide or polypeptide reference sequencecan each have their own percent sequence identity.
- the percent sequence identity valueis rounded to the nearest tenth. For example, 80.11, 80.12, 80.13, and 80.14 are rounded down to 80.1, while 80.15, 80.16, 80.17, 80.18, and 80.19 are rounded up to 80.2.
- the length valuewill always be an integer.
- sequence alignmentscan be generated by integrating sequence data with data from heterogeneous sources such as structural data (e.g., crystallographic protein structures), functional data (e.g., location of mutations), or phylogenetic data.
- a suitable program that integrates heterogeneous data to generate a multiple sequence alignmentis T-Coffee, available at www.tcoffee.org, and alternatively available, e.g., from the EBI.
- T-Coffeeavailable at www.tcoffee.org, and alternatively available, e.g., from the EBI.
- the final alignment used to calculate percent sequence identitycan be curated either automatically or manually.
- Insertional and deletional variantswhen referring to polypeptides are those with one or more amino acids inserted immediately adjacent to an amino acid at a particular position in a native or starting sequence. "Immediately adjacent" to an amino acid means connected to either the alpha-carboxy or alpha-amino functional group of the amino acid. "Deletional variants” when referring to polypeptides are those with one or more amino acids in the native or starting amino acid sequence removed. Ordinarily, deletional variants will have one or more amino acids deleted in a particular region of the molecule.
- IntactAs used herein, in the context of a polypeptide, the term “intact” means retaining an amino acid corresponding to the wild type protein, e.g., not mutating or substituting the wild type amino acid. Conversely, in the context of a nucleic acid, the term “intact” means retaining a nucleobase corresponding to the wild type nucleic acid, e.g., not mutating or substituting the wild type nucleobase.
- Ionizable amino lipidincludes those lipids having one, two, three, or more fatty acid or fatty alkyl chains and a pH-titratable amino head group (e.g., an alkylamino or dialkylamino head group).
- An ionizable amino lipidis typically protonated (i.e., positively charged) at a pH below the pKa of the amino head group and is substantially not charged at a pH above the pKa.
- Such ionizable amino lipidsinclude, but are not limited to DLin-MC3-DMA (MC3), (13Z,165Z)-N,N-dimethyl-3-nonydocosa-13-16- dien-1-amine (L608), and a compound of any one of Formula I, II, and II described herein (e.g., any one of Compound II, Compound VI, and Compound B).
- Linkerrefers to a group of atoms, e.g., 10-1,000 atoms, and can be comprised of the atoms or groups such as, but not limited to, carbon, amino, alkylamino, oxygen, sulfur, sulfoxide, sulfonyl, carbonyl, and imine.
- the linkercan be attached to a modified nucleoside or nucleotide on the nucleobase or sugar moiety at a first end, and to a payload, e.g., a detectable or therapeutic agent, at a second end.
- the linkercan be of sufficient length as to not interfere with incorporation into a nucleic acid sequence.
- the linkercan be used for any useful purpose, such as to form polynucleotide multimers (e.g., through linkage of two or more chimeric polynucleotides molecules or IVT polynucleotides) or polynucleotides conjugates, as well as to administer a payload, as described herein.
- Examples of chemical groups that can be incorporated into the linkerinclude, but are not limited to, alkyl, alkenyl, alkynyl, amido, amino, ether, thioether, ester, alkylene, heteroalkylene, aryl, or heterocyclyl, each of which can be optionally substituted, as described herein.
- linkersinclude, but are not limited to, unsaturated alkanes, polyethylene glycols (e.g., ethylene or propylene glycol monomeric units, e.g., diethylene glycol, dipropylene glycol, triethylene glycol, tripropylene glycol, tetraethylene glycol, or tetraethylene glycol), and dextran polymers and derivatives thereof.
- Non-limiting examples of a selectively cleavable bondinclude an amido bond can be cleaved for example by the use of tris(2-carboxyethyl)phosphine (TCEP), or other reducing agents, and/or photolysis, as well as an ester bond can be cleaved for example by acidic or basic hydrolysis.
- Methods of Administrationcan include intravenous, intramuscular, intradermal, subcutaneous, or other methods of delivering a composition to a subject.
- a method of administrationcan be selected to target delivery (e.g., to specifically deliver) to a specific region or system of a body.
- Modifiedrefers to a changed state or structure of a molecule of this disclosure. Molecules can be modified in many ways including chemically, structurally, and functionally.
- the mRNA molecules of the present inventionare modified by the introduction of non-natural nucleosides and/or nucleotides, e.g., as it relates to the natural ribonucleotides A, U, G, and C.
- Noncanonical nucleotidessuch as the cap structures are not considered “modified” although they differ from the chemical structure of the A, C, G, U ribonucleotides.
- Nanoparticle Compositionis a composition comprising one or more lipids. Nanoparticle compositions are typically sized on the order of micrometers or smaller and can include a lipid bilayer. Nanoparticle compositions encompass lipid nanoparticles (LNPs), liposomes (e.g., lipid vesicles), and lipoplexes. For example, a nanoparticle composition can be a liposome having a lipid bilayer with a diameter of 500 nm or less.
- Naturally occurringAs used herein, "naturally occurring” means existing in nature without artificial aid.
- nucleic acid sequenceThe terms “nucleic acid sequence,” “nucleotide sequence,” or “polynucleotide sequence” are used interchangeably and refer to a contiguous nucleic acid sequence. The sequence can be either single stranded or double stranded DNA or RNA, e.g., an mRNA.
- nucleic acidin its broadest sense, includes any compound and/or substance that comprises a polymer of nucleotides. These polymers are often referred to as polynucleotides.
- nucleic acids or polynucleotides of this disclosureinclude, but are not limited to, ribonucleic acids (RNAs), deoxyribonucleic acids (DNAs), threose nucleic acids (TNAs), glycol nucleic acids (GNAs), peptide nucleic acids (PNAs), locked nucleic acids (LNAs, including LNA having a ⁇ - D-ribo configuration, ⁇ -LNA having an ⁇ -L-ribo configuration (a diastereomer of LNA), 2′-amino-LNA having a 2′-amino functionalization, and 2′-amino- ⁇ -LNA having a 2′-amino functionalization), ethylene nucleic acids (ENA), cyclohexenyl nucleic acids (CeNA) or hybrids or combinations thereof.
- RNAsribonucleic acids
- DNAsdeoxyribonucleic acids
- TAAsthreose nucleic acids
- nucleotide sequence encodingrefers to the nucleic acid (e.g., an mRNA or DNA molecule) coding sequence which encodes a polypeptide.
- the coding sequencecan further include initiation and termination signals operably linked to regulatory elements including a promoter and polyadenylation signal capable of directing expression in the cells of an individual or mammal to which the nucleic acid is administered.
- the coding sequencecan further include sequences that encode signal peptides.
- Operably linkedrefers to a functional connection between two or more molecules, constructs, transcripts, entities, moieties or the like.
- Optionally substituteda phrase of the form "optionally substituted X" (e.g., optionally substituted alkyl) is intended to be equivalent to "X, wherein X is optionally substituted” (e.g., "alkyl, wherein said alkyl is optionally substituted”). It is not intended to mean that the feature "X” (e.g., alkyl) per se is optional.
- a "part" or "region" of a polynucleotideis defined as any portion of the polynucleotide that is less than the entire length of the polynucleotide.
- patientrefers to a subject who can seek or be in need of treatment, requires treatment, is receiving treatment, will receive treatment, or a subject who is under care by a trained professional for a particular disease or condition. In some embodiments, the treatment is needed, required, or received to prevent or decrease the risk of developing acute disease, i.e., it is a prophylactic treatment.
- pharmaceutically acceptableThe phrase “pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms that are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- compositionsrefers any ingredient other than the compounds described herein (for example, a vehicle capable of suspending or dissolving the active compound) and having the properties of being substantially nontoxic and non-inflammatory in a patient.
- Excipientscan include, for example: antiadherents, antioxidants, binders, coatings, compression aids, disintegrants, dyes (colors), emollients, emulsifiers, fillers (diluents), film formers or coatings, flavors, fragrances, glidants (flow enhancers), lubricants, preservatives, printing inks, sorbents, suspension or dispersing agents, sweeteners, and waters of hydration.
- antiadherentsantioxidants, binders, coatings, compression aids, disintegrants, dyes (colors), emollients, emulsifiers, fillers (diluents), film formers or coatings, flavors, fragrances, glidants (flow enhancers), lubricants, preservatives, printing inks, sorbents, suspension or dispersing agents, sweeteners, and waters of hydration.
- excipientsinclude, but are not limited to: butylated hydroxytoluene (BHT), calcium carbonate, calcium phosphate (dibasic), calcium stearate, croscarmellose, crosslinked polyvinyl pyrrolidone, citric acid, crospovidone, cysteine, ethylcellulose, gelatin, hydroxypropyl cellulose, hydroxypropyl methylcellulose, lactose, magnesium stearate, maltitol, mannitol, methionine, methylcellulose, methyl paraben, microcrystalline cellulose, polyethylene glycol, polyvinyl pyrrolidone, povidone, pregelatinized starch, propyl paraben, retinyl palmitate, shellac, silicon dioxide, sodium carboxymethyl cellulose, sodium citrate, sodium starch glycolate, sorbitol, starch (corn), stearic acid, sucrose, talc, titanium dioxide, vitamin A, vitamin E, vitamin C,
- compositions described hereinalso includes pharmaceutically acceptable salts of the compounds described herein.
- pharmaceutically acceptable saltsrefers to derivatives of the disclosed compounds wherein the parent compound is modified by converting an existing acid or base moiety to its salt form (e.g., by reacting the free base group with a suitable organic acid).
- examples of pharmaceutically acceptable saltsinclude, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- Representative acid addition saltsinclude acetate, acetic acid, adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzene sulfonic acid, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptonate, glycerophosphate, hemisulfate, heptonate, hexanoate, hydrobromide, hydrochloride, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2- naphthalenesulfonate, nicotinate, nitrate, ole
- alkali or alkaline earth metal saltsinclude sodium, lithium, potassium, calcium, magnesium, and the like, as well as nontoxic ammonium, quaternary ammonium, and amine cations, including, but not limited to ammonium, tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, and the like.
- the pharmaceutically acceptable salts of the present disclosureinclude the conventional non-toxic salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- the pharmaceutically acceptable salts of the present disclosurecan be synthesized from the parent compound that contains a basic or acidic moiety by conventional chemical methods.
- such saltscan be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are used.
- nonaqueous medialike ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are used.
- Lists of suitable saltsare found in Remington's Pharmaceutical Sciences, 17 th ed., Mack Publishing Company, Easton, Pa., 1985, p.1418, Pharmaceutical Salts: Properties, Selection, and Use, P.H. Stahl and C.G.
- solvatesmeans a compound of this disclosure wherein molecules of a suitable solvent are incorporated in the crystal lattice.
- a suitable solventis physiologically tolerable at the dosage administered.
- solvatescan be prepared by crystallization, recrystallization, or precipitation from a solution that includes organic solvents, water, or a mixture thereof.
- solventsexamples include ethanol, water (for example, mono-, di-, and tri-hydrates), N-methylpyrrolidinone (NMP), dimethyl sulfoxide (DMSO), N,N'-dimethylformamide (DMF), N,N'-dimethylacetamide (DMAC), 1,3-dimethyl- 2-imidazolidinone (DMEU), 1,3-dimethyl-3,4,5,6-tetrahydro-2-(1H)-pyrimidinone (DMPU), acetonitrile (ACN), propylene glycol, ethyl acetate, benzyl alcohol, 2-pyrrolidone, benzyl benzoate, and the like.
- NMPN-methylpyrrolidinone
- DMSOdimethyl sulfoxide
- DMFN,N'-dimethylformamide
- DMACN,N'-dimethylacetamide
- DMEU1,3-dimethyl- 2-imidazolidinone
- DMPU1,
- Pharmacokineticrefers to any one or more properties of a molecule or compound as it relates to the determination of the fate of substances administered to a living organism. Pharmacokinetics is divided into several areas including the extent and rate of absorption, distribution, metabolism and excretion.
- ADMEThis is commonly referred to as ADME where: (A) Absorption is the process of a substance entering the blood circulation; (D) Distribution is the dispersion or dissemination of substances throughout the fluids and tissues of the body; (M) Metabolism (or Biotransformation) is the irreversible transformation of parent compounds into daughter metabolites; and (E) Excretion (or Elimination) refers to the elimination of the substances from the body. In rare cases, some drugs irreversibly accumulate in body tissue.
- PolynucleotideThe term "polynucleotide” as used herein refers to polymers of nucleotides of any length, including ribonucleotides, deoxyribonucleotides, analogs thereof, or mixtures thereof.
- RNAtriple-, double- and single-stranded ribonucleic acid
- DNAtriple-, double- and single-stranded deoxyribonucleic acid
- RNAtriple-, double- and single-stranded ribonucleic acid
- polynucleotideincludes polydeoxyribonucleotides (containing 2- deoxy-D-ribose), polyribonucleotides (containing D-ribose), including tRNA, rRNA, hRNA, siRNA and mRNA, whether spliced or unspliced, any other type of polynucleotide which is an N- or C-glycoside of a purine or pyrimidine base, and other polymers containing normucleotidic backbones, for example, polyamide (e.g., peptide nucleic acids "PNAs”) and polymorpholino polymers, and other synthetic sequence-specific nucleic acid polymers providing that the polymers contain nucleobases in a configuration which allows for base pairing and base stacking, such as is found in DNA and RNA.
- PNAspeptide nucleic acids
- the polynucleotidecomprises an mRNA.
- the mRNAis a synthetic mRNA.
- the synthetic mRNAcomprises at least one unnatural nucleobase.
- all nucleobases of a certain classhave been replaced with unnatural nucleobases (e.g., all uridines in a polynucleotide disclosed herein can be replaced with an unnatural nucleobase, e.g., 5-methoxyuridine).
- the polynucleotide(e.g., a synthetic RNA or a synthetic DNA) comprises only natural nucleobases, i.e., A (adenosine), G (guanosine), C (cytidine), and T (thymidine) in the case of a synthetic DNA, or A, C, G, and U (uridine) in the case of a synthetic RNA.
- Aadenosine
- Gguanosine
- Ccytidine
- Tthymidine
- A, C, G, and Uuridine
- a codon-nucleotide sequence disclosed herein in DNA forme.g., a vector or an in-vitro translation (IVT) template
- IVTin-vitro translation
- a codon-optimized DNA sequences (comprising T) and their corresponding mRNA sequences (comprising U)are considered codon-optimized nucleotide sequence of the present invention.
- a skilled artisanwould also understand that equivalent codon-maps can be generated by replaced one or more bases with non-natural bases.
- a TTC codon(DNA map) would correspond to a UUC codon (RNA map), which in turn would correspond to a ⁇ C codon (RNA map in which U has been replaced with pseudouridine).
- Standard A-T and G-C base pairsform under conditions which allow the formation of hydrogen bonds between the N3-H and C4-oxy of thymidine and the N1 and C6-NH2, respectively, of adenosine and between the C2-oxy, N3 and C4-NH2, of cytidine and the C2- NH2, N′—H and C6-oxy, respectively, of guanosine.
- guanosine(2- amino-6-oxy-9- ⁇ -D-ribofuranosyl-purine) can be modified to form isoguanosine (2-oxy-6- amino-9- ⁇ -D-ribofuranosyl-purine).
- Such modificationresults in a nucleoside base which will no longer effectively form a standard base pair with cytosine.
- cytosine (1- ⁇ -D-ribofuranosyl-2-oxy-4-amino-pyrimidine)modification of cytosine (1- ⁇ -D-ribofuranosyl-2-oxy-4-amino-pyrimidine) to form isocytosine (1- ⁇ -D- ribofuranosyl-2-amino-4-oxy-pyrimidine-) results in a modified nucleotide which will not effectively base pair with guanosine but will form a base pair with isoguanosine (U.S. Pat. No.5,681,702 to Collins et al.). Isocytosine is available from Sigma Chemical Co. (St. Louis, Mo.); isocytidine can be prepared by the method described by Switzer et al.
- Nonnatural base pairscan be synthesized by the method described in Piccirilli et al., 1990, Nature 343:33-37, for the synthesis of 2,6-diaminopyrimidine and its complement (1-methylpyrazolo- [4,3]pyrimidine-5,7-(4H,6H)-dione.
- Other such modified nucleotide units which form unique base pairsare known, such as those described in Leach et al. (1992) J. Am. Chem. Soc. 114:3675-3683 and Switzer et al., supra.
- PolypeptideThe terms "polypeptide,” “peptide,” and “protein” are used interchangeably herein to refer to polymers of amino acids of any length.
- the polymercan comprise modified amino acids.
- the termsalso encompass an amino acid polymer that has been modified naturally or by intervention; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, or any other manipulation or modification, such as conjugation with a labeling component.
- polypeptides containing one or more analogs of an amino acidincluding, for example, unnatural amino acids such as homocysteine, ornithine, p- acetylphenylalanine, D-amino acids, and creatine
- the term, as used herein,refers to proteins, polypeptides, and peptides of any size, structure, or function.
- Polypeptidesinclude encoded polynucleotide products, naturally occurring polypeptides, synthetic polypeptides, homologs, orthologs, paralogs, fragments and other equivalents, variants, and analogs of the foregoing.
- a polypeptidecan be a monomer or can be a multi-molecular complex such as a dimer, trimer or tetramer. They can also comprise single chain or multichain polypeptides. Most commonly disulfide linkages are found in multichain polypeptides.
- the term polypeptidecan also apply to amino acid polymers in which one or more amino acid residues are an artificial chemical analogue of a corresponding naturally occurring amino acid.
- a "peptide”can be less than or equal to 50 amino acids long, e.g., about 5, 10, 15, 20, 25, 30, 35, 40, 45, or 50 amino acids long.
- Polypeptide variantrefers to molecules that differ in their amino acid sequence from a native or reference sequence. The amino acid sequence variants can possess substitutions, deletions, and/or insertions at certain positions within the amino acid sequence, as compared to a native or reference sequence. Ordinarily, variants will possess at least about 50% identity, at least about 60% identity, at least about 70% identity, at least about 80% identity, at least about 90% identity, at least about 95% identity, at least about 99% identity to a native or reference sequence.
- the term "preventing"refers to partially or completely delaying onset of an infection, disease, disorder and/or condition; partially or completely delaying onset of one or more symptoms, features, or clinical manifestations of a particular infection, disease, disorder, and/or condition; partially or completely delaying onset of one or more symptoms, features, or manifestations of a particular infection, disease, disorder, and/or condition; partially or completely delaying progression from an infection, a particular disease, disorder and/or condition; and/or decreasing the risk of developing pathology associated with the infection, the disease, disorder, and/or condition.
- Prophylacticrefers to a therapeutic or course of action used to prevent the spread of disease.
- Prophylaxisrefers to a measure taken to maintain health and prevent the spread of disease.
- An “immune prophylaxis”refers to a measure to produce active or passive immunity to prevent the spread of disease.
- PseudouridineAs used herein, pseudouridine ( ⁇ ) refers to the C-glycoside isomer of the nucleoside uridine.
- a "pseudouridine analog”is any modification, variant, isoform or derivative of pseudouridine.
- pseudouridine analogsinclude but are not limited to 1-carboxymethyl-pseudouridine, 1-propynyl-pseudouridine, 1-taurinomethyl- pseudouridine, 1-taurinomethyl-4-thio-pseudouridine, 1-methylpseudouridine (m 1 ⁇ ) (also known as N1-methyl-pseudouridine), 1-methyl-4-thio-pseudouridine (m 1 s 4 ⁇ ), 4-thio-1- methyl-pseudouridine, 3-methyl-pseudouridine (m 3 ⁇ ), 2-thio-1-methyl-pseudouridine, 1- methyl-1-deaza-pseudouridine, 2-thio-1-methyl-1-deaza-pseudouridine, dihydropseudouridine, 2-thio-dihydropseudouridine, 2-methoxyuridine, 2-methoxy-4-thio- uridine,
- reference Nucleic Acid Sequencerefers to a starting nucleic acid sequence (e.g., a RNA, e.g., an mRNA sequence) that can be sequence optimized.
- the reference nucleic acid sequenceis a wild type nucleic acid sequence, a fragment or a variant thereof.
- the reference nucleic acid sequenceis a previously sequence optimized nucleic acid sequence.
- samplerefers to a subset of its tissues, cells or component parts (e.g., body fluids, including but not limited to blood, mucus, lymphatic fluid, synovial fluid, cerebrospinal fluid, saliva, amniotic fluid, amniotic cord blood, urine, vaginal fluid and semen).
- body fluidsincluding but not limited to blood, mucus, lymphatic fluid, synovial fluid, cerebrospinal fluid, saliva, amniotic fluid, amniotic cord blood, urine, vaginal fluid and semen).
- a samplefurther can include a homogenate, lysate or extract prepared from a whole organism or a subset of its tissues, cells or component parts, or a fraction or portion thereof, including but not limited to, for example, plasma, serum, spinal fluid, lymph fluid, the external sections of the skin, respiratory, intestinal, and genitourinary tracts, tears, saliva, milk, blood cells, tumors, organs.
- a samplefurther refers to a medium, such as a nutrient broth or gel, which can contain cellular components, such as proteins or nucleic acid molecule.
- Signal SequenceAs used herein, the phrases “signal sequence,” “signal peptide,” and “transit peptide” are used interchangeably and refer to a sequence that can direct the transport or localization of a protein to a certain organelle, cell compartment, or extracellular export. The term encompasses both the signal sequence polypeptide and the nucleic acid sequence encoding the signal sequence. Thus, references to a signal sequence in the context of a nucleic acid refer in fact to the nucleic acid sequence encoding the signal sequence polypeptide. Similarity: As used herein, the term “similarity” refers to the overall relatedness between polymeric molecules, e.g. between polynucleotide molecules (e.g.
- Single unit doseis a dose of any therapeutic administered in one dose/at one time/single route/single point of contact, i.e., single administration event.
- the term “specific delivery,” “specifically deliver,” or “specifically delivering”means delivery of more (e.g., at least 1.5 fold more, at least 2-fold more, at least 3-fold more, at least 4-fold more, at least 5-fold more, at least 6-fold more, at least 7-fold more, at least 8-fold more, at least 9-fold more, at least 10-fold more) of a polynucleotide by a nanoparticle to a target tissue of interest (e.g., mammalian liver) compared to an off-target tissue (e.g., mammalian spleen).
- a target tissue of intereste.g., mammalian liver
- an off-target tissuee.g., mammalian spleen
- the level of delivery of a nanoparticle to a particular tissuecan be measured by comparing the amount of protein produced in a tissue to the weight of said tissue, comparing the amount of polynucleotide in a tissue to the weight of said tissue, comparing the amount of protein produced in a tissue to the amount of total protein in said tissue, or comparing the amount of polynucleotide in a tissue to the amount of total polynucleotide in said tissue.
- a polynucleotideis specifically provided to a mammalian kidney as compared to the liver and spleen if 1.5, 2-fold, 3-fold, 5-fold, 10-fold, 15 fold, or 20 fold more polynucleotide per 1 g of tissue is delivered to a kidney compared to that delivered to the liver or spleen following systemic administration of the polynucleotide.
- a surrogatesuch as an animal model (e.g., a rat model).
- Stablerefers to a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and in some cases capable of Formulation into an efficacious therapeutic agent.
- StabilizedAs used herein, the term “stabilize,” “stabilized,” “stabilized region” means to make or become stable.
- SubjectBy “subject” or “individual” or “animal” or “patient” or “mammal,” is meant any subject, particularly a mammalian subject, for whom diagnosis, prognosis, or therapy is desired.
- Mammalian subjectsinclude, but are not limited to, humans, domestic animals, farm animals, zoo animals, sport animals, pet animals such as dogs, cats, guinea pigs, rabbits, rats, mice, horses, cattle, cows; primates such as apes, monkeys, orangutans, and chimpanzees; canids such as dogs and wolves; felids such as cats, lions, and tigers; equids such as horses, donkeys, and zebras; bears, food animals such as cows, pigs, and sheep; ungulates such as deer and giraffes; rodents such as mice, rats, hamsters and guinea pigs; and so on.
- pet animalssuch as dogs, cats, guinea pigs, rabbits, rats, mice, horses, cattle, cows; primates such as apes, monkeys, orangutans, and chimpanzees; canids such as dogs
- the mammalis a human subject.
- a subjectis a human patient.
- a subjectis a human patient in need of treatment.
- substantiallyrefers to the qualitative condition of exhibiting total or near-total extent or degree of a characteristic or property of interest.
- One of ordinary skill in the biological artswill understand that biological and chemical characteristics rarely, if ever, go to completion and/or proceed to completeness or achieve or avoid an absolute result.
- the term “substantially”is therefore used herein to capture the potential lack of completeness inherent in many biological and chemical characteristics.
- Substantially equalAs used herein as it relates to time differences between doses, the term means plus/minus 2%.
- Suffering fromAn individual who is "suffering from” a disease, disorder, and/or condition has been diagnosed with or displays one or more symptoms of the disease, disorder, and/or condition.
- Susceptible toAn individual who is “susceptible to” a disease, disorder, and/or condition has not been diagnosed with and/or cannot exhibit symptoms of the disease, disorder, and/or condition but harbors a propensity to develop a disease or its symptoms.
- an individual who is susceptible to a disease, disorder, and/or conditioncan be characterized by one or more of the following: (1) a genetic mutation associated with development of the disease, disorder, and/or condition; (2) a genetic polymorphism associated with development of the disease, disorder, and/or condition; (3) increased and/or decreased expression and/or activity of a protein and/or nucleic acid associated with the disease, disorder, and/or condition; (4) habits and/or lifestyles associated with development of the disease, disorder, and/or condition; (5) a family history of the disease, disorder, and/or condition; and (6) exposure to and/or infection with a microbe associated with development of the disease, disorder, and/or condition.
- a genetic mutation associated with development of the disease, disorder, and/or conditionfor example, metabolic disease
- a genetic polymorphismassociated with development of the disease, disorder, and/or condition
- increased and/or decreased expression and/or activity of a protein and/or nucleic acid associated with the disease, disorder, and/or condition(4) habits and
- an individual who is susceptible to a disease, disorder, and/or conditionwill develop the disease, disorder, and/or condition. In some embodiments, an individual who is susceptible to a disease, disorder, and/or condition will not develop the disease, disorder, and/or condition.
- Sustained releaseAs used herein, the term “sustained release” refers to a pharmaceutical composition or compound release profile that conforms to a release rate over a specific period of time.
- SyntheticThe term “synthetic” means produced, prepared, and/or manufactured by the hand of man. Synthesis of polynucleotides or other molecules of the present invention can be chemical or enzymatic.
- Targeted CellsAs used herein, “targeted cells” refers to any one or more cells of interest.
- target tissuerefers to any one or more tissue types of interest in which the delivery of a polynucleotide would result in a desired biological and/or pharmacological effect.
- target tissues of interestinclude specific tissues, organs, and systems or groups thereof.
- a target tissuecan be a liver, a kidney, a lung, a spleen, or a vascular endothelium in vessels (e.g., intra-coronary or intra- femoral).
- an “off-target tissue”refers to any one or more tissue types in which the expression of the encoded protein does not result in a desired biological and/or pharmacological effect.
- the presence of a therapeutic agent in an off-target issuecan be the result of: (i) leakage of a polynucleotide from the administration site to peripheral tissue or distant off- target tissue via diffusion or through the bloodstream (e.g., a polynucleotide intended to express a polypeptide in a certain tissue would reach the off-target tissue and the polypeptide would be expressed in the off-target tissue); or (ii) leakage of an polypeptide after administration of a polynucleotide encoding such polypeptide to peripheral tissue or distant off-target tissue via diffusion or through the bloodstream (e.g., a polynucleotide would expressed a polypeptide in the target tissue, and the polypeptide would diffuse to peripheral tissue).
- Targeting sequencerefers to a sequence that can direct the transport or localization of a protein or polypeptide.
- therapeutic agentrefers to an agent that, when administered to a subject, has a therapeutic, diagnostic, and/or prophylactic effect and/or elicits a desired biological and/or pharmacological effect.
- an mRNA encoding an IL-22 polypeptidecan be a therapeutic agent.
- therapeutically effective amountmeans an amount of an agent to be delivered (e.g., nucleic acid, drug, therapeutic agent, diagnostic agent, prophylactic agent, etc.) that is sufficient, when administered to a subject suffering from or susceptible to an infection, disease, disorder, and/or condition, to treat, improve symptoms of, diagnose, prevent, and/or delay the onset of the infection, disease, disorder, and/or condition.
- therapeutically effective outcomemeans an outcome that is sufficient in a subject suffering from or susceptible to an infection, disease, disorder, and/or condition, to treat, improve symptoms of, diagnose, prevent, and/or delay the onset of the infection, disease, disorder, and/or condition.
- Transcriptionrefers to methods to produce mRNA (e.g., an mRNA sequence or template) from DNA (e.g., a DNA template or sequence).
- Treating, treatment, therapyrefers to partially or completely alleviating, ameliorating, improving, relieving, delaying onset of, inhibiting progression of, reducing severity of, and/or reducing incidence of one or more symptoms or features of a disease, e.g., metabolic disease.
- treating metabolic diseasecan refer to diminishing symptoms associate with the disease, prolong the lifespan (increase the survival rate) of patients, reducing the severity of the disease, preventing or delaying the onset of the disease, etc.
- Treatmentcan be administered to a subject who does not exhibit signs of a disease, disorder, and/or condition and/or to a subject who exhibits only early signs of a disease, disorder, and/or condition for the purpose of decreasing the risk of developing pathology associated with the disease, disorder, and/or condition.
- Unmodifiedrefers to any substance, compound or molecule prior to being changed in some way. Unmodified can, but does not always, refer to the wild type or native form of a biomolecule.
- Uracilis one of the four nucleobases in the nucleic acid of RNA, and it is represented by the letter U.
- Uracilcan be attached to a ribose ring, or more specifically, a ribofuranose via a ⁇ -N 1 -glycosidic bond to yield the nucleoside uridine.
- the nucleoside uridineis also commonly abbreviated according to the one letter code of its nucleobase, i.e., U.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Dispersion Chemistry (AREA)
- Biophysics (AREA)
- Diabetes (AREA)
- General Chemical & Material Sciences (AREA)
- Toxicology (AREA)
- Gastroenterology & Hepatology (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Zoology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Physics & Mathematics (AREA)
- Biomedical Technology (AREA)
- Nanotechnology (AREA)
- Optics & Photonics (AREA)
- Biotechnology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
LIPID NANOPARTICLES AND POLYNUCLEOTIDES ENCODING EXTENDED SERUM HALF-LIFE INTERLEUKIN-22 FOR THE TREATMENT OF METABOLIC DISEASE CROSS-REFERENCE TO RELATED APPLICATIONS This application claims the benefit under 35 U.S.C. § 119(e) of U.S. Provisional Patent Application No.63/433,292, filed December 16, 2022, the entire contents of which are incorporated herein by reference. BACKGROUND The incidence of metabolic diseases, such as obesity, type 2 diabetes, and non- alcoholic fatty liver disorder (NAFLD), is of growing global concern, impacting over 650 million, 438 million, and 882 million individuals globally, respectively. Although both invasive and non-invasive treatment options for metabolic diseases are available, many of these interventions are difficult to maintain or are associated with safety concerns. Growing evidence suggests that, as an alternative, the therapeutic use of endogenous cytokines may alleviate the symptoms associated with metabolic disease with improved safety profiles. A member of the IL-10 cytokine family, Interleukin-22 (IL-22) mediates cellular responses by binding to the IL-22 receptor 1 (IL-22R1), inducing conformational changes that enable binding of the IL-22/IL-22R1 complex by the IL-10 receptor β (IL-10Rβ). This in turn results in downstream activation of the extracellular signal-regulated kinases (ERK), c- Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK) pathways. Through these pathways, IL-22 promotes the expression of downstream innate immune mediators, mitogenic modulators, and antiapoptotic modulators that promote survival, proliferation, and regeneration. However, IL-22 exhibits a short serum half-life, which may limit the use of IL-22 in clinical applications. In view of the significant problems associated with existing treatments for metabolic disease, there is an unmet need for improved treatments for metabolic diseases. SUMMARY In one aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a lipid nanoparticle comprising: (a) an ionizable lipid of Formula (I): or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is: wherein denotes a point of attachmeaα aβ aγ aδ
or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is: wherein denotes a point of attachmeaα aβ aγ aδ
 nt; wherein R , R , R , and R are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
nt; wherein R , R , R , and R are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and , wherein
, wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; wherein n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13; and (b) an mRNA molecule comprising an open reading frame (ORF) encoding an Interleukin-22 (IL- 22) polypeptide or an IL-22 polypeptide-containing fusion protein. In some embodiments, in the compound of Formula (I): R’a is R’branched; R’branched is denotes a point of attacaα aβ aγ aδ
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; wherein n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13; and (b) an mRNA molecule comprising an open reading frame (ORF) encoding an Interleukin-22 (IL- 22) polypeptide or an IL-22 polypeptide-containing fusion protein. In some embodiments, in the compound of Formula (I): R’a is R’branched; R’branched is denotes a point of attacaα aβ aγ aδ hment; and R is C2-12 alkyl; R , R , and R are each H; R2 and R3 are each C5-14 alkyl; R4 is10
hment; and R is C2-12 alkyl; R , R , and R are each H; R2 and R3 are each C5-14 alkyl; R4 is10 ; R is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, in the compound of Formula (I): R’a is R’branched; R’branched is denotesaα aβ aγ aδ 2
; R is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, in the compound of Formula (I): R’a is R’branched; R’branched is denotesaα aβ aγ aδ 2 a point of attachment; R , R , R , and R are each H; R and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, in the compound of Formula (I): R’a is R’branched; R’branched is
a point of attachment; R , R , R , and R are each H; R and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each - C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, in the compound of Formula (I): R’a is R’branched; R’branched is denotes a point of attachment; Raβ and Raδ are each H; Raγ is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the ionizable lipid is:
denotes a point of attachment; Raβ and Raδ are each H; Raγ is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the ionizable lipid is: , or N-oxides, salts, or isomers thereof. In some embodiments, the lipid nanoparticle further comprises: a phospholipid; a structural lipid; and a PEG-lipid. In some embodiments, the lipid nanoparticle comprises: 40-50 mol% of the ionizable lipid, 30-45 mol% of the structural lipid, 5-15 mol% of the phospholipid, and 1-5 mol% of the PEG-lipid. In some embodiments, the lipid nanoparticle comprises: 45-50 mol.% of the ionizable amino lipid, 10-15 mol.% of the phospholipid, 35-40 mol.% of the structural lipid, and 1-3 mol.% of the PEG-modified lipid. In some embodiments, the phospholipid comprises comprises 1,2-distearoyl-sn-glycero- 3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2- dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3- phosphocholine (DPPC), 1,2-diundecanoyl-sn-glycero-phosphocholine (DUPC), 1-palmitoyl-2- oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3- phosphocholine (18:0 Diether PC), 1-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3- phosphocholine (OChemsPC), 1-hexadecyl-sn-glycero-3-phosphocholine (C16 Lyso PC), 1,2- dilinolenoyl-sn-glycero-3-phosphocholine,1,2-diarachidonoyl-sn-glycero-3-phosphocholine, 1,2- didocosahexaenoyl-sn-glycero-3-phosphocholine, 1,2-diphytanoyl-sn-glycero-3- phosphoethanolamine (ME 16.0 PE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine, 1,2- dilinoleoyl-sn-glycero-3-phosphoethanolamine, 1,2-dilinolenoyl-sn-glycero-3- phosphoethanolamine, 1,2-diarachidonoyl-sn-glycero-3-phosphoethanolamine, 1,2- didocosahexaenoyl-sn-glycero-3-phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac- (1-glycerol) sodium salt (DOPG), sphingomyelin, and mixtures thereof. In some embodiments, the phospholipid comprises DSPC. In some embodiments, the structural lipid is selected from the group consisting of: cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, derivatives thereof, and mixtures thereof. In some embodiments, the structural lipid comprises cholesterol or a derivative thereof. In some embodiments, the PEG-lipid is selected from: 1,2-dimyristoyl-sn-glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N- [amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG- dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG-DAG), PEG- dipalmitoyl phosphatidylethanolamine (PEG-DPPE), PEG-l,2-dimyristyloxlpropyl-3-amine (PEG-c-DMA), or combinations thereof. In some embodiments, the PEG-lipid comprises PEG- DMG. In some embodiments, the mRNA molecule, when administered as a single dose to a human subject, is sufficient to increase serum IL-22 levels in the human subject to a level at least 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20-fold, 30-fold, 40-fold, or at least 50-fold higher than the level observed prior to the administration, for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120 hours, or at least one week post-administration. In some embodiments, the mRNA molecule, when administered as a single dose to a human subject, is sufficient to: (i) increase serum IL-22 levels in the human subject by at least about 1.1-fold, at least about 1.2-fold, at least about 1.5-fold, at least about 2.0-fold, at least 5.0- fold, or at least 10-fold, compared to the human subject’s baseline serum IL-22 levels for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120 hours, or at least one week post-administration; or (ii) increase IL-22 activity in the human subject by at least about 1.5-fold, at least bout 2.0-fold, or at least about 2.5-fold, compared to the human subject’s baseline IL-22 activity for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120, or at least one week post-administration. In some embodiments, an mRNA molecule comprises an open reading frame (ORF) encoding an Interleukin-22 (IL-22) polypeptide. In some embodiments, an mRNA molecule comprises an open reading frame (ORF) encoding an IL-22 polypeptide-containing fusion protein. In some embodiments, IL-22 polypeptide encoded by the mRNA molecule has an amino acid sequence identical to any one of SEQ ID NO:1-4. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to any one of SEQ ID NO:28-38. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to any one of SEQ ID NO:28-33. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to any one of SEQ ID NO:34-38. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25, SEQ ID NO:26, SEQ ID NO:27, SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38. In some embodiments, the ORF is at least 65% identical to SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328. In some embodiments, the mRNA molecule further comprises a 5’UTR having a nucleic acid sequence according to SEQ ID NO:50, SEQ ID NO:55, or SEQ ID NO:56. In some embodiments, the mRNA molecule further comprises a 3’UTR having a nucleic acid sequence according to SEQ ID NO:108, SEQ ID NO:125, or SEQ ID NO:139. In some embodiments, the mRNA molecule comprises a poly-A region, wherein the poly-A region about 100 nucleotides in length. In some embodiments, the mRNA molecule comprises at least one chemically modified nucleobase, sugar, backbone, or any combination thereof. In some embodiments, the at least one chemically modified nucleobase is selected from the group consisting of pseudouracil (ψ), N1-methylpseudouracil (m1ψ), 1-ethylpseudouracil, 2- thiouracil (s2U), 4’-thiouracil, 5-methylcytosine, 5-methyluracil, 5-methoxyuracil, and any combination thereof. In some embodiments, the mRNA molecule further comprises a 5′ terminal cap. In some embodiments, the 5′ terminal cap comprises a m7G-ppp-Gm-AG, Cap0, Cap1, ARCA, inosine, N1-methyl-guanosine, 2′-fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino- guanosine, LNA-guanosine, 2-azidoguanosine, Cap2, Cap4, 5′ methylG cap, or an analog thereof. In some embodiments, the 5’ terminal cap comprises m7G-ppp-Gm-AG or Cap1. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a pharmaceutical composition comprising: the lipid nanoparticle according to any of the above embodiments; and a pharmaceutically acceptable carrier. In some embodiments, the pharmaceutical composition is for use in treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject. In some embodiments, the pharmaceutical composition is for use in reducing body weight, reducing total cholesterol levels, reducing total HDL cholesterol levels, reducing total LDL cholesterol levels, reducing total triglyceride levels, reducing liver steatosis, or reducing blood glucose levels in a subject. In another aspect, which may be combined with any other aspect or embodiment, the lipid nanoparticle, IL-22 polypeptide, or mRNA molecule according to any of the above embodiments is for use in the manufacture of a medicament or pharmaceutical composition for use in treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject. In another aspect, which may be combined with any other aspect or embodiment, the lipid nanoparticle, IL-22 polypeptide, or mRNA molecule according to any of the above embodiments is for use in the manufacture of a medicament or pharmaceutical composition for use in reducing body weight, reducing total cholesterol levels, reducing total HDL cholesterol levels, reducing total LDL cholesterol levels, reducing total triglyceride levels, reducing liver steatosis, or reducing blood glucose levels in a subject. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of treating or delaying the onset and/or progression of metabolic disease in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of increasing serum IL-22 levels in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of increasing IL-22 activity in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of increasing one or more of serum IL-22 levels or IL-22 activity in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing body weight of a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing total cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing total HDL cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing total LDL cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing total triglyceride levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing liver steatosis in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing blood glucose levels in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In some embodiments, the administration is a single administration. In some embodiments, the administration is a repeated administration. In some embodiments, the administration is about once per day, about once per week, about once per two weeks, or about once per month. In some embodiments, the pharmaceutical composition is administered intravenously, intramuscularly, or subcutaneously. In one embodiment, the pharmaceutical composition is administered intravenously. In another embodiment, the pharmaceutical composition is administered subcutaneously. In another embodiment, the pharmaceutical composition is administered intramuscularly. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to an IL-22 polypeptide-containing fusion protein having an amino acid sequence identical to SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25, SEQ ID NO:26, SEQ ID NO:27, SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38. In some embodiments, the amino acid sequence is identical to SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a pharmaceutical composition comprising: the IL-22 polypeptide according to any of the above embodiments; and a pharmaceutically acceptable carrier. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a cell comprising the IL-22 polypeptide according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a codon-optimized mRNA molecule encoding the IL-22 polypeptide according to any of the above embodiments. In some embodiments, the codon-optimized mRNA molecule has a nucleic acid sequence having at least about 60% identity to the nucleic acid sequence of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328. In some embodiments, the codon-optimized mRNA molecule has a sequence having at least about 60% identity to SEQ ID NO:350, SEQ ID NO:351, SEQ ID NO:352, SEQ ID NO:353, SEQ ID NO:354, SEQ ID NO:355, SEQ ID NO:356, SEQ ID NO:357, SEQ ID NO:358, SEQ ID NO:359, SEQ ID NO:360, SEQ ID NO:361, SEQ ID NO:362, SEQ ID NO:363, SEQ ID NO:364, SEQ ID NO:365, SEQ ID NO:366, SEQ ID NO:367, or SEQ ID NO:368. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a cell comprising the mRNA molecule according to any of the above embodiments. BRIEF DESCRIPTION OF THE DRAWINGS FIG.1A is a graph showing levels of N- or C-terminally MSA-linked IL-22 in conditioned media determined by ELISA 24 hours after the transfection of an extended serum half-life IL-22 mRNA construct into HeLa cells. FIG.1B is a graph showing detection of STAT3 pathway activation by densitometry in HEK-Blue-IL-22 cells incubated with extended serum half-life IL-22 construct #1- containing media for 24 hours. Results are representative of 3 independent biological replicates (n=3, mean ± SD). FIG.1C is a schematic illustrating a dose time course used in experiments described herein. Briefly, N-terminally MSA-linked IL-22 mRNA (construct #1) was intravenously administered to C57BL/6 female mice from Charles River at 0.5 mg/kg (n=5) or 0.25 mg/kg (n=5). Sera were collected for analysis at 6 h post-administration, and daily from Day 3 through Day 7. FIG.1D is a graph showing IL-22 protein levels in sera obtained from mice administered with construct #1. Protein levels were determined by ELISA. FIG.2A is a schematic illustrating a dose time course used in experiments described herein. Briefly, Male C57BL/6 from Taconic mice were fed on the AMLN diet from 6 weeks of age (Week -18). LNP-encapsulated MSA (n=9) or construct #1 (n=9) mRNA was administered at 0.5 mg/kg weekly over 6 weeks to AMLN fed mice with an untreated group of mice (n=5) maintained in parallel. Sera were collected weekly from Week -1 through Week 6. Age-matched chow diet-fed mice (n=6) were included for comparisons at the end of the treatment period. FIG.2B is a set of graphs showing body weights of mice throughout the treatment period. FIG.2C is a set of graphs showing total serum cholesterol, LDL cholesterol, HDL cholesterol, and triglyceride levels determined at Week -1 and 72 hours after the administration of each dose, expressed as % change above baseline. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test for body weight and the serum lipid analyses. Analyses of total serum cholesterol excluded 2 mice, 1 in each of the construct #1 and untreated groups, due to insufficient volume of pre-bleed samples. FIG.3A is a graph showing liver weights measured 72 hours after the last dose in a 6- week treatment period in which MSA (n=9) or construct #1 (n=9) was administered at 0.5 mg/kg to male AMLN diet-fed C57BL/6Tac mice. An untreated group of mice (n=5) was maintained in parallel. FIG.3B shows representative images of H&E-stained liver and epididymal adipose tissue samples obtained from mice in the various treatment conditions described above. FIG 3C is a set of graphs quantifying the scoring of levels of microvesicular and macrovesicular steatosis, inflammation, and liver and adipose hypertrophy in images of H&E-stained liver and epididymal adipose tissue samples obtained from mice in the various treatment conditions described above. FIG 3D is a set of graphs showing differential expression levels of a select set of fibrosis and lipogenesis associated genes were determined relative to Rpl19 expression using qRT-PCR in MSA and construct #1 treated and untreated AMLN diet-fed mice and matched chow diet-fed control mice. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by one-way ANOVA for histology scoring and gene expression. One construct #1 treated mouse was excluded from analyses due to pathology indicating an infection. FIG.4A is a graph showing serum IL-22 levels measured weekly 72 hours after the administration of LNP-encapsulated construct #1 mRNA for 6 weeks in C57BL/6Tac mice fed the AMLN diet. FIG.4B is a graph showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored every week 72 hours following the administration of construct #1 or MSA mRNA at 0.5 mg/kg to mice fed with the AMLN diet. FIG.4C shows representative images of PSR and F4/80 stained liver tissue at the end of the treatment period in construct #1 treated, MSA treated, and untreated mice fed the AMLN diet and untreated mice fed a chow diet. FIG.4D is a set of graphs quantifying and scoring F4/80-positive macrophages and liver fibrosis level using microscopic evaluation. FIG.5 shows a set of graphs showing the expression of genes involved in fibrosis and lipogenesis in AMLN or chow fed C57BL/6Tac mice treated with weekly administration of LNP-encapsulated mRNA construct #1 or MSA mRNA over 6 weeks. LNP-encapsulated MSA (n=9) or construct #1 (n=9) mRNA (0.5 mg/kg each) were administered weekly for 6 weeks to C57BL/6Tac mice that were fed on the AMLN or chow diet from 6 weeks of age for 18 weeks. The untreated groups of mice (AMLN diet group) was maintained in parallel (n=5). Animals fed chow diet as control (n=6) were added at the end of the study.72 hours after the final dose of LNP-encapsulated mRNA, expression of genes involved in fibrosis and lipogenesis was assessed by RT-qPCR. All the genes were analyzed relative to rpl19 mRNA expression. For the qRT-PCR results, the average of MSA treated mice was set to 1. Significance levels (*p< 0.05; *** p< 0.001) are shown relative to MSA treated mice and were determined by one-way ANOVA and Dunnett’s post-hoc test. Not significant, ns. FIG.6 shows the results of a differential gene expression analysis in liver tissue of construct #1 treated mice. Total RNA was used for quantification of gene expression using the NanoString Metabolic Pathway Panel with these expressed as log2 fold change of construct #1 treated over MSA treated and plotted against log10 adjusted p-value. Genes were classified into categories by functional association. AMP-activated protein kinase (AMPK), glycolysis, cytokine, and chemokine; adj. = adjusted. FIG.7A is a schematic illustrating a dose time course used in experiments described herein. Male C57BL/6 from Taconic mice were fed on the AMLN or chow diet from 6 weeks of age (Week -18). LNP-encapsulated MSA (n=8) or construct #1 (n=11) mRNA was administered at 0.5 mg/kg weekly over 3 weeks to AMLN fed mice. Two untreated groups of mice (1 AMLN diet group (n=9) and 1 chow diet group (n=7)) were maintained in parallel. Sera were collected for ELISA and lipid analysis at the indicated timepoints. FIG.7B is a graph showing levels of IL-22 in sera from treated mice measured 72 hours after each administration. FIG.7C is a graph showing mouse body weight monitored through Day 38 without additional treatment. FIG.7D is a graph showing mouse food intake monitored through Day 38 without additional treatment. FIG.7E is a set of graphs showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels were measured after sera collection. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by Two-way ANOVA and Dunnett’s post-hoc test for the different time points. FIG.8A is a schematic illustrating a dose time course used in experiments described herein. Male C57BL/6mice from Taconic were fed on the AMLN or chow diet from 6 weeks of age (Week -18). LNP-encapsulated MSA (n=9) or construct #1 (n=9) mRNA was administered at 0.5 mg/kg every other week on Days 0, 14, and 28 to AMLN fed mice. Two untreated groups of mice (1 AMLN diet group (n=8) and 1 chow diet group (n=6)) were maintained in parallel. Sera were collected for ELISA and lipid analysis at the indicated timepoints. FIG.8B is a graph showing levels of IL-22 in sera from treated mice were measured 72 h after each administration, on days 3, 17, and 31. FIG.8C is a graph showing mouse body weight monitored through Day 38 without additional treatment. Mice were administered MSA or construct #1 via intravenous (IV) or subcutaneous (SC) administration. FIG.8D is a set of graphs showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels measured after sera collection. Mice were administered MSA or construct #1 via intravenous (IV) or subcutaneous (SC) administration. FIG.8E shows representative images of H&E stained liver and adipose tissue samples. FIG.8F is a set of graphs showing differential expression levels determined relative to Rpl19 expression using qRT-PCR for a select set of fibrosis and lipogenesis associated genes in treated mice and untreated AMLN diet and chow diet fed mice. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test. FIG.9A is a schematic illustrating a dose time course used in experiments described herein. Male CETP/APOB mice from Taconic were fed on the AMLN diet from Day -42. LNP-encapsulated MSA or construct #1 mRNA was administered at 0.5 mg/kg weekly for 6 weeks to AMLN fed mice (n=10 mice/group). Sera were collected for ELISA and lipid analysis at the indicated timepoints. FIG.9B is a set of graphs showing mouse body weight monitored through Day 39. FIG.9C is a set of graphs showing mouse total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored through Day 39. FIG.9D is a panel of representative images of H&E stained liver and adipose tissue samples obtained from MSA and construct #1 treated mice. FIG.9E is a set of graphs quantifying and grading for microvesicular and macrovesicular steatosis, inflammation, and adipose hypertrophy. FIG.9F is a set of graphs showing differential expression levels determined relative to Rpl19 expression using qRT-PCR for a select set of fibrosis and lipogenesis associated genes in MSA and construct #1 treated mice. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test or by t-test for the histology score and the gene expression analyses. FIG.10A is a graph showing IL-22 serum levels in mice after weekly administration of construct #1 or MSA mRNA (0.5 mg/kg) in CETP/APOB mice fed an AMLN diet. FIG.10B is a graph showing IL-22 serum levels in mice after weekly administration of construct #1 or MSA mRNA (0.5 mg/kg) in CETP/APOB mice fed a chow diet. FIG.10C is a panel of representative images of H&E stained liver tissue samples from AMLN fed MSA and construct #1 treated mice. FIG.10D is a graph showing a quantification of F4/80 stained liver macrophages in AMLN fed MSA and construct #1 treated mice. FIG.10E is a set of graphs showing differential expression levels determined using qRT-PCR for a select set of genes of interest in AMLN-fed CETP/APOB mice treated with MSA or construct #1. Expression is expressed as fold change relative to Rpl19. FIG.10F is a graph showing fasting PYY levels determined using ELISA. Serum PYY analyses excluded 3 mice in the MSA group and 1 in the construct #1 group due to insufficient volume after fastig samples. Significance levels (*p<0.05; ***p<0.001) were determined by one-way ANOVA and Dunnett’s post-hoc test. FIG.11A is a schematic illustrating a dose time course used in experiments described herein. LNP-encapsulated MSA or construct #1 mRNA was administered at 0.5 mg/kg weekly for 6 weeks to male chow diet fed CETP/APOB mice (n=10 mice/group). Sera were collected for lipid profile analysis at the indicated timepoints. FIG.11B is a graph showing mouse body weight monitored through Day 39. FIG.11C is a graph showing mouse total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored through Day 39. Significance levels (*p<0.05; ***p<0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test. Not significant, ns. FIG.12A is a schematic illustrating a dose time course used in experiments described herein. Two doses of LNP-encapsulated MSA or construct #1 mRNA (0.5 mg/kg each) were administered 1 week apart to male C57BL/6NTac mice that were fed an AMLN or chow diet from 6 weeks of age (Day -126) (n=10 mice/group). One day after dose 2 (Day 8), mice were fasted for 12 h, followed by a 1 h ad libitum refeeding period. Sera were collected before and after the fasting period and after refeeding. FIG.12B is a graph showing mouse body weight measured on Days 1, 3, 7, 8, and 9. FIG.12C is a set of graphs showing differential weight gain calculated on Day 9, food intake measured as the total intake throughout the duration of the study, energy intake calculated as food intake multiplied by metabolizable energy content in the diet, metabolic efficiency calculated as mg of body weight gain over energy intake (kcal), fasting and refeeding glucose levels determined using Abbott Alphatrak 2 glucometers, and Peptide YY (PYY) levels determined using ELISA. Significance levels (*p<0.05; ***p<0.001; Not significant, ns.) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test. FIG.13A is a schematic illustrating a dose time course used in experiments described herein. Two doses of LNP-encapsulated MSA or construct #1 mRNA (0.5 mg/kg each) were administered 1 week apart to male C57BL/6NTac mice from Taconic that were fed an AMLN or chow diet from 6 weeks of age (Day -126) (n=10 mice/group). One day after dose 2 (Day 8), mice were fasted for 6 hours, followed by administration of 2 g/kg of glucose in sterile 0.9% saline IP in a glucose tolerance test (GTT). Glucose levels were measured before (0 min) as well as after (15min, 30min, 60min and 120min) glucose administration. FIG.13B is a graph showing blood glucose levels in AMLN-fed animals during the course of the GTT. FIG.13C is a graph showing blood glucose levels in chow diet-fed animals during the course of the GTT. FIG.13D is a graph showing the area under the curve (AUC) for the glucose traces in chow and AMLN diet-fed mice shown in (B) and (C). Significance levels (*p<0.05; ***p<0.001; Not significant, ns.) are shown relative to MSA treated mice and were determined by two-way ANOVA and Sidak (B,C) or Tukey (D) correction for multiple comparison. N=8 for chow MSA, n=9 for chow construct #1, n=10 for both AMLN diet groups. FIG.14A is a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various mouse IL-22 mRNA constructs, as measured by ELISA. FIG.14B is a graph showing the bioactivity of mouse IL-22 mRNA constructs as a function of concentration, as measured in conditioned medium samples from HEK-Blue IL- 22 cells treated with IL-22 construct protein. The IL-22 bioactivity in the conditioned medium samples was quantified by measuring the sample OD at 655 nm following incubation in QUANTI-Blue solution. FIG.15A is a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various human IL-22 mRNA constructs, as measured by ELISA. The IL-22 concentration is reported in units of pg/mL. FIG.15B is a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various human IL-22 mRNA constructs, as measured by ELISA. The IL-22 concentration is reported as a concentration in nM. FIG.16 is a graph showing the bioactivity of human IL-22 mRNA constructs as a function of concentration, as measured in conditioned medium samples from HEK-Blue IL- 22 cells treated with IL-22 construct protein. The IL-22 bioactivity in the conditioned medium samples was quantified by measuring the sample OD at 655 nm following incubation in QUANTI-Blue solution. FIG.17 is a schematic illustrating a dose time course used in experiments described herein. Male C57BL/6Tac mice were fed an AMLN diet starting from 6 weeks of age through 40 weeks of age, for a total of 34 weeks of AMLN feeding. LNP-encapsulated MSA mRNA (n=10) or Construct #1 mRNA was administered at 0.5 mg/kg weekly IV (n=10) or 0.1 mg/kg weekly IM (n=10) to mice fed the AMLN diet, with an untreated group of mice (n=8) and mice fed in normal chow diet (n=10) maintained in parallel. Body weight measurements and serum collection occurred at the indicated time points. FIG.18 is a graph showing the % change in mouse bodyweight (BW) over time as a function of treatment condition. FIG.19A is a graph showing the daily caloric intake (kcal) per mouse over time as a function of treatment condition. FIG.19B is a graph showing the daily food intake (g) per mouse over time as a function of treatment condition. FIG.19C is a graph showing the cumulative caloric intake (kcal) per mouse over time as a function of treatment condition. FIG.20A is a graph showing the weight gain (g) per mouse as a function of treatment condition. FIG.20B is a graph showing the metabolic efficiency (g weight gain / MJ caloric intake) per mouse as a function of treatment condition. FIG.21A is a graph showing the liver weight (g) per mouse as a function of treatment condition. FIG.21B is a graph showing the epididymal white adipose tissue (eWAT) weight (g) per mouse as a function of treatment condition. FIG.21C is a graph showing the liver weight (% bodyweight) per mouse as a function of treatment condition. FIG.21D is a graph showing the the epididymal white adipose tissue (eWAT) weight (% bodyweight) per mouse as a function of treatment condition. FIG.22A is a graph showing the total serum cholesterol level (mg/dL) per mouse as a function of treatment condition. FIG.22B is a graph showing the serum HDL cholesterol level (mg/dL) per mouse as a function of treatment condition. FIG.22C is a graph showing the serum LDL cholesterol level (mg/dL) per mouse as a function of treatment condition. FIG.22D is a graph showing the serum triglyceride level (mg/dL) per mouse as a function of treatment condition. FIG.22E is a graph showing the serum alanine aminotransferase (ALT) level (U/L) per mouse as a function of treatment condition. DETAILED DESCRIPTION The present disclosure provides mRNA therapeutics for the treatment of metabolic diseases. The incidence of metabolic diseases, such as obesity, type 2 diabetes, and metabolic associated fatty liver disease (MAFLD) are of growing global concern, impacting over 650 million, 438 million, and 882 million individuals globally, respectively. Although both invasive and non-invasive treatment options for metabolic diseases are available, many of these interventions are difficult to maintain or are associated with negative side effects. As an alternative, growing evidence has suggested that the therapeutic use of endogenous cytokines may alleviate the symptoms associated with metabolic disease with improved side effect profiles. Interleukin-22 (IL-22) has been identified as one such cytokine with therapeutic potential due to its biological impacts on multiple metabolic disease factors. IL-22, a member of the IL-10 cytokine family, mediates cellular responses by binding to the IL-22 receptor 1 (IL-22R1), inducing conformational changes that enable binding of the IL-22/IL-22R1 complex by the IL-10 receptor β (IL-10Rβ). This in turn results in the downstream activation of the extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK). Through these pathways, IL-22 promotes the expression of downstream innate immune mediators, mitogenic modulators, and antiapoptotic modulators that promote survival andproliferation. The production and secretion of IL-22 is primarily facilitated through activated T cells, and unlike other IL-10 members, IL-22 does not target immune-modulatory cells but instead targets epithelial cell populations such as hepatocytes and pancreatic cells. In previous studies, IL-22 targeting of hepatocytes has been implicated in protecting against acute hepatitis, inflammation, and alcohol-induced liver damage, and has been observed to play a potential role in liver regeneration, as well as in decreasing weight gain and improving lipid balance. Although IL-22 has shown promise for the treatment of liver associated metabolic diseases with high tolerability in vivo, its short serum half-life (<2 hours) may limit the use of IL-22 in clinical applications. Thus, manipulations that serve to increase the serum half-life of IL-22 are attractive strategies for the use of IL-22 in clinical applications. The present disclosure addresses and utilizes these strategies. One challenge associated with delivering nucleic acid-based therapeutics (e.g., mRNA therapeutics) in vivo stems from the innate immune response, which can occur when the body’s immune system encounters foreign nucleic acids. Foreign mRNAs can activate the immune system via recognition through toll-like receptors (TLRs), in particular TLR7/8, which is activated by single-stranded RNA (ssRNA). In nonimmune cells, the recognition of foreign mRNA can occur through the retinoic acid-inducible gene I (RIG-I). Immune recognition of foreign mRNAs can result in unwanted cytokine effects including interleukin- 1β (IL-1β) production, tumor necrosis factor-α (TNF-α) distribution and a strong type I interferon (type I IFN) response. This disclosure features the incorporation of different modified nucleotides within therapeutic mRNAs to minimize the immune activation and optimize the translation efficiency of mRNA to protein. Particular aspects feature a combination of nucleotide modification to reduce the innate immune response and sequence optimization, in particular, within the open reading frame (ORF) of therapeutic mRNAs encoding IL-22 to enhance protein expression. Certain embodiments of the mRNA therapeutic technology of the instant disclosure also feature delivery of mRNA encoding IL-22 via a lipid nanoparticle (LNP) delivery system. In some embodiments, mRNA encodes an IL-22 polypeptide with an enhanced serum half-life (i.e., an extended serum half-life IL-22). Lipid nanoparticles (LNPs) are an ideal platform for the safe and effective delivery of mRNAs to target cells. LNPs have the unique ability to deliver nucleic acids by a mechanism involving cellular uptake, intracellular transport and endosomal release or endosomal escape. The instant invention features ionizable amino lipid-based LNPs combined with mRNA encoding extended serum half-life IL-22 which have improved properties when administered in vivo. Without being bound to any particular theory, it is believed that the ionizable amino lipid-based LNP Formulations of this disclosure have improved properties, for example, cellular uptake, intracellular transport and/or endosomal release or endosomal escape. LNPs administered by systemic route (e.g., intravenous (IV) administration), for example, in a first administration, can accelerate the clearance of subsequently injected LNPs, for example, in further administrations. This phenomenon is known as accelerated blood clearance (ABC) and is a key challenge, in particular, when replacing low-level (e.g., low serum half-life) signaling proteins (e.g., IL-22) in a therapeutic context. This is because repeat administration of mRNA therapeutics is in most instances essential to maintain necessary levels of a protein in target tissues in subjects (e.g., subjects suffering from metabolic disease). Repeat dosing challenges can be addressed on multiple levels. mRNA engineering and/or efficient delivery by LNPs can result in increased levels and or enhanced duration of protein (e.g., IL-22) being expressed following a first dose of administration, which in turn, can lengthen the time between first dose and subsequent dosing. It is known that the ABC phenomenon is, at least in part, transient in nature, with the immune responses underlying ABC resolving after sufficient time following systemic administration. Thus, increasing the duration of protein expression and/or activity following systemic delivery of an mRNA therapeutic of the disclosure in one aspect, combats the ABC phenomenon. Moreover, LNPs can be engineered to avoid immune sensing and/or recognition and can thus further avoid ABC upon subsequent or repeat dosing. An exemplary aspect of the disclosure features LNPs which have been engineered to have reduced ABC. 1. Lipid Nanoparticle (LNP) Compositions The present disclosure provides LNP compositions compositions with advantageous properties. The lipid nanoparticle compositions described herein may be used for the delivery of therapeutic and/or prophylactic agents, e.g., mRNAs, to mammalian cells or organs. For example, the lipid nanoparticles described herein have little or no immunogenicity. For example, the lipid compounds disclosed herein have a lower immunogenicity as compared to a reference lipid (e.g., MC3, KC2, or DLinDMA). For example, a formulation comprising a lipid disclosed herein and a therapeutic or prophylactic agent, e.g., mRNA, has an increased therapeutic index as compared to a corresponding formulation which comprises a reference lipid (e.g., MC3, KC2, or DLinDMA) and the same therapeutic or prophylactic agent. In some embodiments, the present application provides pharmaceutical compositions comprising: (a) a delivery agent comprising a lipid nanoparticle; and (b) a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide. a. Lipid Nanoparticles In some embodiments, polynucleotides of the present disclosure (e.g., IL-22 mRNA, e.g., extended serum half-life IL-22 mRNA) are included in a lipid nanoparticle (LNP). Lipid nanoparticles according to the present disclosure may comprise: (i) an ionizable lipid (e.g., an ionizable amino lipid); (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-modified lipid. In some embodiments, lipid nanoparticles according to the present disclosure further comprise one or more polynucleotides of the present disclosure (e.g., IL-22 mRNA, e.g., extended serum half-life IL-22 mRNA). The lipid nanoparticles according to the present disclosure can be generated using components, compositions, and methods as are generally known in the art, see for example PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406; PCT/US2016000129; PCT/US2016/014280; PCT/US2016/014280; PCT/US2017/038426; PCT/US2014/027077; PCT/US2014/055394; PCT/US2016/52117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575 and PCT/US2016/069491 all of which are incorporated by reference herein in their entirety. In some embodiments, the lipid nanoparticle comprises an ioniziable cationic lipid (e.g., an ionizable amino lipid) at a content of 20-60 mol.%, 25-60 mol.%, 30-60 mol.%, 35- 60 mol.%, 40-60 mol.%, 45-60 mol.%, 20-55 mol.%, 25-55 mol.%, 30-55 mol.%, 35-55 mol.%, 40-55 mol.%, 45-55 mol.%, 20-50 mol.%, 25-50 mol.%, 30-50 mol.%, 35-50 mol.%, or 40-50 mol.%. For example, the lipid nanoparticle may comprise an ionizable cationic lipid (e.g., an ionizable amino lipid) at a content of 40-50 mol.%, 45-50 mol.%, 45-46 mol.%, 46- 47 mol.%, 47-48 mol.%, 48-49 mol.%, or 49-50 mol.%, for example about 45 mol.%, about 45.5 mol.%, about 46 mol.%, about 46.5 mol.%, about 47 mol.%, about 47.5 mol.%, about 48 mol.%, about 48.5 mol.%, about 49 mol.%, or about 49.5 mol.% ionizable cationic lipid (e.g., an ionizable amino lipid). In some embodiments, the lipid nanoparticle comprises a non-cationic helper lipid or phospholipid at a content of 5-25 mol.%. For example, the lipid nanoparticle may comprise a non-cationic helper lipid or phospholipid at a content of molar ratio of 5-25 mol.%, 5-20 mol.%, 5-15 mol.%, 10-25 mol.%, 10-20 mol.%, 10-15 mol.%, 5-6 mol.%, 6-7 mol.%, 7-8 mol.%, 8-9 mol.%, 9-10 mol.%, 10-11 mol.%, 11-12 mol.%, 12-13 mol.%, 13-14 mol.%, 14- 15 mol.%, 10-14 mol.%, 10-13 mol.%, 10-12 mol.%, 10-11 mol.%, 9-15 mol.%, 9-14 mol.%, 9-13 mol.%, 9-12 mol.%, or 9-11 mol.% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises a sterol or other structural lipid at a content molar ratio of 25-55 mol.%, 25-50 mol.%, 25-45 mol.%, 25-40 mol.%, 25- 35 mol.%, 30-55 mol.%, 30-50 mol.%, 30-45 mol.%, 30-40 mol.%, 30-35 mol.%, 35-55 mol.%, 35-50 mol.%, 35-45 mol.%, 35-40 mol.%, 25-30 mol.%, 30-35 mol.%, 25-28 mol.%, 28-30 mol.%, 30-33 mol.%, 35-38 mol.%, 38-40 mol.%, 36-40 mol.%, 37-40 mol.%, 38-40 mol.%, 38-39 mol.%, 36-40 mol.%, 37-40 mol.%, 36-39 mol.%, or 37-39 mol.%. For example, the lipid nanoparticle may comprise a sterol or other structural lipid at a content of about 30 mol.%, about 30.5 mol.%, about 31.0 mol.%, about 31.5 mol.%, about 32.0 mol.%, about 32.5 mol.%, about 33.0 mol.%, about 33.5 mol.%, about 34.0 mol.%, about 34.5 mol.%, about 35.0 mol.%, about 35.5 mol.%, about 36.0 mol.%, about 36.5 mol.%, about 37.0 mol.%, about 37.5 mol.%, about 38.0 mol.%, about 38.5 mol.%, about 39.0 mol.%, about 39.5 mol.%, about 40.0 mol.%, about 40.5 mol.%, about 41.0 mol.%, about 41.5 mol.%, about 42.0 mol.%, about 42.5 mol.%, about 43.0 mol.%, about 43.5 mol.%, about 44.0 mol.%, about 44.5 mol.%, or about 45.0 mol.%. In some embodiments, the lipid nanoparticle comprises a PEG-modified lipid at a content of 0.5-15 mol.%, 1.0-15 mol.%, 1.5-15 mol.%, 2.0-15 mol.%, 2.5-15 mol.%, 3.0-15 mol.%, 3.5-15 mol.%, 4.0-15 mol.%, 4.5-15 mol.%, 5.0-15 mol.%, 10-15 mol.%, 0.5-10 mol.%, 0.5-5 mol.%, 0.5-4.5 mol.%, 0.5-4.0 mol.%, 0.5-3.5 mol.%, 0.5-3.0 mol.%, 0.5-2.5 mol.%, 0.5-2.0 mol.%, 0.5-1.5 mol.%, 0.5-1.0 mol.%, 1.0-10 mol.%, 1.0-5 mol.%, 1.0-4.5 mol.%, 1.0-4.0 mol.%, 1.0-3.5 mol.%, 1.0-3.0 mol.%, 1.0-2.5 mol.%, 1.0-2.0 mol.%, 1.0-1.5 mol.%, 1.5-5.0 mol.%, 1.5-4.5 mol.%, 1.5-4.0 mol.%, 1.5-3.5 mol.%, 1.5-3.0 mol.%, 1.5-2.5 mol.%, 1.5-2.0 mol.%, 2.0-5.0 mol.%, 2.0-4.5 mol.%, 2.0-4.0 mol.%, 2.0-3.5 mol.%, 2.0-3.0 mol.%, or 2.0-2.5 mol.%. For example, the lipid nanoparticle may comprise a PEG-modified lipid at a content of a about 0.5 mol.%, about 1.0 mol.%, about 1.5 mol.%, about 2.0 mol.%, about 2.5 mol.%, about 3.0 mol.%, about 3.5 mol.%, about 4.0 mol.%, about 4.5 mol.%, about 5.0 mol.%, about 6.0 mol.%, about 7.0 mol.%, about 8.0 mol.%, about 9.0 mol.%, about 10.0 mol.%, or about 15.0 mol.%. In some embodiments, the lipid nanoparticle comprises: (i) 20 to 60 mol.% ionizable cationic lipid (e.g. ionizable amino lipid), (ii) 25 to 55 mol.% sterol or other structural lipid, (iii) 5 to 25 mol.% non-cationic lipid (e.g., phospholipid), and (iv) 0.5 to 15 mol.% PEG- modified lipid. In some embodiments, the lipid nanoparticle comprises: (i) 40 to 50 mol.% ionizable cationic lipid (e.g. ionizable amino lipid), (ii) 30 to 45 mol.% sterol or other structural lipid, (iii) 5 to 15 mol.% non-cationic lipid (e.g., phospholipid), and (iv) 1 to 5 mol.% PEG- modified lipid. In some embodiments, the lipid nanoparticle comprises: (i) 45 to 50 mol.% ionizable cationic lipid (e.g. ionizable amino lipid), (ii) 35 to 45 mol.% sterol or other structural lipid, (iii) 8 to 12 mol.% non-cationic lipid (e.g., phospholipid), and (iv) 1.5 to 3.5 mol.% PEG- modified lipid. In the following sections, “Compounds” numbered with an “I-” prefix (e.g., “Compound I-1,” “Compound I-2,” “Compound I-3,” “Compound I-VI,” etc., indicate specific ionizable lipid compounds. Likewise, compounds numbered with a “P-” prefix (e.g., “Compound P-I,” etc.) indicate a specific PEG-modified lipid compound. b. Ionizable amino lipids In some embodiments, the lipid nanoparticle of the present disclosure comprises an ionizable cationic lipid (e.g., an ionizable amino lipid) that is a compound of Formula (I):
, or N-oxides, salts, or isomers thereof. In some embodiments, the lipid nanoparticle further comprises: a phospholipid; a structural lipid; and a PEG-lipid. In some embodiments, the lipid nanoparticle comprises: 40-50 mol% of the ionizable lipid, 30-45 mol% of the structural lipid, 5-15 mol% of the phospholipid, and 1-5 mol% of the PEG-lipid. In some embodiments, the lipid nanoparticle comprises: 45-50 mol.% of the ionizable amino lipid, 10-15 mol.% of the phospholipid, 35-40 mol.% of the structural lipid, and 1-3 mol.% of the PEG-modified lipid. In some embodiments, the phospholipid comprises comprises 1,2-distearoyl-sn-glycero- 3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2- dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3- phosphocholine (DPPC), 1,2-diundecanoyl-sn-glycero-phosphocholine (DUPC), 1-palmitoyl-2- oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3- phosphocholine (18:0 Diether PC), 1-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3- phosphocholine (OChemsPC), 1-hexadecyl-sn-glycero-3-phosphocholine (C16 Lyso PC), 1,2- dilinolenoyl-sn-glycero-3-phosphocholine,1,2-diarachidonoyl-sn-glycero-3-phosphocholine, 1,2- didocosahexaenoyl-sn-glycero-3-phosphocholine, 1,2-diphytanoyl-sn-glycero-3- phosphoethanolamine (ME 16.0 PE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine, 1,2- dilinoleoyl-sn-glycero-3-phosphoethanolamine, 1,2-dilinolenoyl-sn-glycero-3- phosphoethanolamine, 1,2-diarachidonoyl-sn-glycero-3-phosphoethanolamine, 1,2- didocosahexaenoyl-sn-glycero-3-phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac- (1-glycerol) sodium salt (DOPG), sphingomyelin, and mixtures thereof. In some embodiments, the phospholipid comprises DSPC. In some embodiments, the structural lipid is selected from the group consisting of: cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, derivatives thereof, and mixtures thereof. In some embodiments, the structural lipid comprises cholesterol or a derivative thereof. In some embodiments, the PEG-lipid is selected from: 1,2-dimyristoyl-sn-glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N- [amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG- dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG-DAG), PEG- dipalmitoyl phosphatidylethanolamine (PEG-DPPE), PEG-l,2-dimyristyloxlpropyl-3-amine (PEG-c-DMA), or combinations thereof. In some embodiments, the PEG-lipid comprises PEG- DMG. In some embodiments, the mRNA molecule, when administered as a single dose to a human subject, is sufficient to increase serum IL-22 levels in the human subject to a level at least 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20-fold, 30-fold, 40-fold, or at least 50-fold higher than the level observed prior to the administration, for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120 hours, or at least one week post-administration. In some embodiments, the mRNA molecule, when administered as a single dose to a human subject, is sufficient to: (i) increase serum IL-22 levels in the human subject by at least about 1.1-fold, at least about 1.2-fold, at least about 1.5-fold, at least about 2.0-fold, at least 5.0- fold, or at least 10-fold, compared to the human subject’s baseline serum IL-22 levels for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120 hours, or at least one week post-administration; or (ii) increase IL-22 activity in the human subject by at least about 1.5-fold, at least bout 2.0-fold, or at least about 2.5-fold, compared to the human subject’s baseline IL-22 activity for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120, or at least one week post-administration. In some embodiments, an mRNA molecule comprises an open reading frame (ORF) encoding an Interleukin-22 (IL-22) polypeptide. In some embodiments, an mRNA molecule comprises an open reading frame (ORF) encoding an IL-22 polypeptide-containing fusion protein. In some embodiments, IL-22 polypeptide encoded by the mRNA molecule has an amino acid sequence identical to any one of SEQ ID NO:1-4. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to any one of SEQ ID NO:28-38. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to any one of SEQ ID NO:28-33. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to any one of SEQ ID NO:34-38. In some embodiments, the IL-22 polypeptide-containing fusion protein has an amino acid sequence identical to SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25, SEQ ID NO:26, SEQ ID NO:27, SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38. In some embodiments, the ORF is at least 65% identical to SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328. In some embodiments, the mRNA molecule further comprises a 5’UTR having a nucleic acid sequence according to SEQ ID NO:50, SEQ ID NO:55, or SEQ ID NO:56. In some embodiments, the mRNA molecule further comprises a 3’UTR having a nucleic acid sequence according to SEQ ID NO:108, SEQ ID NO:125, or SEQ ID NO:139. In some embodiments, the mRNA molecule comprises a poly-A region, wherein the poly-A region about 100 nucleotides in length. In some embodiments, the mRNA molecule comprises at least one chemically modified nucleobase, sugar, backbone, or any combination thereof. In some embodiments, the at least one chemically modified nucleobase is selected from the group consisting of pseudouracil (ψ), N1-methylpseudouracil (m1ψ), 1-ethylpseudouracil, 2- thiouracil (s2U), 4’-thiouracil, 5-methylcytosine, 5-methyluracil, 5-methoxyuracil, and any combination thereof. In some embodiments, the mRNA molecule further comprises a 5′ terminal cap. In some embodiments, the 5′ terminal cap comprises a m7G-ppp-Gm-AG, Cap0, Cap1, ARCA, inosine, N1-methyl-guanosine, 2′-fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino- guanosine, LNA-guanosine, 2-azidoguanosine, Cap2, Cap4, 5′ methylG cap, or an analog thereof. In some embodiments, the 5’ terminal cap comprises m7G-ppp-Gm-AG or Cap1. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a pharmaceutical composition comprising: the lipid nanoparticle according to any of the above embodiments; and a pharmaceutically acceptable carrier. In some embodiments, the pharmaceutical composition is for use in treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject. In some embodiments, the pharmaceutical composition is for use in reducing body weight, reducing total cholesterol levels, reducing total HDL cholesterol levels, reducing total LDL cholesterol levels, reducing total triglyceride levels, reducing liver steatosis, or reducing blood glucose levels in a subject. In another aspect, which may be combined with any other aspect or embodiment, the lipid nanoparticle, IL-22 polypeptide, or mRNA molecule according to any of the above embodiments is for use in the manufacture of a medicament or pharmaceutical composition for use in treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject. In another aspect, which may be combined with any other aspect or embodiment, the lipid nanoparticle, IL-22 polypeptide, or mRNA molecule according to any of the above embodiments is for use in the manufacture of a medicament or pharmaceutical composition for use in reducing body weight, reducing total cholesterol levels, reducing total HDL cholesterol levels, reducing total LDL cholesterol levels, reducing total triglyceride levels, reducing liver steatosis, or reducing blood glucose levels in a subject. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of treating or delaying the onset and/or progression of metabolic disease in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of increasing serum IL-22 levels in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of increasing IL-22 activity in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of increasing one or more of serum IL-22 levels or IL-22 activity in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing body weight of a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing total cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing total HDL cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing total LDL cholesterol levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing total triglyceride levels a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing liver steatosis in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a method of reducing blood glucose levels in a human subject, comprising: administering to the human subject an effective amount of a pharmaceutical composition according to any of the above embodiments. In some embodiments, the administration is a single administration. In some embodiments, the administration is a repeated administration. In some embodiments, the administration is about once per day, about once per week, about once per two weeks, or about once per month. In some embodiments, the pharmaceutical composition is administered intravenously, intramuscularly, or subcutaneously. In one embodiment, the pharmaceutical composition is administered intravenously. In another embodiment, the pharmaceutical composition is administered subcutaneously. In another embodiment, the pharmaceutical composition is administered intramuscularly. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to an IL-22 polypeptide-containing fusion protein having an amino acid sequence identical to SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25, SEQ ID NO:26, SEQ ID NO:27, SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38. In some embodiments, the amino acid sequence is identical to SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a pharmaceutical composition comprising: the IL-22 polypeptide according to any of the above embodiments; and a pharmaceutically acceptable carrier. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a cell comprising the IL-22 polypeptide according to any of the above embodiments. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a codon-optimized mRNA molecule encoding the IL-22 polypeptide according to any of the above embodiments. In some embodiments, the codon-optimized mRNA molecule has a nucleic acid sequence having at least about 60% identity to the nucleic acid sequence of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328. In some embodiments, the codon-optimized mRNA molecule has a sequence having at least about 60% identity to SEQ ID NO:350, SEQ ID NO:351, SEQ ID NO:352, SEQ ID NO:353, SEQ ID NO:354, SEQ ID NO:355, SEQ ID NO:356, SEQ ID NO:357, SEQ ID NO:358, SEQ ID NO:359, SEQ ID NO:360, SEQ ID NO:361, SEQ ID NO:362, SEQ ID NO:363, SEQ ID NO:364, SEQ ID NO:365, SEQ ID NO:366, SEQ ID NO:367, or SEQ ID NO:368. In another aspect, which may be combined with any other aspect or embodiment, the present disclosure relates to a cell comprising the mRNA molecule according to any of the above embodiments. BRIEF DESCRIPTION OF THE DRAWINGS FIG.1A is a graph showing levels of N- or C-terminally MSA-linked IL-22 in conditioned media determined by ELISA 24 hours after the transfection of an extended serum half-life IL-22 mRNA construct into HeLa cells. FIG.1B is a graph showing detection of STAT3 pathway activation by densitometry in HEK-Blue-IL-22 cells incubated with extended serum half-life IL-22 construct #1- containing media for 24 hours. Results are representative of 3 independent biological replicates (n=3, mean ± SD). FIG.1C is a schematic illustrating a dose time course used in experiments described herein. Briefly, N-terminally MSA-linked IL-22 mRNA (construct #1) was intravenously administered to C57BL/6 female mice from Charles River at 0.5 mg/kg (n=5) or 0.25 mg/kg (n=5). Sera were collected for analysis at 6 h post-administration, and daily from Day 3 through Day 7. FIG.1D is a graph showing IL-22 protein levels in sera obtained from mice administered with construct #1. Protein levels were determined by ELISA. FIG.2A is a schematic illustrating a dose time course used in experiments described herein. Briefly, Male C57BL/6 from Taconic mice were fed on the AMLN diet from 6 weeks of age (Week -18). LNP-encapsulated MSA (n=9) or construct #1 (n=9) mRNA was administered at 0.5 mg/kg weekly over 6 weeks to AMLN fed mice with an untreated group of mice (n=5) maintained in parallel. Sera were collected weekly from Week -1 through Week 6. Age-matched chow diet-fed mice (n=6) were included for comparisons at the end of the treatment period. FIG.2B is a set of graphs showing body weights of mice throughout the treatment period. FIG.2C is a set of graphs showing total serum cholesterol, LDL cholesterol, HDL cholesterol, and triglyceride levels determined at Week -1 and 72 hours after the administration of each dose, expressed as % change above baseline. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test for body weight and the serum lipid analyses. Analyses of total serum cholesterol excluded 2 mice, 1 in each of the construct #1 and untreated groups, due to insufficient volume of pre-bleed samples. FIG.3A is a graph showing liver weights measured 72 hours after the last dose in a 6- week treatment period in which MSA (n=9) or construct #1 (n=9) was administered at 0.5 mg/kg to male AMLN diet-fed C57BL/6Tac mice. An untreated group of mice (n=5) was maintained in parallel. FIG.3B shows representative images of H&E-stained liver and epididymal adipose tissue samples obtained from mice in the various treatment conditions described above. FIG 3C is a set of graphs quantifying the scoring of levels of microvesicular and macrovesicular steatosis, inflammation, and liver and adipose hypertrophy in images of H&E-stained liver and epididymal adipose tissue samples obtained from mice in the various treatment conditions described above. FIG 3D is a set of graphs showing differential expression levels of a select set of fibrosis and lipogenesis associated genes were determined relative to Rpl19 expression using qRT-PCR in MSA and construct #1 treated and untreated AMLN diet-fed mice and matched chow diet-fed control mice. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by one-way ANOVA for histology scoring and gene expression. One construct #1 treated mouse was excluded from analyses due to pathology indicating an infection. FIG.4A is a graph showing serum IL-22 levels measured weekly 72 hours after the administration of LNP-encapsulated construct #1 mRNA for 6 weeks in C57BL/6Tac mice fed the AMLN diet. FIG.4B is a graph showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored every week 72 hours following the administration of construct #1 or MSA mRNA at 0.5 mg/kg to mice fed with the AMLN diet. FIG.4C shows representative images of PSR and F4/80 stained liver tissue at the end of the treatment period in construct #1 treated, MSA treated, and untreated mice fed the AMLN diet and untreated mice fed a chow diet. FIG.4D is a set of graphs quantifying and scoring F4/80-positive macrophages and liver fibrosis level using microscopic evaluation. FIG.5 shows a set of graphs showing the expression of genes involved in fibrosis and lipogenesis in AMLN or chow fed C57BL/6Tac mice treated with weekly administration of LNP-encapsulated mRNA construct #1 or MSA mRNA over 6 weeks. LNP-encapsulated MSA (n=9) or construct #1 (n=9) mRNA (0.5 mg/kg each) were administered weekly for 6 weeks to C57BL/6Tac mice that were fed on the AMLN or chow diet from 6 weeks of age for 18 weeks. The untreated groups of mice (AMLN diet group) was maintained in parallel (n=5). Animals fed chow diet as control (n=6) were added at the end of the study.72 hours after the final dose of LNP-encapsulated mRNA, expression of genes involved in fibrosis and lipogenesis was assessed by RT-qPCR. All the genes were analyzed relative to rpl19 mRNA expression. For the qRT-PCR results, the average of MSA treated mice was set to 1. Significance levels (*p< 0.05; *** p< 0.001) are shown relative to MSA treated mice and were determined by one-way ANOVA and Dunnett’s post-hoc test. Not significant, ns. FIG.6 shows the results of a differential gene expression analysis in liver tissue of construct #1 treated mice. Total RNA was used for quantification of gene expression using the NanoString Metabolic Pathway Panel with these expressed as log2 fold change of construct #1 treated over MSA treated and plotted against log10 adjusted p-value. Genes were classified into categories by functional association. AMP-activated protein kinase (AMPK), glycolysis, cytokine, and chemokine; adj. = adjusted. FIG.7A is a schematic illustrating a dose time course used in experiments described herein. Male C57BL/6 from Taconic mice were fed on the AMLN or chow diet from 6 weeks of age (Week -18). LNP-encapsulated MSA (n=8) or construct #1 (n=11) mRNA was administered at 0.5 mg/kg weekly over 3 weeks to AMLN fed mice. Two untreated groups of mice (1 AMLN diet group (n=9) and 1 chow diet group (n=7)) were maintained in parallel. Sera were collected for ELISA and lipid analysis at the indicated timepoints. FIG.7B is a graph showing levels of IL-22 in sera from treated mice measured 72 hours after each administration. FIG.7C is a graph showing mouse body weight monitored through Day 38 without additional treatment. FIG.7D is a graph showing mouse food intake monitored through Day 38 without additional treatment. FIG.7E is a set of graphs showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels were measured after sera collection. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by Two-way ANOVA and Dunnett’s post-hoc test for the different time points. FIG.8A is a schematic illustrating a dose time course used in experiments described herein. Male C57BL/6mice from Taconic were fed on the AMLN or chow diet from 6 weeks of age (Week -18). LNP-encapsulated MSA (n=9) or construct #1 (n=9) mRNA was administered at 0.5 mg/kg every other week on Days 0, 14, and 28 to AMLN fed mice. Two untreated groups of mice (1 AMLN diet group (n=8) and 1 chow diet group (n=6)) were maintained in parallel. Sera were collected for ELISA and lipid analysis at the indicated timepoints. FIG.8B is a graph showing levels of IL-22 in sera from treated mice were measured 72 h after each administration, on days 3, 17, and 31. FIG.8C is a graph showing mouse body weight monitored through Day 38 without additional treatment. Mice were administered MSA or construct #1 via intravenous (IV) or subcutaneous (SC) administration. FIG.8D is a set of graphs showing total cholesterol, LDL and HDL cholesterol, and triglyceride levels measured after sera collection. Mice were administered MSA or construct #1 via intravenous (IV) or subcutaneous (SC) administration. FIG.8E shows representative images of H&E stained liver and adipose tissue samples. FIG.8F is a set of graphs showing differential expression levels determined relative to Rpl19 expression using qRT-PCR for a select set of fibrosis and lipogenesis associated genes in treated mice and untreated AMLN diet and chow diet fed mice. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test. FIG.9A is a schematic illustrating a dose time course used in experiments described herein. Male CETP/APOB mice from Taconic were fed on the AMLN diet from Day -42. LNP-encapsulated MSA or construct #1 mRNA was administered at 0.5 mg/kg weekly for 6 weeks to AMLN fed mice (n=10 mice/group). Sera were collected for ELISA and lipid analysis at the indicated timepoints. FIG.9B is a set of graphs showing mouse body weight monitored through Day 39. FIG.9C is a set of graphs showing mouse total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored through Day 39. FIG.9D is a panel of representative images of H&E stained liver and adipose tissue samples obtained from MSA and construct #1 treated mice. FIG.9E is a set of graphs quantifying and grading for microvesicular and macrovesicular steatosis, inflammation, and adipose hypertrophy. FIG.9F is a set of graphs showing differential expression levels determined relative to Rpl19 expression using qRT-PCR for a select set of fibrosis and lipogenesis associated genes in MSA and construct #1 treated mice. Significance levels (*p<0.05; **p<0.01; ***p<0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test or by t-test for the histology score and the gene expression analyses. FIG.10A is a graph showing IL-22 serum levels in mice after weekly administration of construct #1 or MSA mRNA (0.5 mg/kg) in CETP/APOB mice fed an AMLN diet. FIG.10B is a graph showing IL-22 serum levels in mice after weekly administration of construct #1 or MSA mRNA (0.5 mg/kg) in CETP/APOB mice fed a chow diet. FIG.10C is a panel of representative images of H&E stained liver tissue samples from AMLN fed MSA and construct #1 treated mice. FIG.10D is a graph showing a quantification of F4/80 stained liver macrophages in AMLN fed MSA and construct #1 treated mice. FIG.10E is a set of graphs showing differential expression levels determined using qRT-PCR for a select set of genes of interest in AMLN-fed CETP/APOB mice treated with MSA or construct #1. Expression is expressed as fold change relative to Rpl19. FIG.10F is a graph showing fasting PYY levels determined using ELISA. Serum PYY analyses excluded 3 mice in the MSA group and 1 in the construct #1 group due to insufficient volume after fastig samples. Significance levels (*p<0.05; ***p<0.001) were determined by one-way ANOVA and Dunnett’s post-hoc test. FIG.11A is a schematic illustrating a dose time course used in experiments described herein. LNP-encapsulated MSA or construct #1 mRNA was administered at 0.5 mg/kg weekly for 6 weeks to male chow diet fed CETP/APOB mice (n=10 mice/group). Sera were collected for lipid profile analysis at the indicated timepoints. FIG.11B is a graph showing mouse body weight monitored through Day 39. FIG.11C is a graph showing mouse total cholesterol, LDL and HDL cholesterol, and triglyceride levels monitored through Day 39. Significance levels (*p<0.05; ***p<0.001) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test. Not significant, ns. FIG.12A is a schematic illustrating a dose time course used in experiments described herein. Two doses of LNP-encapsulated MSA or construct #1 mRNA (0.5 mg/kg each) were administered 1 week apart to male C57BL/6NTac mice that were fed an AMLN or chow diet from 6 weeks of age (Day -126) (n=10 mice/group). One day after dose 2 (Day 8), mice were fasted for 12 h, followed by a 1 h ad libitum refeeding period. Sera were collected before and after the fasting period and after refeeding. FIG.12B is a graph showing mouse body weight measured on Days 1, 3, 7, 8, and 9. FIG.12C is a set of graphs showing differential weight gain calculated on Day 9, food intake measured as the total intake throughout the duration of the study, energy intake calculated as food intake multiplied by metabolizable energy content in the diet, metabolic efficiency calculated as mg of body weight gain over energy intake (kcal), fasting and refeeding glucose levels determined using Abbott Alphatrak 2 glucometers, and Peptide YY (PYY) levels determined using ELISA. Significance levels (*p<0.05; ***p<0.001; Not significant, ns.) are shown relative to MSA treated mice and were determined by two-way ANOVA and Dunnett’s post-hoc test. FIG.13A is a schematic illustrating a dose time course used in experiments described herein. Two doses of LNP-encapsulated MSA or construct #1 mRNA (0.5 mg/kg each) were administered 1 week apart to male C57BL/6NTac mice from Taconic that were fed an AMLN or chow diet from 6 weeks of age (Day -126) (n=10 mice/group). One day after dose 2 (Day 8), mice were fasted for 6 hours, followed by administration of 2 g/kg of glucose in sterile 0.9% saline IP in a glucose tolerance test (GTT). Glucose levels were measured before (0 min) as well as after (15min, 30min, 60min and 120min) glucose administration. FIG.13B is a graph showing blood glucose levels in AMLN-fed animals during the course of the GTT. FIG.13C is a graph showing blood glucose levels in chow diet-fed animals during the course of the GTT. FIG.13D is a graph showing the area under the curve (AUC) for the glucose traces in chow and AMLN diet-fed mice shown in (B) and (C). Significance levels (*p<0.05; ***p<0.001; Not significant, ns.) are shown relative to MSA treated mice and were determined by two-way ANOVA and Sidak (B,C) or Tukey (D) correction for multiple comparison. N=8 for chow MSA, n=9 for chow construct #1, n=10 for both AMLN diet groups. FIG.14A is a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various mouse IL-22 mRNA constructs, as measured by ELISA. FIG.14B is a graph showing the bioactivity of mouse IL-22 mRNA constructs as a function of concentration, as measured in conditioned medium samples from HEK-Blue IL- 22 cells treated with IL-22 construct protein. The IL-22 bioactivity in the conditioned medium samples was quantified by measuring the sample OD at 655 nm following incubation in QUANTI-Blue solution. FIG.15A is a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various human IL-22 mRNA constructs, as measured by ELISA. The IL-22 concentration is reported in units of pg/mL. FIG.15B is a graph showing the concentration of IL-22 protein in the medium of cultured HeLa cells transfected with various human IL-22 mRNA constructs, as measured by ELISA. The IL-22 concentration is reported as a concentration in nM. FIG.16 is a graph showing the bioactivity of human IL-22 mRNA constructs as a function of concentration, as measured in conditioned medium samples from HEK-Blue IL- 22 cells treated with IL-22 construct protein. The IL-22 bioactivity in the conditioned medium samples was quantified by measuring the sample OD at 655 nm following incubation in QUANTI-Blue solution. FIG.17 is a schematic illustrating a dose time course used in experiments described herein. Male C57BL/6Tac mice were fed an AMLN diet starting from 6 weeks of age through 40 weeks of age, for a total of 34 weeks of AMLN feeding. LNP-encapsulated MSA mRNA (n=10) or Construct #1 mRNA was administered at 0.5 mg/kg weekly IV (n=10) or 0.1 mg/kg weekly IM (n=10) to mice fed the AMLN diet, with an untreated group of mice (n=8) and mice fed in normal chow diet (n=10) maintained in parallel. Body weight measurements and serum collection occurred at the indicated time points. FIG.18 is a graph showing the % change in mouse bodyweight (BW) over time as a function of treatment condition. FIG.19A is a graph showing the daily caloric intake (kcal) per mouse over time as a function of treatment condition. FIG.19B is a graph showing the daily food intake (g) per mouse over time as a function of treatment condition. FIG.19C is a graph showing the cumulative caloric intake (kcal) per mouse over time as a function of treatment condition. FIG.20A is a graph showing the weight gain (g) per mouse as a function of treatment condition. FIG.20B is a graph showing the metabolic efficiency (g weight gain / MJ caloric intake) per mouse as a function of treatment condition. FIG.21A is a graph showing the liver weight (g) per mouse as a function of treatment condition. FIG.21B is a graph showing the epididymal white adipose tissue (eWAT) weight (g) per mouse as a function of treatment condition. FIG.21C is a graph showing the liver weight (% bodyweight) per mouse as a function of treatment condition. FIG.21D is a graph showing the the epididymal white adipose tissue (eWAT) weight (% bodyweight) per mouse as a function of treatment condition. FIG.22A is a graph showing the total serum cholesterol level (mg/dL) per mouse as a function of treatment condition. FIG.22B is a graph showing the serum HDL cholesterol level (mg/dL) per mouse as a function of treatment condition. FIG.22C is a graph showing the serum LDL cholesterol level (mg/dL) per mouse as a function of treatment condition. FIG.22D is a graph showing the serum triglyceride level (mg/dL) per mouse as a function of treatment condition. FIG.22E is a graph showing the serum alanine aminotransferase (ALT) level (U/L) per mouse as a function of treatment condition. DETAILED DESCRIPTION The present disclosure provides mRNA therapeutics for the treatment of metabolic diseases. The incidence of metabolic diseases, such as obesity, type 2 diabetes, and metabolic associated fatty liver disease (MAFLD) are of growing global concern, impacting over 650 million, 438 million, and 882 million individuals globally, respectively. Although both invasive and non-invasive treatment options for metabolic diseases are available, many of these interventions are difficult to maintain or are associated with negative side effects. As an alternative, growing evidence has suggested that the therapeutic use of endogenous cytokines may alleviate the symptoms associated with metabolic disease with improved side effect profiles. Interleukin-22 (IL-22) has been identified as one such cytokine with therapeutic potential due to its biological impacts on multiple metabolic disease factors. IL-22, a member of the IL-10 cytokine family, mediates cellular responses by binding to the IL-22 receptor 1 (IL-22R1), inducing conformational changes that enable binding of the IL-22/IL-22R1 complex by the IL-10 receptor β (IL-10Rβ). This in turn results in the downstream activation of the extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK). Through these pathways, IL-22 promotes the expression of downstream innate immune mediators, mitogenic modulators, and antiapoptotic modulators that promote survival andproliferation. The production and secretion of IL-22 is primarily facilitated through activated T cells, and unlike other IL-10 members, IL-22 does not target immune-modulatory cells but instead targets epithelial cell populations such as hepatocytes and pancreatic cells. In previous studies, IL-22 targeting of hepatocytes has been implicated in protecting against acute hepatitis, inflammation, and alcohol-induced liver damage, and has been observed to play a potential role in liver regeneration, as well as in decreasing weight gain and improving lipid balance. Although IL-22 has shown promise for the treatment of liver associated metabolic diseases with high tolerability in vivo, its short serum half-life (<2 hours) may limit the use of IL-22 in clinical applications. Thus, manipulations that serve to increase the serum half-life of IL-22 are attractive strategies for the use of IL-22 in clinical applications. The present disclosure addresses and utilizes these strategies. One challenge associated with delivering nucleic acid-based therapeutics (e.g., mRNA therapeutics) in vivo stems from the innate immune response, which can occur when the body’s immune system encounters foreign nucleic acids. Foreign mRNAs can activate the immune system via recognition through toll-like receptors (TLRs), in particular TLR7/8, which is activated by single-stranded RNA (ssRNA). In nonimmune cells, the recognition of foreign mRNA can occur through the retinoic acid-inducible gene I (RIG-I). Immune recognition of foreign mRNAs can result in unwanted cytokine effects including interleukin- 1β (IL-1β) production, tumor necrosis factor-α (TNF-α) distribution and a strong type I interferon (type I IFN) response. This disclosure features the incorporation of different modified nucleotides within therapeutic mRNAs to minimize the immune activation and optimize the translation efficiency of mRNA to protein. Particular aspects feature a combination of nucleotide modification to reduce the innate immune response and sequence optimization, in particular, within the open reading frame (ORF) of therapeutic mRNAs encoding IL-22 to enhance protein expression. Certain embodiments of the mRNA therapeutic technology of the instant disclosure also feature delivery of mRNA encoding IL-22 via a lipid nanoparticle (LNP) delivery system. In some embodiments, mRNA encodes an IL-22 polypeptide with an enhanced serum half-life (i.e., an extended serum half-life IL-22). Lipid nanoparticles (LNPs) are an ideal platform for the safe and effective delivery of mRNAs to target cells. LNPs have the unique ability to deliver nucleic acids by a mechanism involving cellular uptake, intracellular transport and endosomal release or endosomal escape. The instant invention features ionizable amino lipid-based LNPs combined with mRNA encoding extended serum half-life IL-22 which have improved properties when administered in vivo. Without being bound to any particular theory, it is believed that the ionizable amino lipid-based LNP Formulations of this disclosure have improved properties, for example, cellular uptake, intracellular transport and/or endosomal release or endosomal escape. LNPs administered by systemic route (e.g., intravenous (IV) administration), for example, in a first administration, can accelerate the clearance of subsequently injected LNPs, for example, in further administrations. This phenomenon is known as accelerated blood clearance (ABC) and is a key challenge, in particular, when replacing low-level (e.g., low serum half-life) signaling proteins (e.g., IL-22) in a therapeutic context. This is because repeat administration of mRNA therapeutics is in most instances essential to maintain necessary levels of a protein in target tissues in subjects (e.g., subjects suffering from metabolic disease). Repeat dosing challenges can be addressed on multiple levels. mRNA engineering and/or efficient delivery by LNPs can result in increased levels and or enhanced duration of protein (e.g., IL-22) being expressed following a first dose of administration, which in turn, can lengthen the time between first dose and subsequent dosing. It is known that the ABC phenomenon is, at least in part, transient in nature, with the immune responses underlying ABC resolving after sufficient time following systemic administration. Thus, increasing the duration of protein expression and/or activity following systemic delivery of an mRNA therapeutic of the disclosure in one aspect, combats the ABC phenomenon. Moreover, LNPs can be engineered to avoid immune sensing and/or recognition and can thus further avoid ABC upon subsequent or repeat dosing. An exemplary aspect of the disclosure features LNPs which have been engineered to have reduced ABC. 1. Lipid Nanoparticle (LNP) Compositions The present disclosure provides LNP compositions compositions with advantageous properties. The lipid nanoparticle compositions described herein may be used for the delivery of therapeutic and/or prophylactic agents, e.g., mRNAs, to mammalian cells or organs. For example, the lipid nanoparticles described herein have little or no immunogenicity. For example, the lipid compounds disclosed herein have a lower immunogenicity as compared to a reference lipid (e.g., MC3, KC2, or DLinDMA). For example, a formulation comprising a lipid disclosed herein and a therapeutic or prophylactic agent, e.g., mRNA, has an increased therapeutic index as compared to a corresponding formulation which comprises a reference lipid (e.g., MC3, KC2, or DLinDMA) and the same therapeutic or prophylactic agent. In some embodiments, the present application provides pharmaceutical compositions comprising: (a) a delivery agent comprising a lipid nanoparticle; and (b) a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide. a. Lipid Nanoparticles In some embodiments, polynucleotides of the present disclosure (e.g., IL-22 mRNA, e.g., extended serum half-life IL-22 mRNA) are included in a lipid nanoparticle (LNP). Lipid nanoparticles according to the present disclosure may comprise: (i) an ionizable lipid (e.g., an ionizable amino lipid); (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-modified lipid. In some embodiments, lipid nanoparticles according to the present disclosure further comprise one or more polynucleotides of the present disclosure (e.g., IL-22 mRNA, e.g., extended serum half-life IL-22 mRNA). The lipid nanoparticles according to the present disclosure can be generated using components, compositions, and methods as are generally known in the art, see for example PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406; PCT/US2016000129; PCT/US2016/014280; PCT/US2016/014280; PCT/US2017/038426; PCT/US2014/027077; PCT/US2014/055394; PCT/US2016/52117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575 and PCT/US2016/069491 all of which are incorporated by reference herein in their entirety. In some embodiments, the lipid nanoparticle comprises an ioniziable cationic lipid (e.g., an ionizable amino lipid) at a content of 20-60 mol.%, 25-60 mol.%, 30-60 mol.%, 35- 60 mol.%, 40-60 mol.%, 45-60 mol.%, 20-55 mol.%, 25-55 mol.%, 30-55 mol.%, 35-55 mol.%, 40-55 mol.%, 45-55 mol.%, 20-50 mol.%, 25-50 mol.%, 30-50 mol.%, 35-50 mol.%, or 40-50 mol.%. For example, the lipid nanoparticle may comprise an ionizable cationic lipid (e.g., an ionizable amino lipid) at a content of 40-50 mol.%, 45-50 mol.%, 45-46 mol.%, 46- 47 mol.%, 47-48 mol.%, 48-49 mol.%, or 49-50 mol.%, for example about 45 mol.%, about 45.5 mol.%, about 46 mol.%, about 46.5 mol.%, about 47 mol.%, about 47.5 mol.%, about 48 mol.%, about 48.5 mol.%, about 49 mol.%, or about 49.5 mol.% ionizable cationic lipid (e.g., an ionizable amino lipid). In some embodiments, the lipid nanoparticle comprises a non-cationic helper lipid or phospholipid at a content of 5-25 mol.%. For example, the lipid nanoparticle may comprise a non-cationic helper lipid or phospholipid at a content of molar ratio of 5-25 mol.%, 5-20 mol.%, 5-15 mol.%, 10-25 mol.%, 10-20 mol.%, 10-15 mol.%, 5-6 mol.%, 6-7 mol.%, 7-8 mol.%, 8-9 mol.%, 9-10 mol.%, 10-11 mol.%, 11-12 mol.%, 12-13 mol.%, 13-14 mol.%, 14- 15 mol.%, 10-14 mol.%, 10-13 mol.%, 10-12 mol.%, 10-11 mol.%, 9-15 mol.%, 9-14 mol.%, 9-13 mol.%, 9-12 mol.%, or 9-11 mol.% non-cationic lipid. In some embodiments, the lipid nanoparticle comprises a sterol or other structural lipid at a content molar ratio of 25-55 mol.%, 25-50 mol.%, 25-45 mol.%, 25-40 mol.%, 25- 35 mol.%, 30-55 mol.%, 30-50 mol.%, 30-45 mol.%, 30-40 mol.%, 30-35 mol.%, 35-55 mol.%, 35-50 mol.%, 35-45 mol.%, 35-40 mol.%, 25-30 mol.%, 30-35 mol.%, 25-28 mol.%, 28-30 mol.%, 30-33 mol.%, 35-38 mol.%, 38-40 mol.%, 36-40 mol.%, 37-40 mol.%, 38-40 mol.%, 38-39 mol.%, 36-40 mol.%, 37-40 mol.%, 36-39 mol.%, or 37-39 mol.%. For example, the lipid nanoparticle may comprise a sterol or other structural lipid at a content of about 30 mol.%, about 30.5 mol.%, about 31.0 mol.%, about 31.5 mol.%, about 32.0 mol.%, about 32.5 mol.%, about 33.0 mol.%, about 33.5 mol.%, about 34.0 mol.%, about 34.5 mol.%, about 35.0 mol.%, about 35.5 mol.%, about 36.0 mol.%, about 36.5 mol.%, about 37.0 mol.%, about 37.5 mol.%, about 38.0 mol.%, about 38.5 mol.%, about 39.0 mol.%, about 39.5 mol.%, about 40.0 mol.%, about 40.5 mol.%, about 41.0 mol.%, about 41.5 mol.%, about 42.0 mol.%, about 42.5 mol.%, about 43.0 mol.%, about 43.5 mol.%, about 44.0 mol.%, about 44.5 mol.%, or about 45.0 mol.%. In some embodiments, the lipid nanoparticle comprises a PEG-modified lipid at a content of 0.5-15 mol.%, 1.0-15 mol.%, 1.5-15 mol.%, 2.0-15 mol.%, 2.5-15 mol.%, 3.0-15 mol.%, 3.5-15 mol.%, 4.0-15 mol.%, 4.5-15 mol.%, 5.0-15 mol.%, 10-15 mol.%, 0.5-10 mol.%, 0.5-5 mol.%, 0.5-4.5 mol.%, 0.5-4.0 mol.%, 0.5-3.5 mol.%, 0.5-3.0 mol.%, 0.5-2.5 mol.%, 0.5-2.0 mol.%, 0.5-1.5 mol.%, 0.5-1.0 mol.%, 1.0-10 mol.%, 1.0-5 mol.%, 1.0-4.5 mol.%, 1.0-4.0 mol.%, 1.0-3.5 mol.%, 1.0-3.0 mol.%, 1.0-2.5 mol.%, 1.0-2.0 mol.%, 1.0-1.5 mol.%, 1.5-5.0 mol.%, 1.5-4.5 mol.%, 1.5-4.0 mol.%, 1.5-3.5 mol.%, 1.5-3.0 mol.%, 1.5-2.5 mol.%, 1.5-2.0 mol.%, 2.0-5.0 mol.%, 2.0-4.5 mol.%, 2.0-4.0 mol.%, 2.0-3.5 mol.%, 2.0-3.0 mol.%, or 2.0-2.5 mol.%. For example, the lipid nanoparticle may comprise a PEG-modified lipid at a content of a about 0.5 mol.%, about 1.0 mol.%, about 1.5 mol.%, about 2.0 mol.%, about 2.5 mol.%, about 3.0 mol.%, about 3.5 mol.%, about 4.0 mol.%, about 4.5 mol.%, about 5.0 mol.%, about 6.0 mol.%, about 7.0 mol.%, about 8.0 mol.%, about 9.0 mol.%, about 10.0 mol.%, or about 15.0 mol.%. In some embodiments, the lipid nanoparticle comprises: (i) 20 to 60 mol.% ionizable cationic lipid (e.g. ionizable amino lipid), (ii) 25 to 55 mol.% sterol or other structural lipid, (iii) 5 to 25 mol.% non-cationic lipid (e.g., phospholipid), and (iv) 0.5 to 15 mol.% PEG- modified lipid. In some embodiments, the lipid nanoparticle comprises: (i) 40 to 50 mol.% ionizable cationic lipid (e.g. ionizable amino lipid), (ii) 30 to 45 mol.% sterol or other structural lipid, (iii) 5 to 15 mol.% non-cationic lipid (e.g., phospholipid), and (iv) 1 to 5 mol.% PEG- modified lipid. In some embodiments, the lipid nanoparticle comprises: (i) 45 to 50 mol.% ionizable cationic lipid (e.g. ionizable amino lipid), (ii) 35 to 45 mol.% sterol or other structural lipid, (iii) 8 to 12 mol.% non-cationic lipid (e.g., phospholipid), and (iv) 1.5 to 3.5 mol.% PEG- modified lipid. In the following sections, “Compounds” numbered with an “I-” prefix (e.g., “Compound I-1,” “Compound I-2,” “Compound I-3,” “Compound I-VI,” etc., indicate specific ionizable lipid compounds. Likewise, compounds numbered with a “P-” prefix (e.g., “Compound P-I,” etc.) indicate a specific PEG-modified lipid compound. b. Ionizable amino lipids In some embodiments, the lipid nanoparticle of the present disclosure comprises an ionizable cationic lipid (e.g., an ionizable amino lipid) that is a compound of Formula (I): (I) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is:
(I) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is: ; wherein
; wherein denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, andR
denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, andR wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, in Formula (I), R’a is R’branched; R’branched is
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, in Formula (I), R’a is R’branched; R’branched is denotes a point of attachment; Raα, Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, in Formula (I), R’a is R’branched; R’branched is denotes a point of attaα aβ aγ aδ
denotes a point of attachment; Raα, Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, in Formula (I), R’a is R’branched; R’branched is denotes a point of attaα aβ aγ aδ achment; R , R , R , and R are each H; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 3; and m is 7. In some embodiments of the compounds of Formula (I), R’a is R’branched; R’branched is
achment; R , R , R , and R are each H; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 3; and m is 7. In some embodiments of the compounds of Formula (I), R’a is R’branched; R’branched is denotes a point of attachment; Raα is C2-12 alkyl; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is
denotes a point of attachment; Raα is C2-12 alkyl; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is ; R10 is NH(C1-6 alkyl); n2 is 2; R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of the compounds of Formula (I), R’a is R’branched; R’branched iaα aβ aδ
; R10 is NH(C1-6 alkyl); n2 is 2; R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of the compounds of Formula (I), R’a is R’branched; R’branched iaα aβ aδ denotes a point of attachment; R , R , and R are each H; Raγ is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the compound of Formula (I) is selected from:
denotes a point of attachment; R , R , and R are each H; Raγ is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the compound of Formula (I) is selected from: In some embodiments, the compound of Formula (I) is:
In some embodiments, the compound of Formula (I) is: (Compound I-1). In some embodiments, the compound of Formula (I) is:
(Compound I-1). In some embodiments, the compound of Formula (I) is: (Compound I-2). In some embodiments, the compound of Formula (I) is:
(Compound I-2). In some embodiments, the compound of Formula (I) is: (Compound I-3). In some aspects, the disclosure relates to a compound of Formula (Ia):
(Compound I-3). In some aspects, the disclosure relates to a compound of Formula (Ia): (Ia) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is:
(Ia) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is: ; wherein
; wherein denotes a point of attachment; wherein Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
denotes a point of attachment; wherein Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some aspects, the disclosure relates to a compound of Formula (Ib):
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some aspects, the disclosure relates to a compound of Formula (Ib): (Ib) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is:
(Ib) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is: ; wherein
; wherein denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments of Formula (I) or (Ib), R’a is R’branched; R’branched is denotes a point of attachmentaβ aγ aδ 2
denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments of Formula (I) or (Ib), R’a is R’branched; R’branched is denotes a point of attachmentaβ aγ aδ 2 ; R , R , and R are each H; R and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of Formula (I) or (Ib), R’a is R’branched; R’branched is denotes a point of attachment;aβ aδ aγ
; R , R , and R are each H; R and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments of Formula (I) or (Ib), R’a is R’branched; R’branched is denotes a point of attachment;aβ aδ aγ R and R are each H; R is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the disclosure relates to a compound of Formula (Ic):
R and R are each H; R is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the disclosure relates to a compound of Formula (Ic): (Ic) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is:
(Ic) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is: ; wherein
; wherein denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is
denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is , wherein
, wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, R’a is R’branched; R’branched is
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13. In some embodiments, R’a is R’branched; R’branched is ; denotes a point of attachment; Raβ, Raγ, and Raδ are each H; Raα is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is deno10
; denotes a point of attachment; Raβ, Raγ, and Raδ are each H; Raα is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is deno10 tes a point of attachment; R is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the compound of Formula (Ic) is:
tes a point of attachment; R is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. In some embodiments, the compound of Formula (Ic) is: (Compound I-2). In some aspects, the disclosure relates to a compound of Formula (II):
(Compound I-2). In some aspects, the disclosure relates to a compound of Formula (II): (II) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein
(II) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein wherein
wherein denotes a point of attachment; Raγ and Raδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Raγ and Raδ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; Rbγ and Rbδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Rbγ and Rbδ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
denotes a point of attachment; Raγ and Raδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Raγ and Raδ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; Rbγ and Rbδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Rbγ and Rbδ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; Ya is a C3-6 carbocycle; R*”a is selected from the group consisting of C1-15 alkyl and C2-15 alkenyl; and s is 2 or 3; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-a):
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; Ya is a C3-6 carbocycle; R*”a is selected from the group consisting of C1-15 alkyl and C2-15 alkenyl; and s is 2 or 3; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-a): (II-a) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is: anb
(II-a) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is: anb d R’ is:
d R’ is: wherein
wherein denotes a point of attachment; Raγ and Raδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Raγ and Raδ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; Rbγ and Rbδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Rbγ and Rbδ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
denotes a point of attachment; Raγ and Raδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Raγ and Raδ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; Rbγ and Rbδ are each independently selected from the group consisting of H, C1-12 alkyl, and C2-12 alkenyl, wherein at least one of Rbγ and Rbδ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-b):
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-b): (II-b) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is:
(II-b) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is: and R’b is:
and R’b is: wherein
wherein denotes a point of attachment; Raγ and Rbγ are each independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
denotes a point of attachment; Raγ and Rbγ are each independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-c):
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-c): (II-c) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is:b
(II-c) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is:b and R’ is:
and R’ is: wherein
wherein denotes a point of attachment; wherein Raγ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
denotes a point of attachment; wherein Raγ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; R’ is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-d):
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; R’ is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-d): (II-d) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is:b
(II-d) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is:b and R’ is:
and R’ is: wherein
wherein denotes a point of attachment; wherein Raγ and Rbγ are each independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
denotes a point of attachment; wherein Raγ and Rbγ are each independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and , wherein
, wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-e):
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1- 6 alkyl, C2-3 alkenyl, and H; and n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R’ independently is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some aspects, the disclosure relates to a compound of Formula (II-e): (II-e) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is:
(II-e) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein R’branched is: and R’b is:
and R’b is: ; wherein
; wherein denotes a point of attachment; wherein Raγ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), m and l are each independently selected from 4, 5, and 6. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), m and l are each 5. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), each R’ independently is a C2-5 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’b is:R
denotes a point of attachment; wherein Raγ is selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C1-14 alkyl and C2-14 alkenyl; R4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl or C2-12 alkenyl; m is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9; l is selected from 1, 2, 3, 4, 5, 6, 7, 8, and 9. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), m and l are each independently selected from 4, 5, and 6. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), m and l are each 5. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), each R’ independently is a C2-5 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’b is:R and R2 and R3 are each independently a C1-14 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’b is:
and R2 and R3 are each independently a C1-14 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’b is: and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’b is:R
and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’b is:R and R2 and R3 are each a C8 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:
and R2 and R3 are each a C8 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: and R’b is:aγ 2 3
and R’b is:aγ 2 3 R is a C1-12 alkyl and R and R are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:b aγ
R is a C1-12 alkyl and R and R are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:b aγ and R’ is:
and R’ is: , R is a C2-6 alkyl and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:
, R is a C2-6 alkyl and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: and R’b is: ,aγ 2 3
and R’b is: ,aγ 2 3 R is a C2-6 alkyl, and R and R are each a C8 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:aγ bγ
R is a C2-6 alkyl, and R and R are each a C8 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:aγ bγ , and R and R are each a C1-12 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II- d), or (II-e), R’branched is:aγ bγ
, and R and R are each a C1-12 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II- d), or (II-e), R’branched is:aγ bγ , and R and R are each a C2-6 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), m and l are each independently selected from 4, 5, and 6 and each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II- d), or (II-e), m and l are each 5 and each R’ independently is a C2-5 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:
, and R and R are each a C2-6 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), m and l are each independently selected from 4, 5, and 6 and each R’ independently is a C1-12 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II- d), or (II-e), m and l are each 5 and each R’ independently is a C2-5 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: , R’b is:
, R’b is: , m and l are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, and Raγ and Rbγ are each a C1-12 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II- Raγ b), (II-c), (II-d), or (II-e), R’branched is:b
, m and l are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, and Raγ and Rbγ are each a C1-12 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II- Raγ b), (II-c), (II-d), or (II-e), R’branched is:b R’ is:
R’ is: , m and l are each 5, each R’ independently is a C2-5 alkyl, and Raγ and Rbγ are each a C2-6 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: andb
, m and l are each 5, each R’ independently is a C2-5 alkyl, and Raγ and Rbγ are each a C2-6 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: andb R’ is:
R’ is: , m and l are each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, Raγ is a C1-12 alkyl and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:b
, m and l are each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, Raγ is a C1-12 alkyl and R2 and R3 are each independently a C6-10 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:b and R’ is:
and R’ is: , m and l are each 5, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C8 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R4 is
, m and l are each 5, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C8 alkyl. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R4 is wherein R10 is NH(C1-6 alkyl) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R4 is , wherei10
wherein R10 is NH(C1-6 alkyl) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R4 is , wherei10 n R is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:
n R is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: , R’b is:
, R’b is: , m and l are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, Raγ and Rbγ are each a C1-12 alkyl, and R4 is
, m and l are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, Raγ and Rbγ are each a C1-12 alkyl, and R4 is , wherein R10 is NH(C1-6 alkyl), and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:
, wherein R10 is NH(C1-6 alkyl), and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: , R’b is:
, R’b is: , m and l are each 5, each R’ independently is a C2 alkyl, Raγ and Rbγ are each a C alkyl, and R4 isR -5 2-6
, m and l are each 5, each R’ independently is a C2 alkyl, Raγ and Rbγ are each a C alkyl, and R4 isR -5 2-6 wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:
wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: and R’b is:
and R’b is: , m and l are each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, R2 and R3 are each independently a C6-10 alkyl, Raγ is a C1-12 alkyl, and R4 is
, m and l are each independently selected from 4, 5, and 6, R’ is a C1-12 alkyl, R2 and R3 are each independently a C6-10 alkyl, Raγ is a C1-12 alkyl, and R4 is , wherein R10 is NH(C1-6 alkyl) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), Raγ R’branched is:
, wherein R10 is NH(C1-6 alkyl) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), Raγ R’branched is: and R’b is:
and R’b is: , m and l are each 5, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, R2 and R3 are each a C8 alkyl, and R4 is
, m and l are each 5, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, R2 and R3 are each a C8 alkyl, and R4 is , wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R4 is -(CH2)nOH and n is 2, 3, or 4. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R4 is -(CH2)nOH and n is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or
, wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R4 is -(CH2)nOH and n is 2, 3, or 4. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R4 is -(CH2)nOH and n is 2. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:b
(II-e), R’branched is:b , R’ is:
, R’ is: , m and l are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, Raγ and Rbγ are each a C1-12 alkyl, R4 is -(CH2)nOH, and n is 2, 3, or 4. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is:
, m and l are each independently selected from 4, 5, and 6, each R’ independently is a C1-12 alkyl, Raγ and Rbγ are each a C1-12 alkyl, R4 is -(CH2)nOH, and n is 2, 3, or 4. In some embodiments of the compound of Formula (II), (II-a), (II-b), (II-c), (II-d), or (II-e), R’branched is: , R’b is: , m and l are each 5, each R’ independently isaγ
, R’b is: , m and l are each 5, each R’ independently isaγ a C2-5 alkyl, R and Rbγ are each a C2-6 alkyl, R4 is -(CH2)nOH, and n is 2. In some aspects, the disclosure relates to a compound of Formula (II-f):
a C2-5 alkyl, R and Rbγ are each a C2-6 alkyl, R4 is -(CH2)nOH, and n is 2. In some aspects, the disclosure relates to a compound of Formula (II-f): (II-f) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein Raγ R’branched is: ab
(II-f) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched or R’cyclic; wherein Raγ R’branched is: ab nd R’ is:
nd R’ is: wherein
wherein denotes a point of attachment; Raγ is a C1-12 alkyl; R2 and R3 are each independently a C1-14 alkyl; R4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl; m is selected from 4, 5, and 6; and l is selected from 4, 5, and 6. In some embodiments of the compound of Formula (II-f), m and l are each 5, and n is 2, 3, or 4. In some embodiments of the compound of Formula (II-f) R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C6-10 alkyl. In some embodiments of the compound of Formula (II-f), m and l are each 5, n is 2, 3, or 4, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C6-10 alkyl. In some aspects, the disclosure relates to a compound of Formula (II-g): 4
denotes a point of attachment; Raγ is a C1-12 alkyl; R2 and R3 are each independently a C1-14 alkyl; R4 is -(CH2)nOH wherein n is selected from the group consisting of 1, 2, 3, 4, and 5; R’ is a C1-12 alkyl; m is selected from 4, 5, and 6; and l is selected from 4, 5, and 6. In some embodiments of the compound of Formula (II-f), m and l are each 5, and n is 2, 3, or 4. In some embodiments of the compound of Formula (II-f) R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C6-10 alkyl. In some embodiments of the compound of Formula (II-f), m and l are each 5, n is 2, 3, or 4, R’ is a C2-5 alkyl, Raγ is a C2-6 alkyl, and R2 and R3 are each a C6-10 alkyl. In some aspects, the disclosure relates to a compound of Formula (II-g): 4 (II-g), wherein Raγ is a C2-6 alkyl; R’ is a C2-5 alkyl; and R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 3, 4, and 5, and
(II-g), wherein Raγ is a C2-6 alkyl; R’ is a C2-5 alkyl; and R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 3, 4, and 5, and wherein
wherein denotes a point of attachment, R10 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3. In some aspects, the disclosure relates to a compound of Formula (II-h):
denotes a point of attachment, R10 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3. In some aspects, the disclosure relates to a compound of Formula (II-h): (II-h), wherein Raγ and Rbγ are each independently a C2-6 alkyl; each R’ independently is a C2-5 alkyl; and R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 3, 4, and 5, and
(II-h), wherein Raγ and Rbγ are each independently a C2-6 alkyl; each R’ independently is a C2-5 alkyl; and R4 is selected from the group consisting of -(CH2)nOH wherein n is selected from the group consisting of 3, 4, and 5, and wherein1
wherein1 denotes a point of attachment, R0 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3. In some embodiments of the compound of Formula (II-g) or (II-h), R4 is
denotes a point of attachment, R0 is NH(C1-6 alkyl), and n2 is selected from the group consisting of 1, 2, and 3. In some embodiments of the compound of Formula (II-g) or (II-h), R4 is , wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (II-g) or (II-h), R4 is -(CH2)2OH. In some aspects, the disclosure relates to a compound having the Formula (III):
, wherein R10 is NH(CH3) and n2 is 2. In some embodiments of the compound of Formula (II-g) or (II-h), R4 is -(CH2)2OH. In some aspects, the disclosure relates to a compound having the Formula (III): or a salt or isomer thereof, wherein R1, R2, R3, R4, and R5 are independently selected from the group consisting of C5-20 alkyl, C5-20 alkenyl, -R”MR’, -R*YR”, -YR”, and -R*OR”; each M is independently selected from the group consisting of -C(O)O-, -OC(O)-, -OC(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S) -, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, an aryl group, and a heteroaryl group; X1, X2, and X3 are independently selected from the group consisting of a bond, -CH2-, -(CH2)2-, -CHR-, -CHY-, -C(O)-, -C(O)O-, -OC(O)-, -C(O)-CH2-, -CH2-C(O)-, -C(O)O-CH2-, -OC(O)-CH2-, -CH2-C(O)O-, -CH2-OC(O)-, -CH(OH)-, -C(S)-, and -CH(SH)-; each Y is independently a C3-6 carbocycle; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each R is independently selected from the group consisting of C1-3 alkyl and a C3-6 carbocycle; each R’ is independently selected from the group consisting of C1-12 alkyl, C2-12 alkenyl, and H; and each R” is independently selected from the group consisting of C3-12 alkyl and C3-12 alkenyl, and wherein: i) at least one of X1, X2, and X3 is not -CH2-; and/or ii) at least one of R1, R2, R3, R4, and R5 is -R”MR’. In some embodiments, R1, R2, R3, R4, and R5 are each C5-20 alkyl; X1 is -CH2-; and X2 and X3 are each -C(O)-. In some embodiments, the compound of Formula (III) is:
or a salt or isomer thereof, wherein R1, R2, R3, R4, and R5 are independently selected from the group consisting of C5-20 alkyl, C5-20 alkenyl, -R”MR’, -R*YR”, -YR”, and -R*OR”; each M is independently selected from the group consisting of -C(O)O-, -OC(O)-, -OC(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S) -, -CH(OH)-, -P(O)(OR’)O-, -S(O)2-, an aryl group, and a heteroaryl group; X1, X2, and X3 are independently selected from the group consisting of a bond, -CH2-, -(CH2)2-, -CHR-, -CHY-, -C(O)-, -C(O)O-, -OC(O)-, -C(O)-CH2-, -CH2-C(O)-, -C(O)O-CH2-, -OC(O)-CH2-, -CH2-C(O)O-, -CH2-OC(O)-, -CH(OH)-, -C(S)-, and -CH(SH)-; each Y is independently a C3-6 carbocycle; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl; each R is independently selected from the group consisting of C1-3 alkyl and a C3-6 carbocycle; each R’ is independently selected from the group consisting of C1-12 alkyl, C2-12 alkenyl, and H; and each R” is independently selected from the group consisting of C3-12 alkyl and C3-12 alkenyl, and wherein: i) at least one of X1, X2, and X3 is not -CH2-; and/or ii) at least one of R1, R2, R3, R4, and R5 is -R”MR’. In some embodiments, R1, R2, R3, R4, and R5 are each C5-20 alkyl; X1 is -CH2-; and X2 and X3 are each -C(O)-. In some embodiments, the compound of Formula (III) is: (Compound I-VI), or a salt or isomer thereof. c. Phospholipids The lipid composition of the lipid nanoparticle composition disclosed herein can comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof. In general, phospholipids comprise a phospholipid moiety and one or more fatty acid moieties. A phospholipid moiety can be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin. A fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid. Particular phospholipids can facilitate fusion to a membrane. For example, a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue. Non-natural phospholipid species including natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated. For example, a phospholipid can be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond). Under appropriate reaction conditions, an alkyne group can undergo a copper- catalyzed cycloaddition upon exposure to an azide. Such reactions can be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye). Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin. In some embodiments, a phospholipid of the present disclosure comprises 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2- dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-diundecanoyl-sn- glycero-phosphocholine (DUPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (OChemsPC), 1-hexadecyl-sn- glycero-3-phosphocholine (C16 Lyso PC), 1,2-dilinolenoyl-sn-glycero-3-phosphocholine,1,2- diarachidonoyl-sn-glycero-3-phosphocholine, 1,2-didocosahexaenoyl-sn-glycero-3- phosphocholine, 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (ME 16.0 PE), 1,2- distearoyl-sn-glycero-3-phosphoethanolamine, 1,2-dilinoleoyl-sn-glycero-3- phosphoethanolamine, 1,2-dilinolenoyl-sn-glycero-3-phosphoethanolamine, 1,2- diarachidonoyl-sn-glycero-3-phosphoethanolamine, 1,2-didocosahexaenoyl-sn-glycero-3- phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DOPG), sphingomyelin, and mixtures thereof. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure is an analog or variant of DSPC. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure is a compound of Formula (IV):
(Compound I-VI), or a salt or isomer thereof. c. Phospholipids The lipid composition of the lipid nanoparticle composition disclosed herein can comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof. In general, phospholipids comprise a phospholipid moiety and one or more fatty acid moieties. A phospholipid moiety can be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin. A fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid. Particular phospholipids can facilitate fusion to a membrane. For example, a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue. Non-natural phospholipid species including natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated. For example, a phospholipid can be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond). Under appropriate reaction conditions, an alkyne group can undergo a copper- catalyzed cycloaddition upon exposure to an azide. Such reactions can be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye). Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin. In some embodiments, a phospholipid of the present disclosure comprises 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), 1,2-dilinoleoyl-sn-glycero-3-phosphocholine (DLPC), 1,2- dimyristoyl-sn-gly cero-phosphocholine (DMPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-diundecanoyl-sn- glycero-phosphocholine (DUPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (OChemsPC), 1-hexadecyl-sn- glycero-3-phosphocholine (C16 Lyso PC), 1,2-dilinolenoyl-sn-glycero-3-phosphocholine,1,2- diarachidonoyl-sn-glycero-3-phosphocholine, 1,2-didocosahexaenoyl-sn-glycero-3- phosphocholine, 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine (ME 16.0 PE), 1,2- distearoyl-sn-glycero-3-phosphoethanolamine, 1,2-dilinoleoyl-sn-glycero-3- phosphoethanolamine, 1,2-dilinolenoyl-sn-glycero-3-phosphoethanolamine, 1,2- diarachidonoyl-sn-glycero-3-phosphoethanolamine, 1,2-didocosahexaenoyl-sn-glycero-3- phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DOPG), sphingomyelin, and mixtures thereof. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure is an analog or variant of DSPC. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure is a compound of Formula (IV): (IV), or a salt thereof, wherein: each R1 is independently optionally substituted alkyl; or optionally two R1 are joined together with the intervening atoms to form optionally substituted monocyclic carbocyclyl or optionally substituted monocyclic heterocyclyl; or optionally three R1 are joined together with the intervening atoms to form optionally substituted bicyclic carbocyclyl or optionally substitute bicyclic heterocyclyl; n is 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; A is of the Formula:
(IV), or a salt thereof, wherein: each R1 is independently optionally substituted alkyl; or optionally two R1 are joined together with the intervening atoms to form optionally substituted monocyclic carbocyclyl or optionally substituted monocyclic heterocyclyl; or optionally three R1 are joined together with the intervening atoms to form optionally substituted bicyclic carbocyclyl or optionally substitute bicyclic heterocyclyl; n is 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; A is of the Formula: each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), - NRNC(O)O, or NRNC(O)N(RN); each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), - NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), - S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or N(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group; Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2; provided that the compound is not of the Formula:
each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), - NRNC(O)O, or NRNC(O)N(RN); each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), - NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), - S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or N(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group; Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2; provided that the compound is not of the Formula: , wherein each instance of R2 is independently unsubstituted alkyl, unsubstituted alkenyl, or unsubstituted alkynyl. In some embodiments, the phospholipids may be one or more of the phospholipids described in U.S. Application No.62/520,530. i. Phospholipid Head Modifications In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a modified phospholipid head (e.g., a modified choline group). In certain embodiments, a phospholipid with a modified head is DSPC, or analog thereof, with a modified quaternary amine. For example, in embodiments of Formula (IV), at least one of R1 is not methyl. In certain embodiments, at least one of R1 is not hydrogen or methyl. In certain embodiments, the compound of Formula (IV) is of one of the following Formulae: ,
, wherein each instance of R2 is independently unsubstituted alkyl, unsubstituted alkenyl, or unsubstituted alkynyl. In some embodiments, the phospholipids may be one or more of the phospholipids described in U.S. Application No.62/520,530. i. Phospholipid Head Modifications In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a modified phospholipid head (e.g., a modified choline group). In certain embodiments, a phospholipid with a modified head is DSPC, or analog thereof, with a modified quaternary amine. For example, in embodiments of Formula (IV), at least one of R1 is not methyl. In certain embodiments, at least one of R1 is not hydrogen or methyl. In certain embodiments, the compound of Formula (IV) is of one of the following Formulae: , , , or a salt thereof, wherein: each t is independently 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; each u is independently 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; and each v is independently 1, 2, or 3. In certain embodiments, a compound of Formula (IV) is of Formula (IV-a):
, , or a salt thereof, wherein: each t is independently 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; each u is independently 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; and each v is independently 1, 2, or 3. In certain embodiments, a compound of Formula (IV) is of Formula (IV-a): (IV-a), or a salt thereof. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a cyclic moiety in place of the glyceride moiety. In certain embodiments, a phospholipid useful in the present disclosure is DSPC, or analog thereof, with a cyclic moiety in place of the glyceride moiety. In certain embodiments, the compound of Formula (IV) is of Formula (IV-b):
(IV-a), or a salt thereof. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a cyclic moiety in place of the glyceride moiety. In certain embodiments, a phospholipid useful in the present disclosure is DSPC, or analog thereof, with a cyclic moiety in place of the glyceride moiety. In certain embodiments, the compound of Formula (IV) is of Formula (IV-b): , (IV-b), or a salt thereof. ii. Phospholipid Tail Modifications In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a modified tail. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure is DSPC, or analog thereof, with a modified tail. As described herein, a “modified tail” may be a tail with shorter or longer aliphatic chains, aliphatic chains with branching introduced, aliphatic chains with substituents introduced, aliphatic chains wherein one or more methylenes are replaced by cyclic or heteroatom groups, or any combination thereof. For example, in certain embodiments, the compound of (IV) is of Formula (IV-a), or a salt thereof, wherein at least one instance of R2 is each instance of R2 is optionally substituted C1-30 alkyl, wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, OS(O)2, - S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or N(RN)S(O)2O. In certain embodiments, the compound of Formula (IV) is of Formula (IV-c):
, (IV-b), or a salt thereof. ii. Phospholipid Tail Modifications In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a modified tail. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure is DSPC, or analog thereof, with a modified tail. As described herein, a “modified tail” may be a tail with shorter or longer aliphatic chains, aliphatic chains with branching introduced, aliphatic chains with substituents introduced, aliphatic chains wherein one or more methylenes are replaced by cyclic or heteroatom groups, or any combination thereof. For example, in certain embodiments, the compound of (IV) is of Formula (IV-a), or a salt thereof, wherein at least one instance of R2 is each instance of R2 is optionally substituted C1-30 alkyl, wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, OS(O)2, - S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or N(RN)S(O)2O. In certain embodiments, the compound of Formula (IV) is of Formula (IV-c): (IV-c), or a salt thereof, wherein: each x is independently an integer between 0-30, inclusive; and each instance is G is independently selected from the group consisting of optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), - NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), - S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or N(RN)S(O)2O. Each possibility represents a separate embodiment of the present disclosure. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful or potentially useful in the present disclosure is a compound of Formula (IV), wherein n is 1, 3, 4, 5, 6, 7, 8, 9, or 10. For example, in certain embodiments, a compound of Formula (IV) is of one of the following Formulae:
(IV-c), or a salt thereof, wherein: each x is independently an integer between 0-30, inclusive; and each instance is G is independently selected from the group consisting of optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), - NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), - S(O)N(RN), N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), or N(RN)S(O)2O. Each possibility represents a separate embodiment of the present disclosure. In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful or potentially useful in the present disclosure is a compound of Formula (IV), wherein n is 1, 3, 4, 5, 6, 7, 8, 9, or 10. For example, in certain embodiments, a compound of Formula (IV) is of one of the following Formulae: , , or a salt thereof. iii. Alternative Lipids In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful. In certain embodiments, an alternative lipid is used in place of a phospholipid of the present disclosure. In certain embodiments, an alternative lipid of the present disclosure is oleic acid. In certain embodiments, the alternative lipid is one of the following: , ,
, , or a salt thereof. iii. Alternative Lipids In certain embodiments, a phospholipid useful or potentially useful in the present disclosure comprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful. In certain embodiments, an alternative lipid is used in place of a phospholipid of the present disclosure. In certain embodiments, an alternative lipid of the present disclosure is oleic acid. In certain embodiments, the alternative lipid is one of the following: , , ,
,





















































































































































Table 1. Amino Acid Sequences encoding serum albumin-IL-22 fusion proteins








An exemplary polynucleotide encoding wild type MSA is set forth in SEQ ID NO: 306. AUGAAGUGGGUGACCUUCCUGCUGCUGCUGUUCGUGAGCGGCUCUGCCUUCAGCCGGGGCGU GUUUCGGCGGGAGGCCCACAAGAGCGAGAUCGCCCACCGGUACAACGACCUGGGCGAGCAGC ACUUCAAGGGCCUGGUGCUGAUCGCCUUCAGCCAGUACCUGCAGAAGUGCAGCUACGACGAG CACGCCAAGCUGGUGCAGGAGGUGACCGACUUCGCCAAGACCUGCGUGGCCGACGAGUCCGC UGCUAACUGCGACAAGAGCCUGCACACCCUGUUCGGCGACAAGCUGUGCGCCAUCCCCAACC UGCGGGAGAACUACGGCGAGCUGGCCGACUGCUGCACCAAGCAGGAGCCCGAGCGGAACGAG UGCUUCCUGCAGCACAAGGACGACAACCCCAGCCUGCCACCCUUCGAGCGGCCAGAAGCCGA GGCCAUGUGCACCAGCUUCAAGGAGAACCCCACCACCUUCAUGGGCCACUACCUCCACGAAG UAGCCAGACGGCACCCCUACUUCUACGCUCCCGAGCUGCUGUACUACGCCGAGCAGUACAAC GAGAUCCUGACCCAGUGCUGCGCUGAGGCCGACAAGGAGAGCUGCCUGACCCCUAAGCUGGA CGGCGUGAAGGAGAAGGCCCUGGUGAGCAGCGUGCGGCAGCGGAUGAAGUGCAGCAGCAUGC AGAAGUUCGGCGAGCGGGCCUUCAAGGCCUGGGCUGUGGCACGGCUGAGCCAGACCUUCCCC AACGCCGACUUCGCCGAGAUCACCAAGCUGGCCACCGACCUGACCAAGGUGAACAAGGAGUG CUGCCACGGCGACCUGCUGGAGUGCGCCGACGACAGAGCCGAGCUGGCCAAGUACAUGUGCG AGAACCAGGCCACCAUCAGCAGCAAGCUGCAGACCUGCUGCGACAAGCCCCUGCUGAAGAAG GCCCACUGCCUGAGCGAGGUGGAGCACGACACCAUGCCCGCCGAUCUGCCAGCCAUCGCCGC CGACUUCGUGGAGGACCAGGAGGUGUGCAAGAACUACGCCGAGGCCAAGGACGUGUUCCUGG GCACCUUCCUGUACGAGUACAGCCGGCGGCACCCAGACUACAGCGUGAGCCUGCUGCUGCGG CUGGCCAAGAAGUACGAGGCCACCCUGGAGAAGUGCUGCGCCGAGGCUAAUCCACCUGCCUG CUACGGCACCGUGCUGGCCGAGUUCCAGCCCCUGGUGGAGGAGCCCAAGAACCUGGUGAAGA CCAACUGCGACCUGUACGAGAAGCUGGGCGAGUACGGCUUCCAGAACGCCAUCCUGGUGCGG UACACCCAGAAGGCCCCACAGGUGAGCACCCCAACCCUGGUUGAGGCCGCCCGUAACCUGGG CCGGGUGGGUACCAAGUGCUGCACCCUGCCCGAGGACCAGCGGCUGCCCUGCGUGGAGGACU ACCUGAGCGCCAUCCUGAACCGGGUGUGCCUGCUGCACGAGAAGACUCCCGUGAGCGAGCAC GUGACCAAGUGCUGCAGCGGCAGCCUGGUCGAGAGACGGCCCUGCUUCAGCGCCCUGACCGU GGACGAGACCUACGUGCCCAAGGAGUUCAAGGCCGAGACCUUCACCUUCCACAGCGACAUCU GCACCCUGCCUGAGAAGGAGAAGCAGAUCAAGAAGCAGACAGCCCUGGCCGAGCUGGUGAAG CACAAGCCCAAGGCCACCGCCGAGCAGCUGAAGACCGUGAUGGACGACUUCGCCCAGUUCCU GGACACCUGCUGCAAGGCCGCCGACAAGGACACCUGCUUCAGCACCGAGGGCCCCAACCUGG UG (SEQ ID NO:306) An exemplary polynucleotide encoding wild type MSA lacking an N-terminal signal peptide is set forth in SEQ ID NO: 307. GCUCACAAGAGCGAGAUCGCCCACCGGUACAACGACCUGGGCGAGCAGCACUUCAAGGGCCU GGUGCUGAUCGCCUUCAGCCAGUACCUGCAGAAGUGCAGCUACGACGAGCACGCCAAGCUGG UGCAGGAGGUGACCGACUUCGCCAAGACCUGCGUCGCCGACGAGAGCGCUGCCAACUGCGAC AAGAGCCUGCACACCCUGUUCGGCGACAAGCUGUGCGCCAUCCCCAACCUGCGGGAGAACUA CGGCGAGCUGGCCGACUGCUGCACCAAGCAGGAGCCCGAGCGGAACGAGUGCUUCCUGCAGC ACAAGGACGACAACCCCAGCCUGCCUCCAUUCGAACGGCCCGAAGCCGAGGCAAUGUGCACC AGCUUCAAGGAGAACCCCACCACCUUCAUGGGCCACUACCUGCACGAGGUCGCCAGGCGGCA UCCCUACUUCUACGCUCCCGAGCUGCUGUACUACGCCGAGCAGUACAACGAGAUCCUGACCC AGUGCUGCGCCGAGGCCGACAAGGAGAGCUGCCUGACUCCCAAGCUGGACGGCGUGAAGGAG AAGGCCCUGGUGAGCAGCGUGCGGCAGCGGAUGAAGUGCAGCAGCAUGCAGAAGUUCGGCGA GCGGGCCUUCAAAGCCUGGGCUGUGGCCAGACUGAGCCAGACCUUCCCCAACGCCGACUUCG CCGAGAUCACCAAGCUGGCCACCGACCUGACCAAGGUGAACAAGGAGUGCUGCCACGGCGAC CUGCUGGAGUGCGCCGACGACAGAGCCGAGCUGGCCAAGUACAUGUGCGAGAACCAGGCCAC CAUCAGCAGCAAGCUGCAGACCUGCUGCGACAAGCCCCUGCUGAAGAAGGCCCACUGCCUGA GCGAGGUGGAGCACGACACCAUGCCUGCCGACCUGCCAGCCAUCGCCGCCGACUUCGUGGAG GACCAGGAGGUGUGCAAGAACUACGCCGAGGCCAAGGACGUGUUCCUGGGCACCUUCCUGUA CGAGUACUCACGGCGGCACCCCGACUACAGCGUGAGCCUGCUGCUGCGGCUGGCCAAGAAGU ACGAGGCCACCCUGGAGAAGUGCUGCGCCGAAGCCAAUCCACCCGCCUGCUACGGCACCGUG CUGGCCGAGUUCCAGCCCCUGGUGGAGGAGCCCAAGAACCUGGUGAAGACCAACUGCGACCU GUACGAGAAGCUGGGCGAGUACGGCUUCCAGAACGCCAUCCUGGUGCGGUACACCCAGAAGG CCCCUCAGGUGAGCACCCCAACCCUCGUGGAGGCCGCACGGAACCUGGGCAGAGUGGGCACC AAGUGCUGUACCCUGCCCGAGGACCAGCGGCUGCCCUGCGUGGAGGACUACCUGAGCGCCAU CCUGAACCGGGUGUGCCUGCUGCACGAGAAGACCCCUGUGAGCGAGCACGUGACCAAGUGCU GCAGCGGCAGCCUGGUGGAGAGGCGGCCCUGCUUCAGCGCCCUGACCGUGGACGAGACCUAC GUGCCCAAGGAGUUCAAGGCCGAGACCUUCACCUUCCACAGCGACAUCUGCACCCUGCCCGA GAAGGAGAAGCAGAUCAAGAAGCAGACCGCUCUGGCCGAGCUCGUGAAGCACAAGCCCAAGG CCACCGCCGAGCAGCUGAAGACCGUGAUGGACGACUUCGCCCAGUUCCUGGACACCUGCUGC AAGGCCGCCGACAAGGACACCUGCUUCAGCACCGAGGGCCCCAACCUGGUG (SEQ ID NO:307) In certain aspects, this disclosure provides polynucleotides (e.g., a RNA such as an mRNA) that comprise a nucleotide sequence (e.g., an ORF) encoding one or more IL-22 polypeptides. In some embodiments, the encoded IL-22 polypeptide of this disclosure can be selected from: (i) a full length IL-22 polypeptide (e.g., having the same or essentially the same length as wild-type IL-22; e.g., SEQ ID NO:1-2); (ii) a functional fragment of IL-22 described herein (e.g., a truncated (e.g., deletion of carboxy, amino terminal, or internal regions) sequence shorter than IL-22; but still retaining IL-22 function) (e.g., SEQ ID NO:3-4); (iii) a variant thereof (e.g., full length or truncated IL-22 protein in which one or more amino acids have been replaced, e.g., variants that retain all or most of the IL-22 activity of the polypeptide with respect to a reference protein (e.g., any natural or artificial variants known in the art or described herein)); or (iv) a fusion protein comprising (i) a full length IL-22 polypeptide (e.g., SEQ ID NO:1-2), a functional fragment of IL-22 described herein, or an isoform thereof or a variant thereof (e.g., SEQ ID NO:3-4), and (ii) a full length serum albumin polypeptide (e.g., SEQ ID NO:5-6), a functional fragment of serum albumin described herein, or an isoform thereof or a variant thereof (e.g., SEQ ID NO:8). In certain embodiments, the encoded IL-22 polypeptide is a mammalian IL-22 polypeptide, such as a human IL-22 polypeptide, a functional fragment or a variant thereof. In certain embodiments, the encoded serum albumin polypeptide is a mammalian serum albumin polypeptide, such as a human serum albumin polypeptide or a mouse serum albumin polypeptide, a functional fragment, or a variant thereof. In some embodiments, the fusion protein comprises a mammalian IL-22 polypeptide and a mammalian serum albumin polypeptide, such as a human IL-22 polypeptide or functional fragment or a variant thereof, and a human serum albumin polypeptide functional fragment or a variant thereof. In some embodiments, the fusion protein comprises an amino acid sequence as set forth in one of SEQ ID NO: 20-38. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure increases IL-22 protein expression levels and/or detectable IL-22 signaling activity levels in cells when introduced in those cells, e.g., by at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 100%, compared to IL-22 protein expression levels and/or detectable IL-22 signaling activity levels in the cells prior to the administration of the polynucleotide of this disclosure. IL-22 protein expression levels and/or IL-22 enzymatic activity can be measured according to methods know in the art. In some embodiments, the polynucleotide is introduced to the cells in vitro. In some embodiments, the polynucleotide is introduced to the cells in vivo. In some embodiments, the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosure comprise a nucleotide sequence (e.g., an ORF) that encodes a wild-type IL-22 polypeptide, e.g., SEQ ID NO:1-2, or variant, fragment, or isoform thereof. In some embodiments, the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosure comprise a nucleotide sequence (e.g., an ORF) that encodes a wild-type IL-22, e.g., SEQ ID NO:3-4, or a fragment, variant, or isoform thereof. In some embodiments, the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosure comprise a nucleotide sequence (e.g., an ORF) that encodes a wild-type serum albumin, e.g., SEQ ID NO:5-8, or a fragment, variant, or isoform thereof. In some embodiments, the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosure comprise a nucleotide sequence (e.g., an ORF) that encodes a variant human IL- 22, or an isoform thereof. In some embodiments, the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosure comprise a nucleotide sequence (e.g., an ORF) that encodes a variant human serum albumin, or an isoform thereof. In some embodiments, the polynucleotides (e.g., a RNA, e.g., an mRNA) of this disclosure comprise a nucleotide sequence that encodes a fusion protein comprising an IL-22 polypeptide and a serum albumin polypeptide. In some embodiments, the polynucleotide comprises a nucleotide sequence that encodes a fusion protein comprising a wild-type IL-22 protein, (e.g., SEQ ID NO:1-2), or a variant, isoform, or fragment thereof (e.g., SEQ ID NO:3-4). In some embodiments, the polynucleotide comprises a nucleotide sequence that encodes a fusion protein comprising a wild-type serum albumin protein, (e.g., SEQ ID NO:5- 8), or a variant, isoform, or fragment thereof. In some embodiments, the polynucleotide comprises a nucleotide sequence that encodes a linker, such as a linker as described herein. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of the present disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22- containing fusion protein (e.g., an IL-22-containing fusion protein comprising the IL-22 wild- type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328, as shown in Table 2 below. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of the present disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22- containing fusion protein (e.g., an IL-22-containing fusion protein comprising the IL-22 wild- type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence has 65% to 100%, 70% to 100%, 75% to 100%, 80% to 100%, 85% to 100%, 90% to 100%, 95% to 100%, 97% to 100%, 98% to 100%, 99% to 10)%, 90% to 95%, 90% to 97%, 90% to 98%, 95% to 97%, 95% to 98%, or 95% to 99% sequence identity to the sequence of of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328, as shown in Table 2 below. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of the present disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22- containing fusion protein (e.g., an IL-22-containing fusion protein comprising the IL-22 wild- type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence is at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% identical to the sequence of of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328, as shown in Table 2 below. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of the present disclosure comprises a nucleotide sequence (e.g., an ORF) encoding an IL-22- containing fusion protein (e.g., an IL-22-containing fusion protein comprising the IL-22 wild- type sequence, functional fragment, or variant thereof), wherein the nucleotide sequence is 65% to 100%, 70% to 100%, 75% to 100%, 80% to 100%, 85% to 100%, 90% to 100%, 95% to 100%, 97% to 100%, 98% to 100%, 99% to 100%, 90% to 95%, 90% to 97%, 90% to 98%, 95% to 97%, 95% to 98%, or 95% to 99% identical to the sequence of of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328, as shown in Table 2 below. Table 2. Nucleotide Sequences encoding serum albumin-IL-22 fusion proteins










 323
323


















Codon options for each amino acid are given in Table 3. Table 3. Codon Options In some embodiments, a polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises a sequence-optimized nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide, a functional fragment, or a variant thereof, or an IL-22-containing fusion protein (e.g., a serum albumin-IL-22 fusion protein), wherein the IL-22 polypeptide, functional fragment, or a variant thereof or fusion protein encoded by the sequence-optimized nucleotide sequence has improved properties (e.g., compared to an IL-22 polypeptide, functional fragment, or a variant thereof or fusion protein encoded by a reference nucleotide sequence that is not sequence optimized), e.g., improved properties related to expression efficacy after administration in vivo. Such properties include, but are not limited to, improving nucleic acid stability (e.g., mRNA stability), increasing translation efficacy in the target tissue, reducing the number of truncated proteins expressed, improving the folding or prevent misfolding of the expressed proteins, reducing toxicity of the expressed products, reducing cell death caused by the expressed products, increasing and/or decreasing protein aggregation. In some embodiments, the sequence-optimized nucleotide sequence (e.g., an ORF) is codon optimized for expression in human subjects, having structural and/or chemical features that avoid one or more of the problems in the art, for example, features which are useful for optimizing Formulation and delivery of nucleic acid-based therapeutics while retaining structural and functional integrity; overcoming a threshold of expression; improving expression rates; half-life and/or protein concentrations; optimizing protein localization; and avoiding deleterious bio-responses such as the immune response and/or degradation pathways. In some embodiments, the polynucleotides of this disclosure comprise a nucleotide sequence (e.g., a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide, a nucleotide sequence (e.g., an ORF) encoding another polypeptide of interest (e.g., a serum albumin polypeptide), a nucleotide sequence encoding a serum albumin-IL-22 fusion protein, a 5′-UTR, a 3′-UTR, a microRNA binding site, a nucleic acid sequence encoding a linker, or any combination thereof) that is sequence-optimized according to a method comprising: (i) substituting at least one codon in a reference nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) with an alternative codon to increase or decrease uridine content to generate a uridine-modified sequence; (ii) substituting at least one codon in a reference nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) with an alternative codon having a higher codon frequency in the synonymous codon set; (iii) substituting at least one codon in a reference nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) with an alternative codon to increase G/C content; or (iv) a combination thereof. In some embodiments, the sequence-optimized nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) has at least one improved property with respect to the reference nucleotide sequence. In some embodiments, the sequence optimization method is multiparametric and comprises one, two, three, four, or more methods disclosed herein and/or other optimization methods known in the art. Features, which can be considered beneficial in some embodiments of this disclosure, can be encoded by or within regions of the polynucleotide and such regions can be upstream (5′) to, downstream (3′) to, or within the region that encodes the IL-22 polypeptide or IL-22- containing fusion protein. These regions can be incorporated into the polynucleotide before and/or after sequence-optimization of the protein encoding region or open reading frame (ORF). Examples of such features include, but are not limited to, untranslated regions (UTRs), microRNA sequences, Kozak sequences, oligo(dT) sequences, poly-A tail, and detectable tags and can include multiple cloning sites that can have XbaI recognition. In some embodiments, the polynucleotide of this disclosure comprises a 5′ UTR, a 3′ UTR and/or a microRNA binding site. In some embodiments, the polynucleotide comprises two or more 5′ UTRs and/or 3′ UTRs, which can be the same or different sequences. In some embodiments, the polynucleotide comprises two or more microRNA binding sites, which can be the same or different sequences. Any portion of the 5′ UTR, 3′ UTR, and/or microRNA binding site, including none, can be sequence-optimized and can independently contain one or more different structural or chemical modifications, before and/or after sequence optimization. In some embodiments, after optimization, the polynucleotide is reconstituted and transformed into a vector such as, but not limited to, plasmids, viruses, cosmids, and artificial chromosomes. For example, the optimized polynucleotide can be reconstituted and transformed into chemically competent E. coli, yeast, neurospora, maize, drosophila, etc. where high copy plasmid-like or chromosome structures occur by methods described herein. 8. Sequence-Optimized Nucleotide Sequences Encoding IL-22 Polypeptides or IL-22-containing Fusion Proteins In some embodiments, the polynucleotide of this disclosure comprises a sequence- optimized nucleotide sequence encoding an IL-22 polypeptide or IL-22-containing fusion protein disclosed herein. In some embodiments, the polynucleotide of this disclosure comprises an open reading frame (ORF) encoding an IL-22 polypeptide or IL-22-containing fusion protein, wherein the ORF has been sequence optimized. An exemplary sequence-optimized nucleotide sequence encoding full length IL-22 is set forth as SEQ ID NO:300-303. An exemplary sequence-optimized nucleotide sequence encoding full length serum albumin is set forth as SEQ ID NO:304-307. Exemplary sequence-optimized nucleotide sequences encoding serum albumin-IL-22 fusion proteins are set forth as SEQ ID NO:310-328 and SEQ ID NO:350-368. In some embodiments, the sequence optimized IL-22 sequence, fragment, variant, and fusion protein thereof are used to practice the methods disclosed herein. In some embodiments, a polynucleotide of the present disclosure, for example a polynucleotide comprising an mRNA nucleotide sequence encoding an IL-22 polypeptide, comprises from 5′ to 3′ end: (i) a 5′ cap such as provided herein, for example, m7Gp-ppGm-A; (ii) a 5′ UTR, such as the sequences provided herein, for example, SEQ ID NO:50 or SEQ ID NO:56; (iii) an open reading frame encoding an IL-22 polypeptide, e.g., a sequence optimized nucleic acid sequence encoding IL-22 set forth as SEQ ID NO:300-303, or a sequence optimized nucleic acid sequence encoding an IL-22-containing fusion protein as set forth in any one of SEQ ID NO:310-328; (iv) at least one stop codon (if not present at 5′ terminus of 3′UTR); (v) a 3′ UTR, such as the sequences provided herein, for example, SEQ ID NO:108; and (vi) a poly-A tail provided above (e.g., SEQ ID NO:195). In some embodiments, a polynucleotide of the present disclosure, for example a polynucleotide comprising an mRNA nucleotide sequence encoding an IL-22 polypeptide, comprises from 5′ to 3′ end: (i) a 5′ cap such as provided herein, for example, m7Gp-ppGm-A; (ii) a 5′ UTR, such as the sequences provided herein, for example, one of SEQ ID NOs:50-79; (iii) an open reading frame encoding an IL-22 polypeptide, e.g., a sequence optimized nucleic acid sequence encoding IL-22 set forth as SEQ ID NO:300-303 or a sequence optimized nucleic acid sequence encoding an IL-22-containing fusion protein as set forth in any one of SEQ ID NO:310-328; (iv) at least one stop codon (if not present at 5′ terminus of 3′UTR); (v) a 3′ UTR, such as the sequences provided herein, for example, one of SEQ ID NOs:100-147; and (vi) a poly-A tail provided above (e.g., SEQ ID NO:195). In certain embodiments, all uracils in the polynucleotide are N1-methylpseudouracil (G5). In certain embodiments, all uracils in the polynucleotide are 5-methoxyuracil (G6). The sequence-optimized nucleotide sequences disclosed herein are distinct from the corresponding wild type nucleotide acid sequences and from other known sequence- optimized nucleotide sequences, e.g., these sequence-optimized nucleic acids have unique compositional characteristics. In some embodiments, the percentage of uracil or thymine nucleobases in a sequence- optimized nucleotide sequence (e.g., encoding an IL-22 polypeptide, a functional fragment, or a variant thereof) is modified (e.g., reduced) with respect to the percentage of uracil or thymine nucleobases in the reference wild-type nucleotide sequence. Such a sequence is referred to as a uracil-modified or thymine-modified sequence. The percentage of uracil or thymine content in a nucleotide sequence can be determined by dividing the number of uracils or thymines in a sequence by the total number of nucleotides and multiplying by 100. In some embodiments, the sequence-optimized nucleotide sequence has a lower uracil or thymine content than the uracil or thymine content in the reference wild-type sequence. In some embodiments, the uracil or thymine content in a sequence-optimized nucleotide sequence of this disclosure is greater than the uracil or thymine content in the reference wild- type sequence and still maintain beneficial effects, e.g., increased expression and/or reduced Toll-Like Receptor (TLR) response when compared to the reference wild-type sequence. Methods for optimizing codon usage are known in the art. For example, an ORF of any one or more of the sequences provided herein may be codon optimized. Codon optimization, in some embodiments, may be used to match codon frequencies in target and host organisms to ensure proper folding; bias GC content to increase mRNA stability or reduce secondary structures; minimize tandem repeat codons or base runs that may impair gene construction or expression; customize transcriptional and translational control regions; insert or remove protein trafficking sequences; remove/add post translation modification sites in encoded protein (e.g., glycosylation sites); add, remove or shuffle protein domains; insert or delete restriction sites; modify ribosome binding sites and mRNA degradation sites; adjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide. Codon optimization tools, algorithms and services are known in the art - non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (Menlo Park CA) and/or proprietary methods. In some embodiments, the open reading frame (ORF) sequence is optimized using optimization algorithms. 9. Characterization of Sequence Optimized Nucleic Acids In some embodiments of this disclosure, the polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a sequence optimized nucleic acid disclosed herein encoding an IL-22 polypeptide or an IL-22-containing fusion protein can be tested to determine whether at least one nucleic acid sequence property (e.g., stability when exposed to nucleases) or expression property has been improved with respect to the non-sequence optimized nucleic acid. As used herein, "expression property" refers to a property of a nucleic acid sequence either in vivo (e.g., translation efficacy of a synthetic mRNA after administration to a subject in need thereof) or in vitro (e.g., translation efficacy of a synthetic mRNA tested in an in vitro model system). Expression properties include but are not limited to the amount of protein produced by an mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein after administration, and the amount of soluble or otherwise functional protein produced. In some embodiments, sequence optimized nucleic acids disclosed herein can be evaluated according to the viability of the cells expressing a protein encoded by a sequence optimized nucleic acid sequence (e.g., a RNA, e.g., an mRNA) encoding an IL-22 polypeptide or an IL- 22-containing fusion protein disclosed herein. In a given embodiment, a plurality of sequence optimized nucleic acids disclosed herein (e.g., a RNA, e.g., an mRNA) containing codon substitutions with respect to the non- optimized reference nucleic acid sequence can be characterized functionally to measure a property of interest, for example an expression property in an in vitro model system, or in vivo in a target tissue or cell. a. Optimization of Nucleic Acid Sequence Intrinsic Properties In some embodiments of this disclosure, the desired property of the polynucleotide is an intrinsic property of the nucleic acid sequence. For example, the nucleotide sequence (e.g., a RNA, e.g., an mRNA) can be sequence optimized for in vivo or in vitro stability. In some embodiments, the nucleotide sequence can be sequence optimized for expression in a given target tissue or cell. In some embodiments, the nucleic acid sequence is sequence optimized to increase its plasma half-life by preventing its degradation by endo and exonucleases. In other embodiments, the nucleic acid sequence is sequence optimized to increase its resistance to hydrolysis in solution, for example, to lengthen the time that the sequence optimized nucleic acid or a pharmaceutical composition comprising the sequence optimized nucleic acid can be stored under aqueous conditions with minimal degradation. In other embodiments, the sequence optimized nucleic acid can be optimized to increase its resistance to hydrolysis in dry storage conditions, for example, to lengthen the time that the sequence optimized nucleic acid can be stored after lyophilization with minimal degradation. b. Nucleic Acids Sequence Optimized for Protein Expression In some embodiments of this disclosure, the desired property of the polynucleotide is the level of expression of an IL-22 polypeptide encoded by a sequence optimized sequence disclosed herein. Protein expression levels can be measured using one or more expression systems. In some embodiments, expression can be measured in cell culture systems, e.g., CHO cells or HEK293 cells. In some embodiments, expression can be measured using in vitro expression systems prepared from extracts of living cells, e.g., rabbit reticulocyte lysates, or in vitro expression systems prepared by assembly of purified individual components. In other embodiments, the protein expression is measured in an in vivo system, e.g., mouse, rabbit, monkey, etc. In some embodiments, protein expression in solution form can be desirable. Accordingly, in some embodiments, a reference sequence can be sequence optimized to yield a sequence optimized nucleic acid sequence having optimized levels of expressed proteins in soluble form. Levels of protein expression and other properties such as solubility, levels of aggregation, and the presence of truncation products (i.e., fragments due to proteolysis, hydrolysis, or defective translation) can be measured according to methods known in the art, for example, using electrophoresis (e.g., native or SDS-PAGE) or chromatographic methods (e.g., HPLC, size exclusion chromatography, etc.). c. Optimization of Target Tissue or Target Cell Viability In some embodiments, the expression of heterologous therapeutic proteins encoded by a nucleic acid sequence can have deleterious effects in the target tissue or cell, reducing protein yield, or reducing the quality of the expressed product (e.g., due to the presence of protein fragments or precipitation of the expressed protein in inclusion bodies), or causing toxicity. Accordingly, in some embodiments of this disclosure, the sequence optimization of a nucleic acid sequence disclosed herein, e.g., a nucleic acid sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein, can be used to increase the viability of target cells expressing the protein encoded by the sequence optimized nucleic acid. Heterologous protein expression can also be deleterious to cells transfected with a nucleic acid sequence for autologous or heterologous transplantation. Accordingly, in some embodiments of the present disclosure the sequence optimization of a nucleic acid sequence disclosed herein can be used to increase the viability of target cells expressing the protein encoded by the sequence optimized nucleic acid sequence. Changes in cell or tissue viability, toxicity, and other physiological reaction can be measured according to methods known in the art. d. Reduction of Immune and/or Inflammatory Response In some cases, the administration of a sequence optimized nucleic acid encoding IL- 22 polypeptide, a functional fragment thereof, or a fusion protein comprising an IL-22 polypeptide or fragment thereof can trigger an immune response, which could be caused by (i) the therapeutic agent (e.g., an mRNA encoding an IL-22 polypeptide or fragment thereof), or (ii) the expression product of such therapeutic agent (e.g., the IL-22 polypeptide or fragment thereof encoded by the mRNA), or (iv) a combination thereof. Accordingly, in some embodiments of the present disclosure the sequence optimization of nucleic acid sequence (e.g., an mRNA) disclosed herein can be used to decrease an immune or inflammatory response triggered by the administration of a nucleic acid encoding an IL-22 polypeptide or by the expression product of IL-22 encoded by such nucleic acid. In some cases, an inflammatory response can be measured by detecting increased levels of one or more inflammatory cytokines using methods known in the art, e.g., ELISA. The term "inflammatory cytokine" refers to cytokines that are elevated in an inflammatory response. Examples of inflammatory cytokines include interleukin-6 (IL-6), CXCL1 (chemokine (C-X-C motif) ligand 1; also known as GROα, interferon-γ (IFNγ), tumor necrosis factor α (TNFα), interferon γ-induced protein 10 (IP-10), or granulocyte-colony stimulating factor (G-CSF). The term inflammatory cytokines includes also other cytokines associated with inflammatory responses known in the art, e.g., interleukin-1 (IL-1), interleukin-8 (IL-8), interleukin-12 (IL-12), interleukin-13 (Il-13), interferon α (IFN-α), etc. 10. Modified Nucleotide Sequences Encoding IL-22 Polypeptides or IL-22- containing Fusion Proteins In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1-methylpseudouracil, 5-methoxyuracil, or the like. In some embodiments, the mRNA is a uracil-modified sequence comprising an ORF encoding an IL- 22 polypeptide or an IL-22-containing fusion protein, wherein the mRNA comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1-methylpseudouracil, or 5-methoxyuracil. In certain aspects of this disclosure, when the modified uracil base is connected to a ribose sugar, as it is in polynucleotides, the resulting modified nucleoside or nucleotide is referred to as modified uridine. In some embodiments, uracil in the polynucleotide is at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least 90%, at least 95%, at least 99%, or about 100% modified uracil. In one embodiment, uracil in the polynucleotide is at least 95% modified uracil. In another embodiment, uracil in the polynucleotide is 100% modified uracil. In embodiments where uracil in the polynucleotide is at least 95% modified uracil overall uracil content can be adjusted such that an mRNA provides suitable protein expression levels while inducing little to no immune response. In some embodiments, the uracil content of the ORF is between about 100% and about 150%, between about 100% and about 110%, between about 105% and about 115%, between about 110% and about 120%, between about 115% and about 125%, between about 120% and about 130%, between about 125% and about 135%, between about 130% and about 140%, between about 135% and about 145%, between about 140% and about 150% of the theoretical minimum uracil content in the corresponding wild-type ORF (%UTM). In other embodiments, the uracil content of the ORF is between about 121% and about 136% or between 123% and 134% of the %UTM. In some embodiments, the uracil content of the ORF encoding an IL-22 polypeptide or an IL- 22-containing fusion protein is about 115%, about 120%, about 125%, about 130%, about 135%, about 140%, about 145%, or about 150% of the %UTM. In this context, the term "uracil" can refer to modified uracil and/or naturally occurring uracil. In some embodiments, the uracil content in the ORF of the mRNA encoding an IL-22 polypeptide of this disclosure is less than about 30%, about 25%, about 20%, about 15%, or about 10% of the total nucleobase content in the ORF. In some embodiments, the uracil content in the ORF is between about 10% and about 20% of the total nucleobase content in the ORF. In other embodiments, the uracil content in the ORF is between about 10% and about 25% of the total nucleobase content in the ORF. In one embodiment, the uracil content in the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein is less than about 20% of the total nucleobase content in the open reading frame. In this context, the term "uracil" can refer to modified uracil and/or naturally occurring uracil. In further embodiments, the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein having modified uracil and adjusted uracil content has increased Cytosine (C), Guanine (G), or Guanine/Cytosine (G/C) content (absolute or relative). In some embodiments, the overall increase in C, G, or G/C content (absolute or relative) of the ORF is at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 10%, at least about 15%, at least about 20%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 100% relative to the G/C content (absolute or relative) of the wild-type ORF. In some embodiments, the G, the C, or the G/C content in the ORF is less than about 100%, less than about 90%, less than about 85%, or less than about 80% of the theoretical maximum G, C, or G/C content of the corresponding wild type nucleotide sequence encoding the IL-22 polypeptide (%GTMX; %CTMX, or %G/CTMX). In some embodiments, the increases in G and/or C content (absolute or relative) described herein can be conducted by replacing synonymous codons with low G, C, or G/C content with synonymous codons having higher G, C, or G/C content. In other embodiments, the increase in G and/or C content (absolute or relative) is conducted by replacing a codon ending with U with a synonymous codon ending with G or C. In further embodiments, the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein of this disclosure comprises modified uracil and has an adjusted uracil content containing less uracil pairs (UU) and/or uracil triplets (UUU) and/or uracil quadruplets (UUUU) than the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide. In some embodiments, the ORF of the mRNA encoding an IL-22 polypeptide of this disclosure contains no uracil pairs and/or uracil triplets and/or uracil quadruplets. In some embodiments, uracil pairs and/or uracil triplets and/or uracil quadruplets are reduced below a certain threshold, e.g., no more than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 occurrences in the ORF of the mRNA encoding the IL-22 polypeptide. In a particular embodiment, the ORF of the mRNA encoding the IL- 22 polypeptide of this disclosure contains less than 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 non-phenylalanine uracil pairs and/or triplets. In another embodiment, the ORF of the mRNA encoding the IL-22 polypeptide contains no non- phenylalanine uracil pairs and/or triplets. In further embodiments, the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein of this disclosure comprises modified uracil and has an adjusted uracil content containing less uracil-rich clusters than the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide. In some embodiments, the ORF of the mRNA encoding the IL-22 polypeptide of this disclosure contains uracil-rich clusters that are shorter in length than corresponding uracil-rich clusters in the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide. In further embodiments, alternative lower frequency codons are employed. At least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, or 100% of the codons in the IL-22 polypeptide–encoding ORF of the modified uracil-comprising mRNA are substituted with alternative codons, each alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set. The ORF also has adjusted uracil content, as described above. In some embodiments, at least one codon in the ORF of the mRNA encoding the IL-22 polypeptide is substituted with an alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set. In some embodiments, the adjusted uracil content, IL-22 polypeptide-encoding ORF of the modified uracil-comprising mRNA exhibits expression levels of IL-22 when administered to a mammalian cell that are higher than expression levels of IL-22 from the corresponding wild-type mRNA. In some embodiments, the mammalian cell is a mouse cell, a rat cell, or a rabbit cell. In other embodiments, the mammalian cell is a monkey cell or a human cell. In some embodiments, the human cell is a HeLa cell, a BJ fibroblast cell, or a peripheral blood mononuclear cell (PBMC). In some embodiments, IL-22 is expressed at a level higher than expression levels of IL-22 from the corresponding wild-type mRNA when the mRNA is administered to a mammalian cell in vivo. In some embodiments, the mRNA is administered to mice, rabbits, rats, monkeys, or humans. In one embodiment, mice are null mice. In some embodiments, the mRNA is administered to mice in an amount of about 0.01 mg/kg, about 0.05 mg/kg, about 0.1 mg/kg, or 0.2 mg/kg or about 0.5 mg/kg. In some embodiments, the mRNA is administered intravenously or intramuscularly. In other embodiments, the IL-22 polypeptide is expressed when the mRNA is administered to a mammalian cell in vitro. In some embodiments, the expression is increased by at least about 2-fold, at least about 5-fold, at least about 10-fold, at least about 50-fold, at least about 500- fold, at least about 1500-fold, or at least about 3000-fold. In other embodiments, the expression is increased by at least about 10%, about 20%, about 30%, about 40%, about 50%, 60%, about 70%, about 80%, about 90%, or about 100%. In some embodiments, adjusted uracil content, IL-22 polypeptide-encoding ORF of the modified uracil-comprising mRNA exhibits increased stability. In some embodiments, the mRNA exhibits increased stability in a cell relative to the stability of a corresponding wild-type mRNA under the same conditions. In some embodiments, the mRNA exhibits increased stability including resistance to nucleases, thermal stability, and/or increased stabilization of secondary structure. In some embodiments, increased stability exhibited by the mRNA is measured by determining the half-life of the mRNA (e.g., in a plasma, serum, cell, or tissue sample) and/or determining the area under the curve (AUC) of the protein expression by the mRNA over time (e.g., in vitro or in vivo). An mRNA is identified as having increased stability if the half-life and/or the AUC is greater than the half-life and/or the AUC of a corresponding wild-type mRNA under the same conditions. In some embodiments, the mRNA of the present invention induces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by a corresponding wild-type mRNA under the same conditions. In other embodiments, the mRNA of the present disclosure induces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by an mRNA that encodes an IL-22 polypeptide but does not comprise modified uracil under the same conditions, or relative to the immune response induced by an mRNA that encodes for an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content under the same conditions. The innate immune response can be manifested by increased expression of pro- inflammatory cytokines, activation of intracellular PRRs (RIG-I, MDA5, etc), cell death, and/or termination or reduction in protein translation. In some embodiments, a reduction in the innate immune response can be measured by expression or activity level of Type 1 interferons (e.g., IFN-α, IFN-β, IFN-κ, IFN-δ, IFN-ε, IFN-τ, IFN-ω, and IFN-ζ) or the expression of interferon-regulated genes such as the toll-like receptors (e.g., TLR7 and TLR8), and/or by decreased cell death following one or more administrations of the mRNA of this disclosure into a cell. In some embodiments, the expression of Type-1 interferons by a mammalian cell in response to the mRNA of the present disclosure is reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99%, 99.9%, or greater than 99.9% relative to a corresponding wild-type mRNA, to an mRNA that encodes an IL-22 polypeptide but does not comprise modified uracil, or to an mRNA that encodes an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content. In some embodiments, the interferon is IFN-β. In some embodiments, cell death frequency caused by administration of mRNA of the present disclosure to a mammalian cell is 10%, 25%, 50%, 75%, 85%, 90%, 95%, or over 95% less than the cell death frequency observed with a corresponding wild-type mRNA, an mRNA that encodes for an IL-22 polypeptide but does not comprise modified uracil, or an mRNA that encodes for an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content. In some embodiments, the mammalian cell is a BJ fibroblast cell. In other embodiments, the mammalian cell is a splenocyte. In some embodiments, the mammalian cell is that of a mouse or a rat. In other embodiments, the mammalian cell is that of a human. In one embodiment, the mRNA of the present disclosure does not substantially induce an innate immune response of a mammalian cell into which the mRNA is introduced. 11. Methods for Modifying Polynucleotides The disclosure includes modified polynucleotides comprising a polynucleotide described herein (e.g., a polynucleotide, e.g. mRNA, comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein). The modified polynucleotides can be chemically modified and/or structurally modified. When the polynucleotides of the present invention are chemically and/or structurally modified the polynucleotides can be referred to as "modified polynucleotides." The present disclosure provides for modified nucleosides and nucleotides of a polynucleotide (e.g., RNA polynucleotides, such as mRNA polynucleotides) encoding an IL- 22 polypeptide. A "nucleoside" refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as "nucleobase"). A “nucleotide" refers to a nucleoside including a phosphate group. Modified nucleotides can be synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Polynucleotides can comprise a region or regions of linked nucleosides. Such regions can have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the polynucleotides would comprise regions of nucleotides. The modified polynucleotides disclosed herein can comprise various distinct modifications. In some embodiments, the modified polynucleotides contain one, two, or more (optionally different) nucleoside or nucleotide modifications. In some embodiments, a modified polynucleotide, introduced to a cell can exhibit one or more desirable properties, e.g., improved protein expression, reduced immunogenicity, or reduced degradation in the cell, as compared to an unmodified polynucleotide. In some embodiments, a polynucleotide of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22- containing fusion protein) is structurally modified. As used herein, a "structural" modification is one in which two or more linked nucleosides are inserted, deleted, duplicated, inverted or randomized in a polynucleotide without significant chemical modification to the nucleotides themselves. Because chemical bonds will necessarily be broken and reformed to effect a structural modification, structural modifications are of a chemical nature and hence are chemical modifications. However, structural modifications will result in a different sequence of nucleotides. For example, the polynucleotide "ATCG" can be chemically modified to "AT- 5meC-G". The same polynucleotide can be structurally modified from "ATCG" to "ATCCCG". Here, the dinucleotide "CC" has been inserted, resulting in a structural modification to the polynucleotide. Therapeutic compositions of the present disclosure comprise, in some embodiments, at least one nucleic acid (e.g., RNA) having an open reading frame encoding IL-22 (e.g., SEQ ID NO: 12), wherein the nucleic acid comprises nucleotides and/or nucleosides that can be standard (unmodified) or modified as is known in the art. In some embodiments, nucleotides and nucleosides of the present disclosure comprise modified nucleotides or nucleosides. Such modified nucleotides and nucleosides can be naturally-occurring modified nucleotides and nucleosides or non-naturally occurring modified nucleotides and nucleosides. Such modifications can include those at the sugar, backbone, or nucleobase portion of the nucleotide and/or nucleoside as are recognized in the art. In some embodiments, a naturally-occurring modified nucleotide or nucleotide of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such naturally occurring modified nucleotides and nucleotides can be found, inter alia, in the widely recognized MODOMICS database. In some embodiments, a non-naturally occurring modified nucleotide or nucleoside of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such non-naturally occurring modified nucleotides and nucleosides can be found, inter alia, in published US application Nos. PCT/US2012/058519; PCT/US2013/075177; PCT/US2014/058897; PCT/US2014/058891; PCT/US2014/070413; PCT/US2015/36773; PCT/US2015/36759; PCT/US2015/36771; or PCT/IB2017/051367 all of which are incorporated by reference herein. In some embodiments, at least one RNA (e.g., mRNA) of the present disclosure is not chemically modified and comprises the standard ribonucleotides consisting of adenosine, guanosine, cytosine and uridine. In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard nucleoside residues such as those present in transcribed RNA (e.g. A, G, C, or U). In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard deoxyribonucleosides such as those present in DNA (e.g. dA, dG, dC, or dT). Hence, nucleic acids of the disclosure (e.g., DNA nucleic acids and RNA nucleic acids, such as mRNA nucleic acids) can comprise standard nucleotides and nucleosides, naturally-occurring nucleotides and nucleosides, non-naturally-occurring nucleotides and nucleosides, or any combination thereof. Nucleic acids of the disclosure (e.g., DNA nucleic acids and RNA nucleic acids, such as mRNA nucleic acids), in some embodiments, comprise various (more than one) different types of standard and/or modified nucleotides and nucleosides. In some embodiments, a particular region of a nucleic acid contains one, two or more (optionally different) types of standard and/or modified nucleotides and nucleosides. In some embodiments, a modified RNA nucleic acid (e.g., a modified mRNA nucleic acid), introduced to a cell or organism, exhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides. In some embodiments, a modified RNA nucleic acid (e.g., a modified mRNA nucleic acid), introduced into a cell or organism, may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides. Nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids), in some embodiments, comprise non-natural modified nucleotides that are introduced during synthesis or post-synthesis of the nucleic acids to achieve desired functions or properties. The modifications may be present on internucleotide linkages, purine or pyrimidine bases, or sugars. The modification may be introduced with chemical synthesis or with a polymerase enzyme at the terminal of a chain or anywhere else in the chain. Any of the regions of a nucleic acid may be chemically modified. The present disclosure provides for modified nucleosides and nucleotides of a nucleic acid (e.g., RNA nucleic acids, such as mRNA nucleic acids). A “nucleoside” refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as “nucleobase”). A “nucleotide” refers to a nucleoside, including a phosphate group. Modified nucleotides may by synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Nucleic acids can comprise a region or regions of linked nucleosides. Such regions may have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the nucleic acids would comprise regions of nucleotides. Modified nucleotide base pairing encompasses not only the standard adenosine- thymine, adenosine-uracil, or guanosine-cytosine base pairs, but also base pairs formed between nucleotides and/or modified nucleotides comprising non-standard or modified bases, wherein the arrangement of hydrogen bond donors and hydrogen bond acceptors permits hydrogen bonding between a non-standard base and a standard base or between two complementary non-standard base structures, such as, for example, in those nucleic acids having at least one chemical modification. One example of such non-standard base pairing is the base pairing between the modified nucleotide inosine and adenine, cytosine or uracil. Any combination of base/sugar or linker may be incorporated into nucleic acids of the present disclosure. In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise N1-methyl-pseudouridine (m1ψ), 1-ethyl- pseudouridine (e1ψ), 5-methoxy-uridine (mo5U), 5-methyl-cytidine (m5C), and/or pseudouridine (ψ). In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise 5-methoxymethyl uridine, 5-methylthio uridine, 1-methoxymethyl pseudouridine, 5-methyl cytidine, and/or 5-methoxy cytidine. In some embodiments, the polyribonucleotide includes a combination of at least two (e.g., 2, 3, 4 or more) of any of the aforementioned modified nucleobases, including but not limited to chemical modifications. In some embodiments, a RNA nucleic acid of the disclosure comprises N1-methyl- pseudouridine (m1ψ) substitutions at one or more or all uridine positions of the nucleic acid. In some embodiments, a RNA nucleic acid of the disclosure comprises N1-methyl- pseudouridine (m1ψ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid. In some embodiments, a RNA nucleic acid of the disclosure comprises pseudouridine (ψ) substitutions at one or more or all uridine positions of the nucleic acid. In some embodiments, a RNA nucleic acid of the disclosure comprises pseudouridine (ψ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid. In some embodiments, a RNA nucleic acid of the disclosure comprises uridine at one or more or all uridine positions of the nucleic acid. In some embodiments, nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) are uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification. For example, a nucleic acid can be uniformly modified with N1-methyl-pseudouridine, meaning that all uridine residues in the mRNA sequence are replaced with N1-methyl-pseudouridine. Similarly, a nucleic acid can be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above. The nucleic acids of the present disclosure may be partially or fully modified along the entire length of the molecule. For example, one or more or all or a given type of nucleotide (e.g., purine or pyrimidine, or any one or more or all of A, G, U, C) may be uniformly modified in a nucleic acid of the disclosure, or in a predetermined sequence region thereof (e.g., in the mRNA including or excluding the polyA tail). In some embodiments, all nucleotides X in a nucleic acid of the present disclosure (or in a sequence region thereof) are modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C or A+G+C. The nucleic acid may contain from about 1% to about 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, i.e., any one or more of A, G, U or C) or any intervening percentage (e.g., from 1% to 20%, from 1% to 25%, from 1% to 50%, from 1% to 60%, from 1% to 70%, from 1% to 80%, from 1% to 90%, from 1% to 95%, from 10% to 20%, from 10% to 25%, from 10% to 50%, from 10% to 60%, from 10% to 70%, from 10% to 80%, from 10% to 90%, from 10% to 95%, from 10% to 100%, from 20% to 25%, from 20% to 50%, from 20% to 60%, from 20% to 70%, from 20% to 80%, from 20% to 90%, from 20% to 95%, from 20% to 100%, from 50% to 60%, from 50% to 70%, from 50% to 80%, from 50% to 90%, from 50% to 95%, from 50% to 100%, from 70% to 80%, from 70% to 90%, from 70% to 95%, from 70% to 100%, from 80% to 90%, from 80% to 95%, from 80% to 100%, from 90% to 95%, from 90% to 100%, and from 95% to 100%). It will be understood that any remaining percentage is accounted for by the presence of unmodified A, G, U, or C. The nucleic acids may contain at a minimum 1% and at maximum 100% modified nucleotides, or any intervening percentage, such as at least 5% modified nucleotides, at least 10% modified nucleotides, at least 25% modified nucleotides, at least 50% modified nucleotides, at least 80% modified nucleotides, or at least 90% modified nucleotides. For example, the nucleic acids may contain a modified pyrimidine such as a modified uracil or cytosine. In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the uracil in the nucleic acid is replaced with a modified uracil (e.g., a 5-substituted uracil). The modified uracil can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the cytosine in the nucleic acid is replaced with a modified cytosine (e.g., a 5-substituted cytosine). The modified cytosine can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). 12. Untranslated Regions (UTRs) Untranslated regions (UTRs) are nucleic acid sections of a polynucleotide before a start codon (5′ UTR) and after a stop codon (3′ UTR) that are not translated. In some embodiments, a polynucleotide (e.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)) of this disclosure comprising an open reading frame (ORF) encoding an IL-22 polypeptide or an IL-22-containing fusion protein further comprises a UTR (e.g., a 5′ UTR or functional fragment thereof, a 3′ UTR or functional fragment thereof, or a combination thereof). A UTR (e.g., 5′ UTR or 3′ UTR) can be homologous or heterologous to the coding region in a polynucleotide. In some embodiments, the UTR is homologous to the ORF encoding the IL-22 polypeptide. In some embodiments, the UTR is heterologous to the ORF encoding the IL-22 polypeptide. In some embodiments, the polynucleotide comprises two or more 5′ UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences. In some embodiments, the polynucleotide comprises two or more 3′ UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences. In some embodiments, the 5′ UTR or functional fragment thereof, 3′ UTR or functional fragment thereof, or any combination thereof is sequence optimized. In some embodiments, the 5′UTR or functional fragment thereof, 3′ UTR or functional fragment thereof, or any combination thereof comprises at least one chemically modified nucleobase, e.g., N1-methylpseudouracil or 5-methoxyuracil. UTRs can have features that provide a regulatory role, e.g., increased or decreased stability, localization and/or translation efficiency. A polynucleotide comprising a UTR can be administered to a cell, tissue, or organism, and one or more regulatory features can be measured using routine methods. In some embodiments, a functional fragment of a 5′ UTR or 3′ UTR comprises one or more regulatory features of a full length 5′ or 3′ UTR, respectively. Natural 5′UTRs bear features that play roles in translation initiation. They harbor signatures like Kozak sequences that are commonly known to be involved in the process by which the ribosome initiates translation of many genes. Kozak sequences have the consensus CCR(A/G)CCAUGG (SEQ ID NO: 214), where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another ‘G’.5′ UTRs also have been known to form secondary structures that are involved in elongation factor binding. By engineering the features typically found in abundantly expressed genes of specific target organs, one can enhance the stability and protein production of a polynucleotide. For example, introduction of 5′ UTR of liver-expressed mRNA, such as albumin, serum amyloid A, Apolipoprotein A/B/E, transferrin, alpha fetoprotein, erythropoietin, or Factor VIII, can enhance expression of polynucleotides in hepatic cell lines or liver. Likewise, use of 5′UTR from other tissue-specific mRNA to improve expression in that tissue is possible for muscle (e.g., MyoD, Myosin, Myoglobin, Myogenin, Herculin), for endothelial cells (e.g., Tie-1, CD36), for myeloid cells (e.g., C/EBP, AML1, G-CSF, GM-CSF, CD11b, MSR, Fr-1, i- NOS), for leukocytes (e.g., CD45, CD18), for adipose tissue (e.g., CD36, GLUT4, ACRP30, adiponectin) and for lung epithelial cells (e.g., SP-A/B/C/D). In some embodiments, UTRs are selected from a family of transcripts whose proteins share a common function, structure, feature or property. For example, an encoded polypeptide can belong to a family of proteins (i.e., that share at least one function, structure, feature, localization, origin, or expression pattern), which are expressed in a particular cell, tissue or at some time during development. The UTRs from any of the genes or mRNA can be swapped for any other UTR of the same or different family of proteins to create a new polynucleotide. In some embodiments, the 5′ UTR and the 3′ UTR can be heterologous. In some embodiments, the 5′ UTR can be derived from a different species than the 3′ UTR. In some embodiments, the 3′ UTR can be derived from a different species than the 5′ UTR. Co-owned International Patent Application No. PCT/US2014/021522 (Publ. No. WO/2014/164253, incorporated herein by reference in its entirety) provides a listing of exemplary UTRs that can be utilized in the polynucleotide of the present invention as flanking regions to an ORF. Additional exemplary UTRs of the application include, but are not limited to, one or more 5′UTR and/or 3′UTR derived from the nucleic acid sequence of: a globin, such as an α- or β-globin (e.g., a Xenopus, mouse, rabbit, or human globin); a strong Kozak translational initiation signal; a CYBA (e.g., human cytochrome b-245 α polypeptide); an albumin (e.g., human albumin7); a HSD17B4 (hydroxysteroid (17-β) dehydrogenase); a virus (e.g., a tobacco etch virus (TEV), a Venezuelan equine encephalitis virus (VEEV), a Dengue virus, a cytomegalovirus (CMV) (e.g., CMV immediate early 1 (IE1)), a hepatitis virus (e.g., hepatitis B virus), a sindbis virus, or a PAV barley yellow dwarf virus); a heat shock protein (e.g., hsp70); a translation initiation factor (e.g., elF4G); a glucose transporter (e.g., hGLUT1 (human glucose transporter 1)); an actin (e.g., human α or β actin); a GAPDH; a tubulin; a histone; a citric acid cycle enzyme; a topoisomerase (e.g., a 5′UTR of a TOP gene lacking the 5′ TOP motif (the oligopyrimidine tract)); a ribosomal protein Large 32 (L32); a ribosomal protein (e.g., human or mouse ribosomal protein, such as, for example, rps9); an ATP synthase (e.g., ATP5A1 or the β subunit of mitochondrial H+-ATP synthase); a growth hormone e (e.g., bovine (bGH) or human (hGH)); an elongation factor (e.g., elongation factor 1 α1 (EEF1A1)); a manganese superoxide dismutase (MnSOD); a myocyte enhancer factor 2A (MEF2A); a β-F1-ATPase, a creatine kinase, a myoglobin, a granulocyte-colony stimulating factor (G-CSF); a collagen (e.g., collagen type I, alpha 2 (Col1A2), collagen type I, alpha 1 (Col1A1), collagen type VI, alpha 2 (Col6A2), collagen type VI, alpha 1 (Col6A1)); a ribophorin (e.g., ribophorin I (RPNI)); a low density lipoprotein receptor-related protein (e.g., LRP1); a cardiotrophin-like cytokine factor (e.g., Nnt1); calreticulin (Calr); a procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (Plod1); and a nucleobindin (e.g., Nucb1). In some embodiments, the 5′ UTR is selected from the group consisting of a β-globin 5′ UTR; a 5′UTR containing a strong Kozak translational initiation signal; a cytochrome b- 245 α polypeptide (CYBA) 5′ UTR; a hydroxysteroid (17-β) dehydrogenase (HSD17B4) 5′ UTR; a Tobacco etch virus (TEV) 5′ UTR; a Venezuelen equine encephalitis virus (TEEV) 5′ UTR; a 5′ proximal open reading frame of rubella virus (RV) RNA encoding nonstructural proteins; a Dengue virus (DEN) 5′ UTR; a heat shock protein 70 (Hsp70) 5′ UTR; a eIF4G 5′ UTR; a GLUT15′ UTR; functional fragments thereof and any combination thereof. In some embodiments, the 3′ UTR is selected from the group consisting of a β-globin 3′ UTR; a CYBA 3′ UTR; an albumin 3′ UTR; a growth hormone (GH) 3′ UTR; a VEEV 3′ UTR; a hepatitis B virus (HBV) 3′ UTR; α-globin 3′UTR; a DEN 3′ UTR; a PAV barley yellow dwarf virus (BYDV-PAV) 3′ UTR; an elongation factor 1 α1 (EEF1A1) 3′ UTR; a manganese superoxide dismutase (MnSOD) 3′ UTR; a β subunit of mitochondrial H(+)-ATP synthase (β-mRNA) 3′ UTR; a GLUT13′ UTR; a MEF2A 3′ UTR; a β-F1-ATPase 3′ UTR; functional fragments thereof and combinations thereof. Wild-type UTRs derived from any gene or mRNA can be incorporated into the polynucleotides of this disclosure. In some embodiments, a UTR can be altered relative to a wild type or native UTR to produce a variant UTR, e.g., by changing the orientation or location of the UTR relative to the ORF; or by inclusion of additional nucleotides, deletion of nucleotides, swapping or transposition of nucleotides. In some embodiments, variants of 5′ or 3′ UTRs can be utilized, for example, mutants of wild type UTRs, or variants wherein one or more nucleotides are added to or removed from a terminus of the UTR. Additionally, one or more synthetic UTRs can be used in combination with one or more non-synthetic UTRs. See, e.g., Mandal and Rossi, Nat. Protoc.20138(3):568-82, the contents of which are incorporated herein by reference in their entirety. UTRs or portions thereof can be placed in the same orientation as in the transcript from which they were selected or can be altered in orientation or location. Hence, a 5′ and/or 3′ UTR can be inverted, shortened, lengthened, or combined with one or more other 5′ UTRs or 3′ UTRs. In some embodiments, the polynucleotide comprises multiple UTRs, e.g., a double, a triple or a quadruple 5′ UTR or 3′ UTR. For example, a double UTR comprises two copies of the same UTR either in series or substantially in series. For example, a double beta-globin 3′UTR can be used (see US2010/0129877, the contents of which are incorporated herein by reference in its entirety). The polynucleotides of this disclosure can comprise combinations of features. For example, the ORF can be flanked by a 5′UTR that comprises a strong Kozak translational initiation signal and/or a 3′UTR comprising an oligo(dT) sequence for templated addition of a poly-A tail. A 5′UTR can comprise a first polynucleotide fragment and a second polynucleotide fragment from the same and/or different UTRs (see, e.g., US2010/0293625, herein incorporated by reference in its entirety). Other non-UTR sequences can be used as regions or subregions within the polynucleotides of this disclosure. For example, introns or portions of intron sequences can be incorporated into the polynucleotides of this disclosure. Incorporation of intronic sequences can increase protein production as well as polynucleotide expression levels. In some embodiments, the polynucleotide of this disclosure comprises an internal ribosome entry site (IRES) instead of or in addition to a UTR (see, e.g., Yakubov et al., Biochem. Biophys. Res. Commun.2010394(1):189-193, the contents of which are incorporated herein by reference in their entirety). In some embodiments, the polynucleotide comprises an IRES instead of a 5′ UTR sequence. In some embodiments, the polynucleotide comprises an ORF and a viral capsid sequence. In some embodiments, the polynucleotide comprises a synthetic 5′ UTR in combination with a non-synthetic 3′ UTR. In some embodiments, the UTR can also include at least one translation enhancer polynucleotide, translation enhancer element, or translational enhancer elements (collectively, "TEE," which refers to nucleic acid sequences that increase the amount of polypeptide or protein produced from a polynucleotide. As a non-limiting example, the TEE can be located between the transcription promoter and the start codon. In some embodiments, the 5′ UTR comprises a TEE. In one aspect, a TEE is a conserved element in a UTR that can promote translational activity of a nucleic acid such as, but not limited to, cap-dependent or cap-independent translation. a.5′ UTR sequences 5′ UTR sequences are important for ribosome recruitment to the mRNA and have been reported to play a role in translation (Hinnebusch A, et al., (2016) Science, 352:6292: 1413-6). Disclosed herein, inter alia, is a polynucleotide, e.g., mRNA, comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1-4) or an IL-22-containing fusion protein (e.g., SEQ ID NO: 20-38), which polynucleotide has a 5′ UTR that confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself. In an embodiment, a polynucleotide disclosed herein comprises: (a) a 5′-UTR (e.g., as provided in Table 4 or a variant or fragment thereof); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as described herein), and LNP compositions comprising the same. In an embodiment, the polynucleotide comprises a 5′-UTR comprising a sequence provided in Table 4 or a variant or fragment thereof (e.g., a functional variant or fragment thereof). In an embodiment, the polynucleotide having a 5′ UTR sequence provided in Table 4 or a variant or fragment thereof, has an increase in the half-life of the polynucleotide, e.g., about 1.5-20-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more. In an embodiment, the increase in half life is about 1.5-fold or more. In an embodiment, the increase in half life is about 2-fold or more. In an embodiment, the increase in half life is about 3-fold or more. In an embodiment, the increase in half life is about 4-fold or more. In an embodiment, the increase in half life is about 5-fold or more. In an embodiment, the polynucleotide having a 5′ UTR sequence provided in Table 4 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the 5′UTR results in about 1.5-20-fold increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase in level and/or activity is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more. In an embodiment, the increase in level and/or activity is about 1.5-fold or more. In an embodiment, the increase in level and/or activity is about 2-fold or more. In an embodiment, the increase in level and/or activity is about 3-fold or more. In an embodiment, the increase in level and/or activity is about 4-fold or more. In an embodiment, the increase in level and/or activity is about 5-fold or more. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 5′ UTR, has a different 5′ UTR, or does not have a 5′ UTR described in Table 4 or a variant or fragment thereof. In an embodiment, the increase in half-life of the polynucleotide is measured according to an assay that measures the half-life of a polynucleotide. In an embodiment, the increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide is measured according to an assay that measures the level and/or activity of a polypeptide. In an embodiment, the 5′ UTR comprises a sequence provided in Table 4 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 5′ UTR sequence provided in Table 4, or a variant or a fragment thereof. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 50, SEQ ID NO: 51, SEQ ID NO: 52, SEQ ID NO: 53, SEQ ID NO: 54, SEQ ID NO: 55, SEQ ID NO: 56, SEQ ID NO: 57 or SEQ ID NO: 58. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 50. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 51. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 52. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 53. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 54. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 55. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 56. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 57. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 58. In an embodiment, the 5′ UTR comprises the sequence of SEQ ID NO:58. In an embodiment, the 5′ UTR consists of the sequence of SEQ ID NO:58. In an embodiment, a 5′ UTR sequence provided in Table 4 has a first nucleotide which is an A. In an embodiment, a 5′ UTR sequence provided in Table 4 has a first nucleotide which is a G. Table 4: 5′ UTR sequences
In some embodiments, a polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises a sequence-optimized nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide, a functional fragment, or a variant thereof, or an IL-22-containing fusion protein (e.g., a serum albumin-IL-22 fusion protein), wherein the IL-22 polypeptide, functional fragment, or a variant thereof or fusion protein encoded by the sequence-optimized nucleotide sequence has improved properties (e.g., compared to an IL-22 polypeptide, functional fragment, or a variant thereof or fusion protein encoded by a reference nucleotide sequence that is not sequence optimized), e.g., improved properties related to expression efficacy after administration in vivo. Such properties include, but are not limited to, improving nucleic acid stability (e.g., mRNA stability), increasing translation efficacy in the target tissue, reducing the number of truncated proteins expressed, improving the folding or prevent misfolding of the expressed proteins, reducing toxicity of the expressed products, reducing cell death caused by the expressed products, increasing and/or decreasing protein aggregation. In some embodiments, the sequence-optimized nucleotide sequence (e.g., an ORF) is codon optimized for expression in human subjects, having structural and/or chemical features that avoid one or more of the problems in the art, for example, features which are useful for optimizing Formulation and delivery of nucleic acid-based therapeutics while retaining structural and functional integrity; overcoming a threshold of expression; improving expression rates; half-life and/or protein concentrations; optimizing protein localization; and avoiding deleterious bio-responses such as the immune response and/or degradation pathways. In some embodiments, the polynucleotides of this disclosure comprise a nucleotide sequence (e.g., a nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide, a nucleotide sequence (e.g., an ORF) encoding another polypeptide of interest (e.g., a serum albumin polypeptide), a nucleotide sequence encoding a serum albumin-IL-22 fusion protein, a 5′-UTR, a 3′-UTR, a microRNA binding site, a nucleic acid sequence encoding a linker, or any combination thereof) that is sequence-optimized according to a method comprising: (i) substituting at least one codon in a reference nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) with an alternative codon to increase or decrease uridine content to generate a uridine-modified sequence; (ii) substituting at least one codon in a reference nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) with an alternative codon having a higher codon frequency in the synonymous codon set; (iii) substituting at least one codon in a reference nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) with an alternative codon to increase G/C content; or (iv) a combination thereof. In some embodiments, the sequence-optimized nucleotide sequence (e.g., an ORF encoding an IL-22 polypeptide) has at least one improved property with respect to the reference nucleotide sequence. In some embodiments, the sequence optimization method is multiparametric and comprises one, two, three, four, or more methods disclosed herein and/or other optimization methods known in the art. Features, which can be considered beneficial in some embodiments of this disclosure, can be encoded by or within regions of the polynucleotide and such regions can be upstream (5′) to, downstream (3′) to, or within the region that encodes the IL-22 polypeptide or IL-22- containing fusion protein. These regions can be incorporated into the polynucleotide before and/or after sequence-optimization of the protein encoding region or open reading frame (ORF). Examples of such features include, but are not limited to, untranslated regions (UTRs), microRNA sequences, Kozak sequences, oligo(dT) sequences, poly-A tail, and detectable tags and can include multiple cloning sites that can have XbaI recognition. In some embodiments, the polynucleotide of this disclosure comprises a 5′ UTR, a 3′ UTR and/or a microRNA binding site. In some embodiments, the polynucleotide comprises two or more 5′ UTRs and/or 3′ UTRs, which can be the same or different sequences. In some embodiments, the polynucleotide comprises two or more microRNA binding sites, which can be the same or different sequences. Any portion of the 5′ UTR, 3′ UTR, and/or microRNA binding site, including none, can be sequence-optimized and can independently contain one or more different structural or chemical modifications, before and/or after sequence optimization. In some embodiments, after optimization, the polynucleotide is reconstituted and transformed into a vector such as, but not limited to, plasmids, viruses, cosmids, and artificial chromosomes. For example, the optimized polynucleotide can be reconstituted and transformed into chemically competent E. coli, yeast, neurospora, maize, drosophila, etc. where high copy plasmid-like or chromosome structures occur by methods described herein. 8. Sequence-Optimized Nucleotide Sequences Encoding IL-22 Polypeptides or IL-22-containing Fusion Proteins In some embodiments, the polynucleotide of this disclosure comprises a sequence- optimized nucleotide sequence encoding an IL-22 polypeptide or IL-22-containing fusion protein disclosed herein. In some embodiments, the polynucleotide of this disclosure comprises an open reading frame (ORF) encoding an IL-22 polypeptide or IL-22-containing fusion protein, wherein the ORF has been sequence optimized. An exemplary sequence-optimized nucleotide sequence encoding full length IL-22 is set forth as SEQ ID NO:300-303. An exemplary sequence-optimized nucleotide sequence encoding full length serum albumin is set forth as SEQ ID NO:304-307. Exemplary sequence-optimized nucleotide sequences encoding serum albumin-IL-22 fusion proteins are set forth as SEQ ID NO:310-328 and SEQ ID NO:350-368. In some embodiments, the sequence optimized IL-22 sequence, fragment, variant, and fusion protein thereof are used to practice the methods disclosed herein. In some embodiments, a polynucleotide of the present disclosure, for example a polynucleotide comprising an mRNA nucleotide sequence encoding an IL-22 polypeptide, comprises from 5′ to 3′ end: (i) a 5′ cap such as provided herein, for example, m7Gp-ppGm-A; (ii) a 5′ UTR, such as the sequences provided herein, for example, SEQ ID NO:50 or SEQ ID NO:56; (iii) an open reading frame encoding an IL-22 polypeptide, e.g., a sequence optimized nucleic acid sequence encoding IL-22 set forth as SEQ ID NO:300-303, or a sequence optimized nucleic acid sequence encoding an IL-22-containing fusion protein as set forth in any one of SEQ ID NO:310-328; (iv) at least one stop codon (if not present at 5′ terminus of 3′UTR); (v) a 3′ UTR, such as the sequences provided herein, for example, SEQ ID NO:108; and (vi) a poly-A tail provided above (e.g., SEQ ID NO:195). In some embodiments, a polynucleotide of the present disclosure, for example a polynucleotide comprising an mRNA nucleotide sequence encoding an IL-22 polypeptide, comprises from 5′ to 3′ end: (i) a 5′ cap such as provided herein, for example, m7Gp-ppGm-A; (ii) a 5′ UTR, such as the sequences provided herein, for example, one of SEQ ID NOs:50-79; (iii) an open reading frame encoding an IL-22 polypeptide, e.g., a sequence optimized nucleic acid sequence encoding IL-22 set forth as SEQ ID NO:300-303 or a sequence optimized nucleic acid sequence encoding an IL-22-containing fusion protein as set forth in any one of SEQ ID NO:310-328; (iv) at least one stop codon (if not present at 5′ terminus of 3′UTR); (v) a 3′ UTR, such as the sequences provided herein, for example, one of SEQ ID NOs:100-147; and (vi) a poly-A tail provided above (e.g., SEQ ID NO:195). In certain embodiments, all uracils in the polynucleotide are N1-methylpseudouracil (G5). In certain embodiments, all uracils in the polynucleotide are 5-methoxyuracil (G6). The sequence-optimized nucleotide sequences disclosed herein are distinct from the corresponding wild type nucleotide acid sequences and from other known sequence- optimized nucleotide sequences, e.g., these sequence-optimized nucleic acids have unique compositional characteristics. In some embodiments, the percentage of uracil or thymine nucleobases in a sequence- optimized nucleotide sequence (e.g., encoding an IL-22 polypeptide, a functional fragment, or a variant thereof) is modified (e.g., reduced) with respect to the percentage of uracil or thymine nucleobases in the reference wild-type nucleotide sequence. Such a sequence is referred to as a uracil-modified or thymine-modified sequence. The percentage of uracil or thymine content in a nucleotide sequence can be determined by dividing the number of uracils or thymines in a sequence by the total number of nucleotides and multiplying by 100. In some embodiments, the sequence-optimized nucleotide sequence has a lower uracil or thymine content than the uracil or thymine content in the reference wild-type sequence. In some embodiments, the uracil or thymine content in a sequence-optimized nucleotide sequence of this disclosure is greater than the uracil or thymine content in the reference wild- type sequence and still maintain beneficial effects, e.g., increased expression and/or reduced Toll-Like Receptor (TLR) response when compared to the reference wild-type sequence. Methods for optimizing codon usage are known in the art. For example, an ORF of any one or more of the sequences provided herein may be codon optimized. Codon optimization, in some embodiments, may be used to match codon frequencies in target and host organisms to ensure proper folding; bias GC content to increase mRNA stability or reduce secondary structures; minimize tandem repeat codons or base runs that may impair gene construction or expression; customize transcriptional and translational control regions; insert or remove protein trafficking sequences; remove/add post translation modification sites in encoded protein (e.g., glycosylation sites); add, remove or shuffle protein domains; insert or delete restriction sites; modify ribosome binding sites and mRNA degradation sites; adjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide. Codon optimization tools, algorithms and services are known in the art - non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (Menlo Park CA) and/or proprietary methods. In some embodiments, the open reading frame (ORF) sequence is optimized using optimization algorithms. 9. Characterization of Sequence Optimized Nucleic Acids In some embodiments of this disclosure, the polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a sequence optimized nucleic acid disclosed herein encoding an IL-22 polypeptide or an IL-22-containing fusion protein can be tested to determine whether at least one nucleic acid sequence property (e.g., stability when exposed to nucleases) or expression property has been improved with respect to the non-sequence optimized nucleic acid. As used herein, "expression property" refers to a property of a nucleic acid sequence either in vivo (e.g., translation efficacy of a synthetic mRNA after administration to a subject in need thereof) or in vitro (e.g., translation efficacy of a synthetic mRNA tested in an in vitro model system). Expression properties include but are not limited to the amount of protein produced by an mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein after administration, and the amount of soluble or otherwise functional protein produced. In some embodiments, sequence optimized nucleic acids disclosed herein can be evaluated according to the viability of the cells expressing a protein encoded by a sequence optimized nucleic acid sequence (e.g., a RNA, e.g., an mRNA) encoding an IL-22 polypeptide or an IL- 22-containing fusion protein disclosed herein. In a given embodiment, a plurality of sequence optimized nucleic acids disclosed herein (e.g., a RNA, e.g., an mRNA) containing codon substitutions with respect to the non- optimized reference nucleic acid sequence can be characterized functionally to measure a property of interest, for example an expression property in an in vitro model system, or in vivo in a target tissue or cell. a. Optimization of Nucleic Acid Sequence Intrinsic Properties In some embodiments of this disclosure, the desired property of the polynucleotide is an intrinsic property of the nucleic acid sequence. For example, the nucleotide sequence (e.g., a RNA, e.g., an mRNA) can be sequence optimized for in vivo or in vitro stability. In some embodiments, the nucleotide sequence can be sequence optimized for expression in a given target tissue or cell. In some embodiments, the nucleic acid sequence is sequence optimized to increase its plasma half-life by preventing its degradation by endo and exonucleases. In other embodiments, the nucleic acid sequence is sequence optimized to increase its resistance to hydrolysis in solution, for example, to lengthen the time that the sequence optimized nucleic acid or a pharmaceutical composition comprising the sequence optimized nucleic acid can be stored under aqueous conditions with minimal degradation. In other embodiments, the sequence optimized nucleic acid can be optimized to increase its resistance to hydrolysis in dry storage conditions, for example, to lengthen the time that the sequence optimized nucleic acid can be stored after lyophilization with minimal degradation. b. Nucleic Acids Sequence Optimized for Protein Expression In some embodiments of this disclosure, the desired property of the polynucleotide is the level of expression of an IL-22 polypeptide encoded by a sequence optimized sequence disclosed herein. Protein expression levels can be measured using one or more expression systems. In some embodiments, expression can be measured in cell culture systems, e.g., CHO cells or HEK293 cells. In some embodiments, expression can be measured using in vitro expression systems prepared from extracts of living cells, e.g., rabbit reticulocyte lysates, or in vitro expression systems prepared by assembly of purified individual components. In other embodiments, the protein expression is measured in an in vivo system, e.g., mouse, rabbit, monkey, etc. In some embodiments, protein expression in solution form can be desirable. Accordingly, in some embodiments, a reference sequence can be sequence optimized to yield a sequence optimized nucleic acid sequence having optimized levels of expressed proteins in soluble form. Levels of protein expression and other properties such as solubility, levels of aggregation, and the presence of truncation products (i.e., fragments due to proteolysis, hydrolysis, or defective translation) can be measured according to methods known in the art, for example, using electrophoresis (e.g., native or SDS-PAGE) or chromatographic methods (e.g., HPLC, size exclusion chromatography, etc.). c. Optimization of Target Tissue or Target Cell Viability In some embodiments, the expression of heterologous therapeutic proteins encoded by a nucleic acid sequence can have deleterious effects in the target tissue or cell, reducing protein yield, or reducing the quality of the expressed product (e.g., due to the presence of protein fragments or precipitation of the expressed protein in inclusion bodies), or causing toxicity. Accordingly, in some embodiments of this disclosure, the sequence optimization of a nucleic acid sequence disclosed herein, e.g., a nucleic acid sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein, can be used to increase the viability of target cells expressing the protein encoded by the sequence optimized nucleic acid. Heterologous protein expression can also be deleterious to cells transfected with a nucleic acid sequence for autologous or heterologous transplantation. Accordingly, in some embodiments of the present disclosure the sequence optimization of a nucleic acid sequence disclosed herein can be used to increase the viability of target cells expressing the protein encoded by the sequence optimized nucleic acid sequence. Changes in cell or tissue viability, toxicity, and other physiological reaction can be measured according to methods known in the art. d. Reduction of Immune and/or Inflammatory Response In some cases, the administration of a sequence optimized nucleic acid encoding IL- 22 polypeptide, a functional fragment thereof, or a fusion protein comprising an IL-22 polypeptide or fragment thereof can trigger an immune response, which could be caused by (i) the therapeutic agent (e.g., an mRNA encoding an IL-22 polypeptide or fragment thereof), or (ii) the expression product of such therapeutic agent (e.g., the IL-22 polypeptide or fragment thereof encoded by the mRNA), or (iv) a combination thereof. Accordingly, in some embodiments of the present disclosure the sequence optimization of nucleic acid sequence (e.g., an mRNA) disclosed herein can be used to decrease an immune or inflammatory response triggered by the administration of a nucleic acid encoding an IL-22 polypeptide or by the expression product of IL-22 encoded by such nucleic acid. In some cases, an inflammatory response can be measured by detecting increased levels of one or more inflammatory cytokines using methods known in the art, e.g., ELISA. The term "inflammatory cytokine" refers to cytokines that are elevated in an inflammatory response. Examples of inflammatory cytokines include interleukin-6 (IL-6), CXCL1 (chemokine (C-X-C motif) ligand 1; also known as GROα, interferon-γ (IFNγ), tumor necrosis factor α (TNFα), interferon γ-induced protein 10 (IP-10), or granulocyte-colony stimulating factor (G-CSF). The term inflammatory cytokines includes also other cytokines associated with inflammatory responses known in the art, e.g., interleukin-1 (IL-1), interleukin-8 (IL-8), interleukin-12 (IL-12), interleukin-13 (Il-13), interferon α (IFN-α), etc. 10. Modified Nucleotide Sequences Encoding IL-22 Polypeptides or IL-22- containing Fusion Proteins In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of this disclosure comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1-methylpseudouracil, 5-methoxyuracil, or the like. In some embodiments, the mRNA is a uracil-modified sequence comprising an ORF encoding an IL- 22 polypeptide or an IL-22-containing fusion protein, wherein the mRNA comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1-methylpseudouracil, or 5-methoxyuracil. In certain aspects of this disclosure, when the modified uracil base is connected to a ribose sugar, as it is in polynucleotides, the resulting modified nucleoside or nucleotide is referred to as modified uridine. In some embodiments, uracil in the polynucleotide is at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least 90%, at least 95%, at least 99%, or about 100% modified uracil. In one embodiment, uracil in the polynucleotide is at least 95% modified uracil. In another embodiment, uracil in the polynucleotide is 100% modified uracil. In embodiments where uracil in the polynucleotide is at least 95% modified uracil overall uracil content can be adjusted such that an mRNA provides suitable protein expression levels while inducing little to no immune response. In some embodiments, the uracil content of the ORF is between about 100% and about 150%, between about 100% and about 110%, between about 105% and about 115%, between about 110% and about 120%, between about 115% and about 125%, between about 120% and about 130%, between about 125% and about 135%, between about 130% and about 140%, between about 135% and about 145%, between about 140% and about 150% of the theoretical minimum uracil content in the corresponding wild-type ORF (%UTM). In other embodiments, the uracil content of the ORF is between about 121% and about 136% or between 123% and 134% of the %UTM. In some embodiments, the uracil content of the ORF encoding an IL-22 polypeptide or an IL- 22-containing fusion protein is about 115%, about 120%, about 125%, about 130%, about 135%, about 140%, about 145%, or about 150% of the %UTM. In this context, the term "uracil" can refer to modified uracil and/or naturally occurring uracil. In some embodiments, the uracil content in the ORF of the mRNA encoding an IL-22 polypeptide of this disclosure is less than about 30%, about 25%, about 20%, about 15%, or about 10% of the total nucleobase content in the ORF. In some embodiments, the uracil content in the ORF is between about 10% and about 20% of the total nucleobase content in the ORF. In other embodiments, the uracil content in the ORF is between about 10% and about 25% of the total nucleobase content in the ORF. In one embodiment, the uracil content in the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein is less than about 20% of the total nucleobase content in the open reading frame. In this context, the term "uracil" can refer to modified uracil and/or naturally occurring uracil. In further embodiments, the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein having modified uracil and adjusted uracil content has increased Cytosine (C), Guanine (G), or Guanine/Cytosine (G/C) content (absolute or relative). In some embodiments, the overall increase in C, G, or G/C content (absolute or relative) of the ORF is at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 10%, at least about 15%, at least about 20%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 100% relative to the G/C content (absolute or relative) of the wild-type ORF. In some embodiments, the G, the C, or the G/C content in the ORF is less than about 100%, less than about 90%, less than about 85%, or less than about 80% of the theoretical maximum G, C, or G/C content of the corresponding wild type nucleotide sequence encoding the IL-22 polypeptide (%GTMX; %CTMX, or %G/CTMX). In some embodiments, the increases in G and/or C content (absolute or relative) described herein can be conducted by replacing synonymous codons with low G, C, or G/C content with synonymous codons having higher G, C, or G/C content. In other embodiments, the increase in G and/or C content (absolute or relative) is conducted by replacing a codon ending with U with a synonymous codon ending with G or C. In further embodiments, the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein of this disclosure comprises modified uracil and has an adjusted uracil content containing less uracil pairs (UU) and/or uracil triplets (UUU) and/or uracil quadruplets (UUUU) than the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide. In some embodiments, the ORF of the mRNA encoding an IL-22 polypeptide of this disclosure contains no uracil pairs and/or uracil triplets and/or uracil quadruplets. In some embodiments, uracil pairs and/or uracil triplets and/or uracil quadruplets are reduced below a certain threshold, e.g., no more than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 occurrences in the ORF of the mRNA encoding the IL-22 polypeptide. In a particular embodiment, the ORF of the mRNA encoding the IL- 22 polypeptide of this disclosure contains less than 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 non-phenylalanine uracil pairs and/or triplets. In another embodiment, the ORF of the mRNA encoding the IL-22 polypeptide contains no non- phenylalanine uracil pairs and/or triplets. In further embodiments, the ORF of the mRNA encoding an IL-22 polypeptide or an IL-22-containing fusion protein of this disclosure comprises modified uracil and has an adjusted uracil content containing less uracil-rich clusters than the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide. In some embodiments, the ORF of the mRNA encoding the IL-22 polypeptide of this disclosure contains uracil-rich clusters that are shorter in length than corresponding uracil-rich clusters in the corresponding wild-type nucleotide sequence encoding the IL-22 polypeptide. In further embodiments, alternative lower frequency codons are employed. At least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, or 100% of the codons in the IL-22 polypeptide–encoding ORF of the modified uracil-comprising mRNA are substituted with alternative codons, each alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set. The ORF also has adjusted uracil content, as described above. In some embodiments, at least one codon in the ORF of the mRNA encoding the IL-22 polypeptide is substituted with an alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set. In some embodiments, the adjusted uracil content, IL-22 polypeptide-encoding ORF of the modified uracil-comprising mRNA exhibits expression levels of IL-22 when administered to a mammalian cell that are higher than expression levels of IL-22 from the corresponding wild-type mRNA. In some embodiments, the mammalian cell is a mouse cell, a rat cell, or a rabbit cell. In other embodiments, the mammalian cell is a monkey cell or a human cell. In some embodiments, the human cell is a HeLa cell, a BJ fibroblast cell, or a peripheral blood mononuclear cell (PBMC). In some embodiments, IL-22 is expressed at a level higher than expression levels of IL-22 from the corresponding wild-type mRNA when the mRNA is administered to a mammalian cell in vivo. In some embodiments, the mRNA is administered to mice, rabbits, rats, monkeys, or humans. In one embodiment, mice are null mice. In some embodiments, the mRNA is administered to mice in an amount of about 0.01 mg/kg, about 0.05 mg/kg, about 0.1 mg/kg, or 0.2 mg/kg or about 0.5 mg/kg. In some embodiments, the mRNA is administered intravenously or intramuscularly. In other embodiments, the IL-22 polypeptide is expressed when the mRNA is administered to a mammalian cell in vitro. In some embodiments, the expression is increased by at least about 2-fold, at least about 5-fold, at least about 10-fold, at least about 50-fold, at least about 500- fold, at least about 1500-fold, or at least about 3000-fold. In other embodiments, the expression is increased by at least about 10%, about 20%, about 30%, about 40%, about 50%, 60%, about 70%, about 80%, about 90%, or about 100%. In some embodiments, adjusted uracil content, IL-22 polypeptide-encoding ORF of the modified uracil-comprising mRNA exhibits increased stability. In some embodiments, the mRNA exhibits increased stability in a cell relative to the stability of a corresponding wild-type mRNA under the same conditions. In some embodiments, the mRNA exhibits increased stability including resistance to nucleases, thermal stability, and/or increased stabilization of secondary structure. In some embodiments, increased stability exhibited by the mRNA is measured by determining the half-life of the mRNA (e.g., in a plasma, serum, cell, or tissue sample) and/or determining the area under the curve (AUC) of the protein expression by the mRNA over time (e.g., in vitro or in vivo). An mRNA is identified as having increased stability if the half-life and/or the AUC is greater than the half-life and/or the AUC of a corresponding wild-type mRNA under the same conditions. In some embodiments, the mRNA of the present invention induces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by a corresponding wild-type mRNA under the same conditions. In other embodiments, the mRNA of the present disclosure induces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by an mRNA that encodes an IL-22 polypeptide but does not comprise modified uracil under the same conditions, or relative to the immune response induced by an mRNA that encodes for an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content under the same conditions. The innate immune response can be manifested by increased expression of pro- inflammatory cytokines, activation of intracellular PRRs (RIG-I, MDA5, etc), cell death, and/or termination or reduction in protein translation. In some embodiments, a reduction in the innate immune response can be measured by expression or activity level of Type 1 interferons (e.g., IFN-α, IFN-β, IFN-κ, IFN-δ, IFN-ε, IFN-τ, IFN-ω, and IFN-ζ) or the expression of interferon-regulated genes such as the toll-like receptors (e.g., TLR7 and TLR8), and/or by decreased cell death following one or more administrations of the mRNA of this disclosure into a cell. In some embodiments, the expression of Type-1 interferons by a mammalian cell in response to the mRNA of the present disclosure is reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99%, 99.9%, or greater than 99.9% relative to a corresponding wild-type mRNA, to an mRNA that encodes an IL-22 polypeptide but does not comprise modified uracil, or to an mRNA that encodes an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content. In some embodiments, the interferon is IFN-β. In some embodiments, cell death frequency caused by administration of mRNA of the present disclosure to a mammalian cell is 10%, 25%, 50%, 75%, 85%, 90%, 95%, or over 95% less than the cell death frequency observed with a corresponding wild-type mRNA, an mRNA that encodes for an IL-22 polypeptide but does not comprise modified uracil, or an mRNA that encodes for an IL-22 polypeptide and that comprises modified uracil but that does not have adjusted uracil content. In some embodiments, the mammalian cell is a BJ fibroblast cell. In other embodiments, the mammalian cell is a splenocyte. In some embodiments, the mammalian cell is that of a mouse or a rat. In other embodiments, the mammalian cell is that of a human. In one embodiment, the mRNA of the present disclosure does not substantially induce an innate immune response of a mammalian cell into which the mRNA is introduced. 11. Methods for Modifying Polynucleotides The disclosure includes modified polynucleotides comprising a polynucleotide described herein (e.g., a polynucleotide, e.g. mRNA, comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22-containing fusion protein). The modified polynucleotides can be chemically modified and/or structurally modified. When the polynucleotides of the present invention are chemically and/or structurally modified the polynucleotides can be referred to as "modified polynucleotides." The present disclosure provides for modified nucleosides and nucleotides of a polynucleotide (e.g., RNA polynucleotides, such as mRNA polynucleotides) encoding an IL- 22 polypeptide. A "nucleoside" refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as "nucleobase"). A “nucleotide" refers to a nucleoside including a phosphate group. Modified nucleotides can be synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Polynucleotides can comprise a region or regions of linked nucleosides. Such regions can have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the polynucleotides would comprise regions of nucleotides. The modified polynucleotides disclosed herein can comprise various distinct modifications. In some embodiments, the modified polynucleotides contain one, two, or more (optionally different) nucleoside or nucleotide modifications. In some embodiments, a modified polynucleotide, introduced to a cell can exhibit one or more desirable properties, e.g., improved protein expression, reduced immunogenicity, or reduced degradation in the cell, as compared to an unmodified polynucleotide. In some embodiments, a polynucleotide of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide or an IL-22- containing fusion protein) is structurally modified. As used herein, a "structural" modification is one in which two or more linked nucleosides are inserted, deleted, duplicated, inverted or randomized in a polynucleotide without significant chemical modification to the nucleotides themselves. Because chemical bonds will necessarily be broken and reformed to effect a structural modification, structural modifications are of a chemical nature and hence are chemical modifications. However, structural modifications will result in a different sequence of nucleotides. For example, the polynucleotide "ATCG" can be chemically modified to "AT- 5meC-G". The same polynucleotide can be structurally modified from "ATCG" to "ATCCCG". Here, the dinucleotide "CC" has been inserted, resulting in a structural modification to the polynucleotide. Therapeutic compositions of the present disclosure comprise, in some embodiments, at least one nucleic acid (e.g., RNA) having an open reading frame encoding IL-22 (e.g., SEQ ID NO: 12), wherein the nucleic acid comprises nucleotides and/or nucleosides that can be standard (unmodified) or modified as is known in the art. In some embodiments, nucleotides and nucleosides of the present disclosure comprise modified nucleotides or nucleosides. Such modified nucleotides and nucleosides can be naturally-occurring modified nucleotides and nucleosides or non-naturally occurring modified nucleotides and nucleosides. Such modifications can include those at the sugar, backbone, or nucleobase portion of the nucleotide and/or nucleoside as are recognized in the art. In some embodiments, a naturally-occurring modified nucleotide or nucleotide of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such naturally occurring modified nucleotides and nucleotides can be found, inter alia, in the widely recognized MODOMICS database. In some embodiments, a non-naturally occurring modified nucleotide or nucleoside of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such non-naturally occurring modified nucleotides and nucleosides can be found, inter alia, in published US application Nos. PCT/US2012/058519; PCT/US2013/075177; PCT/US2014/058897; PCT/US2014/058891; PCT/US2014/070413; PCT/US2015/36773; PCT/US2015/36759; PCT/US2015/36771; or PCT/IB2017/051367 all of which are incorporated by reference herein. In some embodiments, at least one RNA (e.g., mRNA) of the present disclosure is not chemically modified and comprises the standard ribonucleotides consisting of adenosine, guanosine, cytosine and uridine. In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard nucleoside residues such as those present in transcribed RNA (e.g. A, G, C, or U). In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard deoxyribonucleosides such as those present in DNA (e.g. dA, dG, dC, or dT). Hence, nucleic acids of the disclosure (e.g., DNA nucleic acids and RNA nucleic acids, such as mRNA nucleic acids) can comprise standard nucleotides and nucleosides, naturally-occurring nucleotides and nucleosides, non-naturally-occurring nucleotides and nucleosides, or any combination thereof. Nucleic acids of the disclosure (e.g., DNA nucleic acids and RNA nucleic acids, such as mRNA nucleic acids), in some embodiments, comprise various (more than one) different types of standard and/or modified nucleotides and nucleosides. In some embodiments, a particular region of a nucleic acid contains one, two or more (optionally different) types of standard and/or modified nucleotides and nucleosides. In some embodiments, a modified RNA nucleic acid (e.g., a modified mRNA nucleic acid), introduced to a cell or organism, exhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides. In some embodiments, a modified RNA nucleic acid (e.g., a modified mRNA nucleic acid), introduced into a cell or organism, may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides. Nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids), in some embodiments, comprise non-natural modified nucleotides that are introduced during synthesis or post-synthesis of the nucleic acids to achieve desired functions or properties. The modifications may be present on internucleotide linkages, purine or pyrimidine bases, or sugars. The modification may be introduced with chemical synthesis or with a polymerase enzyme at the terminal of a chain or anywhere else in the chain. Any of the regions of a nucleic acid may be chemically modified. The present disclosure provides for modified nucleosides and nucleotides of a nucleic acid (e.g., RNA nucleic acids, such as mRNA nucleic acids). A “nucleoside” refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g., a purine or pyrimidine) or a derivative thereof (also referred to herein as “nucleobase”). A “nucleotide” refers to a nucleoside, including a phosphate group. Modified nucleotides may by synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Nucleic acids can comprise a region or regions of linked nucleosides. Such regions may have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the nucleic acids would comprise regions of nucleotides. Modified nucleotide base pairing encompasses not only the standard adenosine- thymine, adenosine-uracil, or guanosine-cytosine base pairs, but also base pairs formed between nucleotides and/or modified nucleotides comprising non-standard or modified bases, wherein the arrangement of hydrogen bond donors and hydrogen bond acceptors permits hydrogen bonding between a non-standard base and a standard base or between two complementary non-standard base structures, such as, for example, in those nucleic acids having at least one chemical modification. One example of such non-standard base pairing is the base pairing between the modified nucleotide inosine and adenine, cytosine or uracil. Any combination of base/sugar or linker may be incorporated into nucleic acids of the present disclosure. In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise N1-methyl-pseudouridine (m1ψ), 1-ethyl- pseudouridine (e1ψ), 5-methoxy-uridine (mo5U), 5-methyl-cytidine (m5C), and/or pseudouridine (ψ). In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise 5-methoxymethyl uridine, 5-methylthio uridine, 1-methoxymethyl pseudouridine, 5-methyl cytidine, and/or 5-methoxy cytidine. In some embodiments, the polyribonucleotide includes a combination of at least two (e.g., 2, 3, 4 or more) of any of the aforementioned modified nucleobases, including but not limited to chemical modifications. In some embodiments, a RNA nucleic acid of the disclosure comprises N1-methyl- pseudouridine (m1ψ) substitutions at one or more or all uridine positions of the nucleic acid. In some embodiments, a RNA nucleic acid of the disclosure comprises N1-methyl- pseudouridine (m1ψ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid. In some embodiments, a RNA nucleic acid of the disclosure comprises pseudouridine (ψ) substitutions at one or more or all uridine positions of the nucleic acid. In some embodiments, a RNA nucleic acid of the disclosure comprises pseudouridine (ψ) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid. In some embodiments, a RNA nucleic acid of the disclosure comprises uridine at one or more or all uridine positions of the nucleic acid. In some embodiments, nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) are uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification. For example, a nucleic acid can be uniformly modified with N1-methyl-pseudouridine, meaning that all uridine residues in the mRNA sequence are replaced with N1-methyl-pseudouridine. Similarly, a nucleic acid can be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above. The nucleic acids of the present disclosure may be partially or fully modified along the entire length of the molecule. For example, one or more or all or a given type of nucleotide (e.g., purine or pyrimidine, or any one or more or all of A, G, U, C) may be uniformly modified in a nucleic acid of the disclosure, or in a predetermined sequence region thereof (e.g., in the mRNA including or excluding the polyA tail). In some embodiments, all nucleotides X in a nucleic acid of the present disclosure (or in a sequence region thereof) are modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C or A+G+C. The nucleic acid may contain from about 1% to about 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, i.e., any one or more of A, G, U or C) or any intervening percentage (e.g., from 1% to 20%, from 1% to 25%, from 1% to 50%, from 1% to 60%, from 1% to 70%, from 1% to 80%, from 1% to 90%, from 1% to 95%, from 10% to 20%, from 10% to 25%, from 10% to 50%, from 10% to 60%, from 10% to 70%, from 10% to 80%, from 10% to 90%, from 10% to 95%, from 10% to 100%, from 20% to 25%, from 20% to 50%, from 20% to 60%, from 20% to 70%, from 20% to 80%, from 20% to 90%, from 20% to 95%, from 20% to 100%, from 50% to 60%, from 50% to 70%, from 50% to 80%, from 50% to 90%, from 50% to 95%, from 50% to 100%, from 70% to 80%, from 70% to 90%, from 70% to 95%, from 70% to 100%, from 80% to 90%, from 80% to 95%, from 80% to 100%, from 90% to 95%, from 90% to 100%, and from 95% to 100%). It will be understood that any remaining percentage is accounted for by the presence of unmodified A, G, U, or C. The nucleic acids may contain at a minimum 1% and at maximum 100% modified nucleotides, or any intervening percentage, such as at least 5% modified nucleotides, at least 10% modified nucleotides, at least 25% modified nucleotides, at least 50% modified nucleotides, at least 80% modified nucleotides, or at least 90% modified nucleotides. For example, the nucleic acids may contain a modified pyrimidine such as a modified uracil or cytosine. In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the uracil in the nucleic acid is replaced with a modified uracil (e.g., a 5-substituted uracil). The modified uracil can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the cytosine in the nucleic acid is replaced with a modified cytosine (e.g., a 5-substituted cytosine). The modified cytosine can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). 12. Untranslated Regions (UTRs) Untranslated regions (UTRs) are nucleic acid sections of a polynucleotide before a start codon (5′ UTR) and after a stop codon (3′ UTR) that are not translated. In some embodiments, a polynucleotide (e.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)) of this disclosure comprising an open reading frame (ORF) encoding an IL-22 polypeptide or an IL-22-containing fusion protein further comprises a UTR (e.g., a 5′ UTR or functional fragment thereof, a 3′ UTR or functional fragment thereof, or a combination thereof). A UTR (e.g., 5′ UTR or 3′ UTR) can be homologous or heterologous to the coding region in a polynucleotide. In some embodiments, the UTR is homologous to the ORF encoding the IL-22 polypeptide. In some embodiments, the UTR is heterologous to the ORF encoding the IL-22 polypeptide. In some embodiments, the polynucleotide comprises two or more 5′ UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences. In some embodiments, the polynucleotide comprises two or more 3′ UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences. In some embodiments, the 5′ UTR or functional fragment thereof, 3′ UTR or functional fragment thereof, or any combination thereof is sequence optimized. In some embodiments, the 5′UTR or functional fragment thereof, 3′ UTR or functional fragment thereof, or any combination thereof comprises at least one chemically modified nucleobase, e.g., N1-methylpseudouracil or 5-methoxyuracil. UTRs can have features that provide a regulatory role, e.g., increased or decreased stability, localization and/or translation efficiency. A polynucleotide comprising a UTR can be administered to a cell, tissue, or organism, and one or more regulatory features can be measured using routine methods. In some embodiments, a functional fragment of a 5′ UTR or 3′ UTR comprises one or more regulatory features of a full length 5′ or 3′ UTR, respectively. Natural 5′UTRs bear features that play roles in translation initiation. They harbor signatures like Kozak sequences that are commonly known to be involved in the process by which the ribosome initiates translation of many genes. Kozak sequences have the consensus CCR(A/G)CCAUGG (SEQ ID NO: 214), where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another ‘G’.5′ UTRs also have been known to form secondary structures that are involved in elongation factor binding. By engineering the features typically found in abundantly expressed genes of specific target organs, one can enhance the stability and protein production of a polynucleotide. For example, introduction of 5′ UTR of liver-expressed mRNA, such as albumin, serum amyloid A, Apolipoprotein A/B/E, transferrin, alpha fetoprotein, erythropoietin, or Factor VIII, can enhance expression of polynucleotides in hepatic cell lines or liver. Likewise, use of 5′UTR from other tissue-specific mRNA to improve expression in that tissue is possible for muscle (e.g., MyoD, Myosin, Myoglobin, Myogenin, Herculin), for endothelial cells (e.g., Tie-1, CD36), for myeloid cells (e.g., C/EBP, AML1, G-CSF, GM-CSF, CD11b, MSR, Fr-1, i- NOS), for leukocytes (e.g., CD45, CD18), for adipose tissue (e.g., CD36, GLUT4, ACRP30, adiponectin) and for lung epithelial cells (e.g., SP-A/B/C/D). In some embodiments, UTRs are selected from a family of transcripts whose proteins share a common function, structure, feature or property. For example, an encoded polypeptide can belong to a family of proteins (i.e., that share at least one function, structure, feature, localization, origin, or expression pattern), which are expressed in a particular cell, tissue or at some time during development. The UTRs from any of the genes or mRNA can be swapped for any other UTR of the same or different family of proteins to create a new polynucleotide. In some embodiments, the 5′ UTR and the 3′ UTR can be heterologous. In some embodiments, the 5′ UTR can be derived from a different species than the 3′ UTR. In some embodiments, the 3′ UTR can be derived from a different species than the 5′ UTR. Co-owned International Patent Application No. PCT/US2014/021522 (Publ. No. WO/2014/164253, incorporated herein by reference in its entirety) provides a listing of exemplary UTRs that can be utilized in the polynucleotide of the present invention as flanking regions to an ORF. Additional exemplary UTRs of the application include, but are not limited to, one or more 5′UTR and/or 3′UTR derived from the nucleic acid sequence of: a globin, such as an α- or β-globin (e.g., a Xenopus, mouse, rabbit, or human globin); a strong Kozak translational initiation signal; a CYBA (e.g., human cytochrome b-245 α polypeptide); an albumin (e.g., human albumin7); a HSD17B4 (hydroxysteroid (17-β) dehydrogenase); a virus (e.g., a tobacco etch virus (TEV), a Venezuelan equine encephalitis virus (VEEV), a Dengue virus, a cytomegalovirus (CMV) (e.g., CMV immediate early 1 (IE1)), a hepatitis virus (e.g., hepatitis B virus), a sindbis virus, or a PAV barley yellow dwarf virus); a heat shock protein (e.g., hsp70); a translation initiation factor (e.g., elF4G); a glucose transporter (e.g., hGLUT1 (human glucose transporter 1)); an actin (e.g., human α or β actin); a GAPDH; a tubulin; a histone; a citric acid cycle enzyme; a topoisomerase (e.g., a 5′UTR of a TOP gene lacking the 5′ TOP motif (the oligopyrimidine tract)); a ribosomal protein Large 32 (L32); a ribosomal protein (e.g., human or mouse ribosomal protein, such as, for example, rps9); an ATP synthase (e.g., ATP5A1 or the β subunit of mitochondrial H+-ATP synthase); a growth hormone e (e.g., bovine (bGH) or human (hGH)); an elongation factor (e.g., elongation factor 1 α1 (EEF1A1)); a manganese superoxide dismutase (MnSOD); a myocyte enhancer factor 2A (MEF2A); a β-F1-ATPase, a creatine kinase, a myoglobin, a granulocyte-colony stimulating factor (G-CSF); a collagen (e.g., collagen type I, alpha 2 (Col1A2), collagen type I, alpha 1 (Col1A1), collagen type VI, alpha 2 (Col6A2), collagen type VI, alpha 1 (Col6A1)); a ribophorin (e.g., ribophorin I (RPNI)); a low density lipoprotein receptor-related protein (e.g., LRP1); a cardiotrophin-like cytokine factor (e.g., Nnt1); calreticulin (Calr); a procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (Plod1); and a nucleobindin (e.g., Nucb1). In some embodiments, the 5′ UTR is selected from the group consisting of a β-globin 5′ UTR; a 5′UTR containing a strong Kozak translational initiation signal; a cytochrome b- 245 α polypeptide (CYBA) 5′ UTR; a hydroxysteroid (17-β) dehydrogenase (HSD17B4) 5′ UTR; a Tobacco etch virus (TEV) 5′ UTR; a Venezuelen equine encephalitis virus (TEEV) 5′ UTR; a 5′ proximal open reading frame of rubella virus (RV) RNA encoding nonstructural proteins; a Dengue virus (DEN) 5′ UTR; a heat shock protein 70 (Hsp70) 5′ UTR; a eIF4G 5′ UTR; a GLUT15′ UTR; functional fragments thereof and any combination thereof. In some embodiments, the 3′ UTR is selected from the group consisting of a β-globin 3′ UTR; a CYBA 3′ UTR; an albumin 3′ UTR; a growth hormone (GH) 3′ UTR; a VEEV 3′ UTR; a hepatitis B virus (HBV) 3′ UTR; α-globin 3′UTR; a DEN 3′ UTR; a PAV barley yellow dwarf virus (BYDV-PAV) 3′ UTR; an elongation factor 1 α1 (EEF1A1) 3′ UTR; a manganese superoxide dismutase (MnSOD) 3′ UTR; a β subunit of mitochondrial H(+)-ATP synthase (β-mRNA) 3′ UTR; a GLUT13′ UTR; a MEF2A 3′ UTR; a β-F1-ATPase 3′ UTR; functional fragments thereof and combinations thereof. Wild-type UTRs derived from any gene or mRNA can be incorporated into the polynucleotides of this disclosure. In some embodiments, a UTR can be altered relative to a wild type or native UTR to produce a variant UTR, e.g., by changing the orientation or location of the UTR relative to the ORF; or by inclusion of additional nucleotides, deletion of nucleotides, swapping or transposition of nucleotides. In some embodiments, variants of 5′ or 3′ UTRs can be utilized, for example, mutants of wild type UTRs, or variants wherein one or more nucleotides are added to or removed from a terminus of the UTR. Additionally, one or more synthetic UTRs can be used in combination with one or more non-synthetic UTRs. See, e.g., Mandal and Rossi, Nat. Protoc.20138(3):568-82, the contents of which are incorporated herein by reference in their entirety. UTRs or portions thereof can be placed in the same orientation as in the transcript from which they were selected or can be altered in orientation or location. Hence, a 5′ and/or 3′ UTR can be inverted, shortened, lengthened, or combined with one or more other 5′ UTRs or 3′ UTRs. In some embodiments, the polynucleotide comprises multiple UTRs, e.g., a double, a triple or a quadruple 5′ UTR or 3′ UTR. For example, a double UTR comprises two copies of the same UTR either in series or substantially in series. For example, a double beta-globin 3′UTR can be used (see US2010/0129877, the contents of which are incorporated herein by reference in its entirety). The polynucleotides of this disclosure can comprise combinations of features. For example, the ORF can be flanked by a 5′UTR that comprises a strong Kozak translational initiation signal and/or a 3′UTR comprising an oligo(dT) sequence for templated addition of a poly-A tail. A 5′UTR can comprise a first polynucleotide fragment and a second polynucleotide fragment from the same and/or different UTRs (see, e.g., US2010/0293625, herein incorporated by reference in its entirety). Other non-UTR sequences can be used as regions or subregions within the polynucleotides of this disclosure. For example, introns or portions of intron sequences can be incorporated into the polynucleotides of this disclosure. Incorporation of intronic sequences can increase protein production as well as polynucleotide expression levels. In some embodiments, the polynucleotide of this disclosure comprises an internal ribosome entry site (IRES) instead of or in addition to a UTR (see, e.g., Yakubov et al., Biochem. Biophys. Res. Commun.2010394(1):189-193, the contents of which are incorporated herein by reference in their entirety). In some embodiments, the polynucleotide comprises an IRES instead of a 5′ UTR sequence. In some embodiments, the polynucleotide comprises an ORF and a viral capsid sequence. In some embodiments, the polynucleotide comprises a synthetic 5′ UTR in combination with a non-synthetic 3′ UTR. In some embodiments, the UTR can also include at least one translation enhancer polynucleotide, translation enhancer element, or translational enhancer elements (collectively, "TEE," which refers to nucleic acid sequences that increase the amount of polypeptide or protein produced from a polynucleotide. As a non-limiting example, the TEE can be located between the transcription promoter and the start codon. In some embodiments, the 5′ UTR comprises a TEE. In one aspect, a TEE is a conserved element in a UTR that can promote translational activity of a nucleic acid such as, but not limited to, cap-dependent or cap-independent translation. a.5′ UTR sequences 5′ UTR sequences are important for ribosome recruitment to the mRNA and have been reported to play a role in translation (Hinnebusch A, et al., (2016) Science, 352:6292: 1413-6). Disclosed herein, inter alia, is a polynucleotide, e.g., mRNA, comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1-4) or an IL-22-containing fusion protein (e.g., SEQ ID NO: 20-38), which polynucleotide has a 5′ UTR that confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself. In an embodiment, a polynucleotide disclosed herein comprises: (a) a 5′-UTR (e.g., as provided in Table 4 or a variant or fragment thereof); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as described herein), and LNP compositions comprising the same. In an embodiment, the polynucleotide comprises a 5′-UTR comprising a sequence provided in Table 4 or a variant or fragment thereof (e.g., a functional variant or fragment thereof). In an embodiment, the polynucleotide having a 5′ UTR sequence provided in Table 4 or a variant or fragment thereof, has an increase in the half-life of the polynucleotide, e.g., about 1.5-20-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more. In an embodiment, the increase in half life is about 1.5-fold or more. In an embodiment, the increase in half life is about 2-fold or more. In an embodiment, the increase in half life is about 3-fold or more. In an embodiment, the increase in half life is about 4-fold or more. In an embodiment, the increase in half life is about 5-fold or more. In an embodiment, the polynucleotide having a 5′ UTR sequence provided in Table 4 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the 5′UTR results in about 1.5-20-fold increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase in level and/or activity is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more. In an embodiment, the increase in level and/or activity is about 1.5-fold or more. In an embodiment, the increase in level and/or activity is about 2-fold or more. In an embodiment, the increase in level and/or activity is about 3-fold or more. In an embodiment, the increase in level and/or activity is about 4-fold or more. In an embodiment, the increase in level and/or activity is about 5-fold or more. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 5′ UTR, has a different 5′ UTR, or does not have a 5′ UTR described in Table 4 or a variant or fragment thereof. In an embodiment, the increase in half-life of the polynucleotide is measured according to an assay that measures the half-life of a polynucleotide. In an embodiment, the increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide is measured according to an assay that measures the level and/or activity of a polypeptide. In an embodiment, the 5′ UTR comprises a sequence provided in Table 4 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 5′ UTR sequence provided in Table 4, or a variant or a fragment thereof. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 50, SEQ ID NO: 51, SEQ ID NO: 52, SEQ ID NO: 53, SEQ ID NO: 54, SEQ ID NO: 55, SEQ ID NO: 56, SEQ ID NO: 57 or SEQ ID NO: 58. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 50. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 51. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 52. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 53. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 54. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 55. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 56. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 57. In an embodiment, the 5′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 58. In an embodiment, the 5′ UTR comprises the sequence of SEQ ID NO:58. In an embodiment, the 5′ UTR consists of the sequence of SEQ ID NO:58. In an embodiment, a 5′ UTR sequence provided in Table 4 has a first nucleotide which is an A. In an embodiment, a 5′ UTR sequence provided in Table 4 has a first nucleotide which is a G. Table 4: 5′ UTR sequences

 In an embodiment, the 5′ UTR comprises a variant of SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a nucleic acid sequence of Formula A: G G A A A U C G C A A A A (N2)X (N3)X C U (N4)X (N5)X C G C G U U A G A U U U C U U U U A G U U U U C U N6 N7 C A A C U A G C A A G C U U U U U G U U C U C G C C (N8 C C)x (SEQ ID NO: 59), wherein: (N2)x is a uracil and x is an integer from 0 to 5, e.g., wherein x =3 or 4; (N3)x is a guanine and x is an integer from 0 to 1; (N4)x is a cytosine and x is an integer from 0 to 1; (N5)x is a uracil and x is an integer from 0 to 5, e.g., wherein x =2 or 3; N6 is a uracil or cytosine; N7 is a uracil or guanine; N8 is adenine or guanine and x is an integer from 0 to 1. In an embodiment (N2)x is a uracil and x is 0. In an embodiment (N2)x is a uracil and x is 1. In an embodiment (N2)x is a uracil and x is 2. In an embodiment (N2)x is a uracil and x is 3. In an embodiment, (N2)x is a uracil and x is 4. In an embodiment (N2)x is a uracil and x is 5. In an embodiment, (N3)x is a guanine and x is 0. In an embodiment, (N3)x is a guanine and x is 1. In an embodiment, (N4)x is a cytosine and x is 0. In an embodiment, (N4)x is a cytosine and x is 1. In an embodiment (N5)x is a uracil and x is 0. In an embodiment (N5)x is a uracil and x is 1. In an embodiment (N5)x is a uracil and x is 2. In an embodiment (N5)x is a uracil and x is 3. In an embodiment, (N5)x is a uracil and x is 4. In an embodiment (N5)x is a uracil and x is 5. In an embodiment, N6 is a uracil. In an embodiment, N6 is a cytosine. In an embodiment, N7 is a uracil. In an embodiment, N7 is a guanine. In an embodiment, N8 is an adenine and x is 0. In an embodiment, N8 is an adenine and x is 1. In an embodiment, N8 is a guanine and x is 0. In an embodiment, N8 is a guanine and x is 1. In an embodiment, the 5′ UTR comprises a variant of SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 50%, 60%, 70%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 50% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 60% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 70% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 80% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 90% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 95% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 96% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 97% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 98% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 99% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, or 80%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 10%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 20%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 30%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 40%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 50%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 60%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 70%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 80%. In an embodiment, the variant of SEQ ID NO: 50 comprises at least 2, 3, 4, 5, 6 or 7 consecutive uridines (e.g., a polyuridine tract). In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises at least 1-7, 2-7, 3-7, 4-7, 5-7, 6-7, 1-6, 1-5, 1-4, 1-3, 1- 2, 2-6, or 3-5 consecutive uridines. In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises 4 consecutive uridines. In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises 5 consecutive uridines. In an embodiment, the variant of SEQ ID NO: 50 comprises 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 3 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 4 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 5 polyuridine tracts. In an embodiment, one or more of the polyuridine tracts are adjacent to a different polyuridine tract. In an embodiment, each of, e.g., all, the polyuridine tracts are adjacent to each other, e.g., all of the polyuridine tracts are contiguous. In an embodiment, one or more of the polyuridine tracts are separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides. In an embodiment, each of, e.g., all of, the polyuridine tracts are separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides. In an embodiment, a first polyuridine tract and a second polyuridine tract are adjacent to each other. In an embodiment, a subsequent, e.g., third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth, polyuridine tract is separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides from the first polyuridine tract, the second polyuridine tract, or any one of the subsequent polyuridine tracts. In an embodiment, a first polyuridine tract is separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides from a subsequent polyuridine tract, e.g., a second, third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth polyuridine tract. In an embodiment, one or more of the subsequent polyuridine tracts are adjacent to a different polyuridine tract. In an embodiment, the 5′ UTR comprises a Kozak sequence, e.g., a GCCRCC nucleotide sequence (SEQ ID NO: 79) wherein R is an adenine or guanine. In an embodiment, the Kozak sequence is disposed at the 3′ end of the 5′UTR sequence. In an aspect, the polynucleotide (e.g., mRNA) comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1) and comprising a 5′ UTR sequence disclosed herein is formulated as an LNP. In an embodiment, the LNP composition comprises: (i) an ionizable lipid, e.g., an amino lipid; (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-lipid. In another aspect, the LNP compositions of the disclosure are used in a method of treating metabolic disease in a subject. In an aspect, an LNP composition comprising a polynucleotide disclosed herein encoding an IL-22 polypeptide, e.g., as described herein, can be administered with an additional agent, e.g., as described herein. b. 3′ UTR sequences 3′UTR sequences have been shown to influence translation, half-life, and subcellular localization of mRNAs (Mayr C., Cold Spring Harb Persp Biol 2019 Oct 1;11(10):a034728). Disclosed herein, inter alia, is a polynucleotide, e.g., mRNA, comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1), which polynucleotide has a 3′ UTR that confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself. In an embodiment, a polynucleotide disclosed herein comprises: (a) a 5′-UTR (e.g., as described herein); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as provided in Table 5 or a variant or fragment thereof), and LNP compositions comprising the same. In an embodiment, the polynucleotide comprises a 3′-UTR comprising a sequence provided in Table 5 or a variant or fragment thereof. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereof, results in a polynucleotide with a mean half-life score of greater than 10. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of Table 5 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in Table 5 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in Table 5, or a fragment thereof. In an embodiment, the 3′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 100, SEQ ID NO: 101, SEQ ID NO: 102, SEQ ID NO: 103, SEQ ID NO: 104, SEQ ID NO: 105, SEQ ID NO: 106, SEQ ID NO: 107, SEQ ID NO: 108, SEQ ID NO: 109, SEQ ID NO: 110, SEQ ID NO: 111, SEQ ID NO: 112, SEQ ID NO: 113, SEQ ID NO: 114, SEQ ID NO:115, SEQ ID NO:116, SEQ ID NO:117, SEQ ID NO:118, SEQ ID NO:119, SEQ ID NO:120, SEQ ID NO:121, SEQ ID NO:122, SEQ ID NO:123, SEQ ID NO:124, SEQ ID NO:125, SEQ ID NO:126, SEQ ID NO:127, SEQ ID NO:128, SEQ ID NO:129, SEQ ID NO:130, SEQ ID NO:131, SEQ ID NO:132, SEQ ID NO:133, SEQ ID NO:134, SEQ ID NO:135, SEQ ID NO:136, SEQ ID NO:139, SEQ ID NO:140, SEQ ID NO:141, SEQ ID NO:142, SEQ ID NO:143, SEQ ID NO:144, SEQ ID NO:145, SEQ ID NO:146, or SEQ ID NO:147. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 100, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 100. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 101, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 101. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 102, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 102. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 103, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 103. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 104, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 104. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 105, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 105. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 106, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 106. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 107, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 107. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 108, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 108. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 109, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 109. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 110, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 110. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 111, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 111. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 112, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 112. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 113, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 113. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 114, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 114. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 115, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 115. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 116, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 116. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 117, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 117. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 118, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 118. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 119, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 119. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 120, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 120. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 121, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 121. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 122, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 122. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 123, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 123. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 124, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 124. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 125, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 125. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 126, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 126. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 127, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 127. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 128, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 128. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 129, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 129. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 130, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 130. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 131, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 131. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 132, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 132. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 133, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 133. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 134, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 134. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 135, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 135. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 136, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 136. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 139, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 139. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 140, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 140. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 141, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 141. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 142, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 142. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 143, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 143. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 144, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 144. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 145, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 145. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 146, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 146. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 147, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 147. Table 5: 3′ UTR sequences
In an embodiment, the 5′ UTR comprises a variant of SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a nucleic acid sequence of Formula A: G G A A A U C G C A A A A (N2)X (N3)X C U (N4)X (N5)X C G C G U U A G A U U U C U U U U A G U U U U C U N6 N7 C A A C U A G C A A G C U U U U U G U U C U C G C C (N8 C C)x (SEQ ID NO: 59), wherein: (N2)x is a uracil and x is an integer from 0 to 5, e.g., wherein x =3 or 4; (N3)x is a guanine and x is an integer from 0 to 1; (N4)x is a cytosine and x is an integer from 0 to 1; (N5)x is a uracil and x is an integer from 0 to 5, e.g., wherein x =2 or 3; N6 is a uracil or cytosine; N7 is a uracil or guanine; N8 is adenine or guanine and x is an integer from 0 to 1. In an embodiment (N2)x is a uracil and x is 0. In an embodiment (N2)x is a uracil and x is 1. In an embodiment (N2)x is a uracil and x is 2. In an embodiment (N2)x is a uracil and x is 3. In an embodiment, (N2)x is a uracil and x is 4. In an embodiment (N2)x is a uracil and x is 5. In an embodiment, (N3)x is a guanine and x is 0. In an embodiment, (N3)x is a guanine and x is 1. In an embodiment, (N4)x is a cytosine and x is 0. In an embodiment, (N4)x is a cytosine and x is 1. In an embodiment (N5)x is a uracil and x is 0. In an embodiment (N5)x is a uracil and x is 1. In an embodiment (N5)x is a uracil and x is 2. In an embodiment (N5)x is a uracil and x is 3. In an embodiment, (N5)x is a uracil and x is 4. In an embodiment (N5)x is a uracil and x is 5. In an embodiment, N6 is a uracil. In an embodiment, N6 is a cytosine. In an embodiment, N7 is a uracil. In an embodiment, N7 is a guanine. In an embodiment, N8 is an adenine and x is 0. In an embodiment, N8 is an adenine and x is 1. In an embodiment, N8 is a guanine and x is 0. In an embodiment, N8 is a guanine and x is 1. In an embodiment, the 5′ UTR comprises a variant of SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 50%, 60%, 70%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 50% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 60% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 70% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 80% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 90% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 95% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 96% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 97% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 98% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 99% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, or 80%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 10%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 20%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 30%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 40%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 50%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 60%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 70%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 80%. In an embodiment, the variant of SEQ ID NO: 50 comprises at least 2, 3, 4, 5, 6 or 7 consecutive uridines (e.g., a polyuridine tract). In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises at least 1-7, 2-7, 3-7, 4-7, 5-7, 6-7, 1-6, 1-5, 1-4, 1-3, 1- 2, 2-6, or 3-5 consecutive uridines. In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises 4 consecutive uridines. In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises 5 consecutive uridines. In an embodiment, the variant of SEQ ID NO: 50 comprises 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 3 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 4 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 5 polyuridine tracts. In an embodiment, one or more of the polyuridine tracts are adjacent to a different polyuridine tract. In an embodiment, each of, e.g., all, the polyuridine tracts are adjacent to each other, e.g., all of the polyuridine tracts are contiguous. In an embodiment, one or more of the polyuridine tracts are separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides. In an embodiment, each of, e.g., all of, the polyuridine tracts are separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides. In an embodiment, a first polyuridine tract and a second polyuridine tract are adjacent to each other. In an embodiment, a subsequent, e.g., third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth, polyuridine tract is separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides from the first polyuridine tract, the second polyuridine tract, or any one of the subsequent polyuridine tracts. In an embodiment, a first polyuridine tract is separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18.19, 20, 30, 40, 50 or 60 nucleotides from a subsequent polyuridine tract, e.g., a second, third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth polyuridine tract. In an embodiment, one or more of the subsequent polyuridine tracts are adjacent to a different polyuridine tract. In an embodiment, the 5′ UTR comprises a Kozak sequence, e.g., a GCCRCC nucleotide sequence (SEQ ID NO: 79) wherein R is an adenine or guanine. In an embodiment, the Kozak sequence is disposed at the 3′ end of the 5′UTR sequence. In an aspect, the polynucleotide (e.g., mRNA) comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1) and comprising a 5′ UTR sequence disclosed herein is formulated as an LNP. In an embodiment, the LNP composition comprises: (i) an ionizable lipid, e.g., an amino lipid; (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-lipid. In another aspect, the LNP compositions of the disclosure are used in a method of treating metabolic disease in a subject. In an aspect, an LNP composition comprising a polynucleotide disclosed herein encoding an IL-22 polypeptide, e.g., as described herein, can be administered with an additional agent, e.g., as described herein. b. 3′ UTR sequences 3′UTR sequences have been shown to influence translation, half-life, and subcellular localization of mRNAs (Mayr C., Cold Spring Harb Persp Biol 2019 Oct 1;11(10):a034728). Disclosed herein, inter alia, is a polynucleotide, e.g., mRNA, comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1), which polynucleotide has a 3′ UTR that confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself. In an embodiment, a polynucleotide disclosed herein comprises: (a) a 5′-UTR (e.g., as described herein); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as provided in Table 5 or a variant or fragment thereof), and LNP compositions comprising the same. In an embodiment, the polynucleotide comprises a 3′-UTR comprising a sequence provided in Table 5 or a variant or fragment thereof. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereof, results in a polynucleotide with a mean half-life score of greater than 10. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in Table 5 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of Table 5 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in Table 5 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in Table 5, or a fragment thereof. In an embodiment, the 3′ UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 100, SEQ ID NO: 101, SEQ ID NO: 102, SEQ ID NO: 103, SEQ ID NO: 104, SEQ ID NO: 105, SEQ ID NO: 106, SEQ ID NO: 107, SEQ ID NO: 108, SEQ ID NO: 109, SEQ ID NO: 110, SEQ ID NO: 111, SEQ ID NO: 112, SEQ ID NO: 113, SEQ ID NO: 114, SEQ ID NO:115, SEQ ID NO:116, SEQ ID NO:117, SEQ ID NO:118, SEQ ID NO:119, SEQ ID NO:120, SEQ ID NO:121, SEQ ID NO:122, SEQ ID NO:123, SEQ ID NO:124, SEQ ID NO:125, SEQ ID NO:126, SEQ ID NO:127, SEQ ID NO:128, SEQ ID NO:129, SEQ ID NO:130, SEQ ID NO:131, SEQ ID NO:132, SEQ ID NO:133, SEQ ID NO:134, SEQ ID NO:135, SEQ ID NO:136, SEQ ID NO:139, SEQ ID NO:140, SEQ ID NO:141, SEQ ID NO:142, SEQ ID NO:143, SEQ ID NO:144, SEQ ID NO:145, SEQ ID NO:146, or SEQ ID NO:147. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 100, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 100. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 101, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 101. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 102, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 102. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 103, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 103. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 104, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 104. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 105, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 105. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 106, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 106. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 107, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 107. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 108, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 108. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 109, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 109. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 110, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 110. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 111, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 111. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 112, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 112. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 113, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 113. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 114, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 114. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 115, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 115. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 116, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 116. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 117, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 117. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 118, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 118. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 119, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 119. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 120, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 120. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 121, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 121. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 122, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 122. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 123, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 123. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 124, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 124. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 125, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 125. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 126, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 126. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 127, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 127. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 128, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 128. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 129, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 129. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 130, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 130. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 131, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 131. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 132, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 132. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 133, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 133. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 134, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 134. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 135, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 135. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 136, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 136. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 139, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 139. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 140, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 140. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 141, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 141. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 142, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 142. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 143, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 143. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 144, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 144. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 145, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 145. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 146, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 146. In an embodiment, the 3′ UTR comprises the sequence of SEQ ID NO: 147, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 147. Table 5: 3′ UTR sequences



 In an embodiment, the 3′ UTR comprises a micro RNA (miRNA) binding site, e.g., as described herein, which binds to a miR present in a human cell. In an embodiment, the 3′ UTR comprises a miRNA binding site of SEQ ID NO: 212, SEQ ID NO: 174, SEQ ID NO: 152 or a combination thereof. In an embodiment, the 3′ UTR comprises a plurality of miRNA binding sites, e.g., 2, 3, 4, 5, 6, 7 or 8 miRNA binding sites. In an embodiment, the plurality of miRNA binding sites comprises the same or different miRNA binding sites. miR122 bs = CAAACACCAUUGUCACACUCCA (SEQ ID NO: 212) miR-142-3p bs = UCCAUAAAGUAGGAAACACUACA (SEQ ID NO: 174) miR-126 bs = CGCAUUAUUACUCACGGUACGA (SEQ ID NO: 152) In an aspect, disclosed herein is a polynucleotide encoding a polypeptide, wherein the polynucleotide comprises: (a) a 5′-UTR, e.g., as described herein; (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as described herein). In an aspect, an LNP composition comprising a polynucleotide comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1) and comprising a 3′ UTR disclosed herein comprises: (i) an ionizable lipid, e.g., an amino lipid; (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-lipid. In another aspect, the LNP compositions of the disclosure are used in a method of treating metabolic disease in a subject. In an aspect, an LNP composition comprising a polynucleotide disclosed herein encoding an IL-22 polypeptide, e.g., as described herein, can be administered with an additional agent, e.g., as described herein. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUC UGAGUGGGCGGC (SEQ ID NO:139) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:139. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:139 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:139 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:139 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:139 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:139. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGUCUAAGCUGGAGCCUCCUGAGAGACCUGUGUGAACUAUUGAGAAGAUCG GAACAGCUCCUUACUCUGAGGAAGUUGGUACCCCCGUGGUCUUUGAAUAAAG UCUGAGUGGGCGGC (SEQ ID NO:140) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:140. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:140 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:140 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:140 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:140 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:140. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGCAAACACCAUUGUCACACUCCAGCCUCGGUGGCCUAGCU UCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCCAGCCCCUCC UCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUCCAGUGGUCU UUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:141) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:141. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:141 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:141 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:141 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:141 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:141. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGUCCAUAAAGUAGGAAACACUACAGCUGGAGCCUCGGUG GCCUAGCUUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCC AGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUCU GAGUGGGCGGC (SEQ ID NO:142) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:142. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:142 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:142 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:142 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:142 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:142. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUC CAGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:143) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:143. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:143 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:143 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:143 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:143 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:143. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGCAAACACCAUUGUCACACUCCAGCCUCGGUGGCCUAGC UUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCCAGCCCCUC CUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUCCAGUGGUCU UUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:144) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:144. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:144 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:144 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:144 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:144 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:144. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGUCCAUAAAGUAGGAAACACUACAGCCUCGGUGGCCUAG CUUCUUGCCCCUUGGGCCUCCAUAAAGUAGGAAACACUACAUCCCCCCAGCCC CUCCUCCCCUUCCUGCACCCGUACCCCCCGCAUUAUUACUCACGGUACGAGUG GUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:145) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:145. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:145 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:145 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:145 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:145 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:145. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGUCCAUAAAGUAGGAAACACUACAGCUGGAGCCUCGGUG GCCUAGCUUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCC AGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUCU GAGUGGGCGGC (SEQ ID NO:146) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:146. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:146 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:146 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:146 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:146 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:146. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUC CAGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:147) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:147. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:147 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:147 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:147 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:147 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:147. 13. MicroRNA (miRNA) Binding Sites Polynucleotides of this disclosure can include regulatory elements, for example, microRNA (miRNA) binding sites, transcription factor binding sites, structured mRNA sequences and/or motifs, artificial binding sites engineered to act as pseudo-receptors for endogenous nucleic acid binding molecules, and combinations thereof. In some embodiments, polynucleotides including such regulatory elements are referred to as including “sensor sequences”. In some embodiments, a polynucleotide (e.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)) of this disclosure comprises an open reading frame (ORF) encoding a polypeptide of interest and further comprises one or more miRNA binding site(s). Inclusion or incorporation of miRNA binding site(s) provides for regulation of polynucleotides of this disclosure, and in turn, of the polypeptides encoded therefrom, based on tissue-specific and/or cell-type specific expression of naturally-occurring miRNAs. The present invention also provides pharmaceutical compositions and Formulations that comprise any of the polynucleotides described above. In some embodiments, the composition or Formulation further comprises a delivery agent. In some embodiments, the composition or Formulation can contain a polynucleotide comprising a sequence optimized nucleic acid sequence disclosed herein which encodes a polypeptide. In some embodiments, the composition or Formulation can contain a polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a polynucleotide (e.g., an ORF) having significant sequence identity to a sequence optimized nucleic acid sequence disclosed herein which encodes a polypeptide. In some embodiments, the polynucleotide further comprises a miRNA binding site, e.g., a miRNA binding site that binds A miRNA, e.g., a natural-occurring miRNA, is a 19-25 nucleotide long noncoding RNA that binds to a polynucleotide and down-regulates gene expression either by reducing stability or by inhibiting translation of the polynucleotide. A miRNA sequence comprises a “seed” region, i.e., a sequence in the region of positions 2-8 of the mature miRNA. A miRNA seed can comprise positions 2-8 or 2-7 of the mature miRNA. microRNAs derive enzymatically from regions of RNA transcripts that fold back on themselves to form short hairpin structures often termed a pre-miRNA (precursor-miRNA). A pre-miRNA typically has a two-nucleotide overhang at its 3′ end, and has 3′ hydroxyl and 5′ phosphate groups. This precursor-mRNA is processed in the nucleus and subsequently transported to the cytoplasm where it is further processed by DICER (a RNase III enzyme), to form a mature microRNA of approximately 22 nucleotides. The mature microRNA is then incorporated into a ribonuclear particle to form the RNA-induced silencing complex, RISC, which mediates gene silencing. Art-recognized nomenclature for mature miRNAs typically designates the arm of the pre-miRNA from which the mature miRNA derives; "5p" means the microRNA is from the 5 prime arm of the pre-miRNA hairpin and "3p" means the microRNA is from the 3 prime end of the pre-miRNA hairpin. A miR referred to by number herein can refer to either of the two mature microRNAs originating from opposite arms of the same pre- miRNA (e.g., either the 3p or 5p microRNA). All miRs referred to herein are intended to include both the 3p and 5p arms/sequences, unless particularly specified by the 3p or 5p designation. As used herein, the term “microRNA (miRNA or miR) binding site” refers to a sequence within a polynucleotide, e.g., within a DNA or within an RNA transcript, including in the 5′UTR and/or 3′UTR, that has sufficient complementarity to all or a region of a miRNA to interact with, associate with or bind to the miRNA. In some embodiments, a polynucleotide of this disclosure comprising an ORF encoding a polypeptide of interest and further comprises one or more miRNA binding site(s). In exemplary embodiments, a 5′ UTR and/or 3′ UTR of the polynucleotide (e.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)) comprises the one or more miRNA binding site(s). A miRNA binding site having sufficient complementarity to a miRNA refers to a degree of complementarity sufficient to facilitate miRNA-mediated regulation of a polynucleotide, e.g., miRNA-mediated translational repression or degradation of the polynucleotide. In exemplary aspects of this disclosure, a miRNA binding site having sufficient complementarity to the miRNA refers to a degree of complementarity sufficient to facilitate miRNA-mediated degradation of the polynucleotide, e.g., miRNA-guided RNA- induced silencing complex (RISC)-mediated cleavage of mRNA. The miRNA binding site can have complementarity to, for example, a 19-25 nucleotide long miRNA sequence, to a 19-23 nucleotide long miRNA sequence, or to a 22 nucleotide long miRNA sequence. A miRNA binding site can be complementary to only a portion of a miRNA, e.g., to a portion less than 1, 2, 3, or 4 nucleotides of the full length of a naturally-occurring miRNA sequence, or to a portion less than 1, 2, 3, or 4 nucleotides shorter than a naturally-occurring miRNA sequence. Full or complete complementarity (e.g., full complementarity or complete complementarity over all or a significant portion of the length of a naturally-occurring miRNA) is preferred when the desired regulation is mRNA degradation. In some embodiments, a miRNA binding site includes a sequence that has complementarity (e.g., partial or complete complementarity) with an miRNA seed sequence. In some embodiments, the miRNA binding site includes a sequence that has complete complementarity with a miRNA seed sequence. In some embodiments, a miRNA binding site includes a sequence that has complementarity (e.g., partial or complete complementarity) with an miRNA sequence. In some embodiments, the miRNA binding site includes a sequence that has complete complementarity with a miRNA sequence. In other embodiments, the sequence is not completely complementary. In some embodiments, a miRNA binding site has complete complementarity with a miRNA sequence but for 1, 2, or 3 nucleotide substitutions, terminal additions, and/or truncations. In some embodiments, the miRNA binding site is the same length as the corresponding miRNA. In other embodiments, the miRNA binding site is one, two, three, four, five, six, seven, eight, nine, ten, eleven or twelve nucleotide(s) shorter than the corresponding miRNA at the 5′ terminus, the 3′ terminus, or both. In still other embodiments, the microRNA binding site is two nucleotides shorter than the corresponding microRNA at the 5′ terminus, the 3′ terminus, or both. The miRNA binding sites that are shorter than the corresponding miRNAs are still capable of degrading the mRNA incorporating one or more of the miRNA binding sites or preventing the mRNA from translation. In some embodiments, the miRNA binding site binds the corresponding mature miRNA that is part of an active RISC containing Dicer. In another embodiment, binding of the miRNA binding site to the corresponding miRNA in RISC degrades the mRNA containing the miRNA binding site or prevents the mRNA from being translated. In some embodiments, the miRNA binding site has sufficient complementarity to miRNA so that a RISC complex comprising the miRNA cleaves the polynucleotide comprising the miRNA binding site. In other embodiments, the miRNA binding site has imperfect complementarity so that a RISC complex comprising the miRNA induces instability in the polynucleotide comprising the miRNA binding site. In another embodiment, the miRNA binding site has imperfect complementarity so that a RISC complex comprising the miRNA represses transcription of the polynucleotide comprising the miRNA binding site. In some embodiments, the miRNA binding site has one, two, three, four, five, six, seven, eight, nine, ten, eleven or twelve mismatch(es) from the corresponding miRNA. In some embodiments, the miRNA binding site has at least about ten, at least about eleven, at least about twelve, at least about thirteen, at least about fourteen, at least about fifteen, at least about sixteen, at least about seventeen, at least about eighteen, at least about nineteen, at least about twenty, or at least about twenty-one contiguous nucleotides complementary to at least about ten, at least about eleven, at least about twelve, at least about thirteen, at least about fourteen, at least about fifteen, at least about sixteen, at least about seventeen, at least about eighteen, at least about nineteen, at least about twenty, or at least about twenty-one, respectively, contiguous nucleotides of the corresponding miRNA. By engineering one or more miRNA binding sites into a polynucleotide of this disclosure, the polynucleotide can be targeted for degradation or reduced translation, provided the miRNA in question is available. This can reduce off-target effects upon delivery of the polynucleotide. For example, if a polynucleotide of this disclosure is not intended to be delivered to a tissue or cell but ends up is said tissue or cell, then a miRNA abundant in the tissue or cell can inhibit the expression of the gene of interest if one or multiple binding sites of the miRNA are engineered into the 5′ UTR and/or 3′ UTR of the polynucleotide. Thus, in some embodiments, incorporation of one or more miRNA binding sites into an mRNA of the disclosure may reduce the hazard of off-target effects upon nucleic acid molecule delivery and/or enable tissue-specific regulation of expression of a polypeptide encoded by the mRNA. In yet other embodiments, incorporation of one or more miRNA binding sites into an mRNA of the disclosure can modulate immune responses upon nucleic acid delivery in vivo. In further embodiments, incorporation of one or more miRNA binding sites into an mRNA of the disclosure can modulate accelerated blood clearance (ABC) of lipid-comprising compounds and compositions described herein. Conversely, miRNA binding sites can be removed from polynucleotide sequences in which they naturally occur to increase protein expression in specific tissues. For example, a binding site for a specific miRNA can be removed from a polynucleotide to improve protein expression in tissues or cells containing the miRNA. Regulation of expression in multiple tissues can be accomplished through introduction or removal of one or more miRNA binding sites, e.g., one or more distinct miRNA binding sites. The decision whether to remove or insert a miRNA binding site can be made based on miRNA expression patterns and/or their profilings in tissues and/or cells in development and/or disease. Identification of miRNAs, miRNA binding sites, and their expression patterns and role in biology have been reported (e.g., Bonauer et al., Curr Drug Targets 2010 11:943-949; Anand and Cheresh Curr Opin Hematol 201118:171-176; Contreras and Rao Leukemia 201226:404-413 (2011 Dec 20. doi: 10.1038/leu.2011.356); Bartel Cell 2009 136:215-233; Landgraf et al, Cell, 2007129:1401-1414; Gentner and Naldini, Tissue Antigens.201280:393-403 and all references therein; each of which is incorporated herein by reference in its entirety). Examples of tissues where miRNA are known to regulate mRNA, and thereby protein expression, include, but are not limited to, liver (miR-122), muscle (miR-133, miR-206, miR- 208), endothelial cells (miR-17-92, miR-126), myeloid cells (miR-142-3p, miR-142-5p, miR- 16, miR-21, miR-223, miR-24, miR-27), adipose tissue (let-7, miR-30c), heart (miR-1d, miR- 149), kidney (miR-192, miR-194, miR-204), and lung epithelial cells (let-7, miR-133, miR- 126). Specifically, miRNAs are known to be differentially expressed in immune cells (also called hematopoietic cells), such as antigen presenting cells (APCs) (e.g., dendritic cells and macrophages), macrophages, monocytes, B lymphocytes, T lymphocytes, granulocytes, natural killer cells, etc. Immune cell specific miRNAs are involved in immunogenicity, autoimmunity, the immune-response to infection, inflammation, as well as unwanted immune response after gene therapy and tissue/organ transplantation. Immune cells specific miRNAs also regulate many aspects of development, proliferation, differentiation and apoptosis of hematopoietic cells (immune cells). For example, miR-142 and miR-146 are exclusively expressed in immune cells, particularly abundant in myeloid dendritic cells. It has been demonstrated that the immune response to a polynucleotide can be shut-off by adding miR- 142 binding sites to the 3′-UTR of the polynucleotide, enabling more stable gene transfer in tissues and cells. miR-142 efficiently degrades exogenous polynucleotides in antigen presenting cells and suppresses cytotoxic elimination of transduced cells (e.g., Annoni A et al., blood, 2009, 114, 5152-5161; Brown BD, et al., Nat med.2006, 12(5), 585-591; Brown BD, et al., blood, 2007, 110(13): 4144-4152, each of which is incorporated herein by reference in its entirety). An antigen-mediated immune response can refer to an immune response triggered by foreign antigens, which, when entering an organism, are processed by the antigen presenting cells and displayed on the surface of the antigen presenting cells. T cells can recognize the presented antigen and induce a cytotoxic elimination of cells that express the antigen. Introducing one or more (e.g., one, two, or three) miR-142 binding sites into the 5′ UTR and/or 3′UTR of a polynucleotide of this disclosure can selectively repress gene expression in antigen presenting cells through miR-142 mediated degradation, limiting antigen presentation in antigen presenting cells (e.g., dendritic cells) and thereby preventing antigen-mediated immune response after the delivery of the polynucleotide. The polynucleotide is then stably expressed in target tissues or cells without triggering cytotoxic elimination. In some embodiments, it may be beneficial to target the same cell type with multiple miRs and to incorporate binding sites to each of the 3p and 5p arm if both are abundant (e.g., both miR-142-3p and miR142-5p are abundant in hematopoietic stem cells). Thus, in certain embodiments, polynucleotides of this disclosure contain two or more (e.g., two, three, four or more) miR bindings sites from: (i) the group consisting of miR-142, miR-144, miR-150, miR-155 and miR-223 (which are expressed in many hematopoietic cells); or (ii) the group consisting of miR-142, miR150, miR-16 and miR-223 (which are expressed in B cells); or the group consisting of miR-223, miR-451, miR-26a, miR-16 (which are expressed in progenitor hematopoietic cells). In some embodiments, it may also be beneficial to combine various miRs such that multiple cell types of interest are targeted at the same time (e.g., miR-142 and miR-126 to target many cells of the hematopoietic lineage and endothelial cells). Thus, for example, in certain embodiments, polynucleotides of this disclosure comprise two or more (e.g., two, three, four or more) miRNA bindings sites, wherein: (i) at least one of the miRs targets cells of the hematopoietic lineage (e.g., miR-142, miR-144, miR-150, miR-155 or miR-223) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or (ii) at least one of the miRs targets B cells (e.g., miR-142, miR150, miR-16 or miR-223) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or (iii) at least one of the miRs targets progenitor hematopoietic cells (e.g., miR-223, miR-451, miR-26a or miR-16) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or (iv) at least one of the miRs targets cells of the hematopoietic lineage (e.g., miR-142, miR- 144, miR-150, miR-155 or miR-223), at least one of the miRs targets B cells (e.g., miR-142, miR150, miR-16 or miR-223) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or any other possible combination of the foregoing four classes of miR binding sites (i.e., those targeting the hematopoietic lineage, those targeting B cells, those targeting progenitor hematopoietic cells and/or those targeting plasmacytoid dendritic cells/platelets/endothelial cells). In one embodiment, to modulate immune responses, polynucleotides of the present invention can comprise one or more miRNA binding sequences that bind to one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro-inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells). It has now been discovered that incorporation into an mRNA of one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro- inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells) reduces or inhibits immune cell activation (e.g., B cell activation, as measured by frequency of activated B cells) and/or cytokine production (e.g., production of IL-6, IFN-γ and/or TNFα). Furthermore, it has now been discovered that incorporation into an mRNA of one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro- inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells) can reduce or inhibit an anti-drug antibody (ADA) response against a protein of interest encoded by the mRNA. In another embodiment, to modulate accelerated blood clearance of a polynucleotide delivered in a lipid-comprising compound or composition, polynucleotides of this disclosure can comprise one or more miR binding sequences that bind to one or more miRNAs expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro-inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells). It has now been discovered that incorporation into an mRNA of one or more miR binding sites reduces or inhibits accelerated blood clearance (ABC) of the lipid-comprising compound or composition for use in delivering the mRNA. Furthermore, it has now been discovered that incorporation of one or more miR binding sites into an mRNA reduces serum levels of anti-PEG anti-IgM (e.g, reduces or inhibits the acute production of IgMs that recognize polyethylene glycol (PEG) by B cells) and/or reduces or inhibits proliferation and/or activation of plasmacytoid dendritic cells following administration of a lipid-comprising compound or composition comprising the mRNA. In some embodiments, miR sequences may correspond to any known microRNA expressed in immune cells, including but not limited to those taught in US Publication US2005/0261218 and US Publication US2005/0059005, the contents of which are incorporated herein by reference in their entirety. Non-limiting examples of miRs expressed in immune cells include those expressed in spleen cells, myeloid cells, dendritic cells, plasmacytoid dendritic cells, B cells, T cells and/or macrophages. For example, miR-142-3p, miR-142-5p, miR-16, miR-21, miR-223, miR-24 and miR-27 are expressed in myeloid cells, miR-155 is expressed in dendritic cells, B cells and T cells, miR-146 is upregulated in macrophages upon TLR stimulation and miR-126 is expressed in plasmacytoid dendritic cells. In certain embodiments, the miR(s) is expressed abundantly or preferentially in immune cells. For example, miR-142 (miR-142-3p and/or miR-142-5p), miR-126 (miR-126- 3p and/or miR-126-5p), miR-146 (miR-146-3p and/or miR-146-5p) and miR-155 (miR-155- 3p and/or miR155-5p) are expressed abundantly in immune cells. These microRNA sequences are known in the art and, thus, one of ordinary skill in the art can readily design binding sequences or target sequences to which these microRNAs will bind based upon Watson-Crick complementarity. In one embodiment, the polynucleotide of this disclosure comprises three copies of the same miRNA binding site. In certain embodiments, use of three copies of the same miR binding site can exhibit beneficial properties as compared to use of a single miRNA binding site. In another embodiment, the polynucleotide of this disclosure comprises two or more (e.g., two, three, four) copies of at least two different miR binding sites expressed in immune cells. In another embodiment, the polynucleotide of this disclosure comprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-142-3p. In various embodiments, the polynucleotide of this disclosure comprises binding sites for miR-142-3p and miR-155 (miR-155-3p or miR-155- 5p), miR-142-3p and miR-146 (miR-146-3 or miR-146-5p), or miR-142-3p and miR-126 (miR-126-3p or miR-126-5p). In another embodiment, the polynucleotide of this disclosure comprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-126-3p. In various embodiments, the polynucleotide of this disclosure comprises binding sites for miR-126-3p and miR-155 (miR-155-3p or miR-155- 5p), miR-126-3p and miR-146 (miR-146-3p or miR-146-5p), or miR-126-3p and miR-142 (miR-142-3p or miR-142-5p). In another embodiment, the polynucleotide of this disclosure comprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-142-5p. In various embodiments, the polynucleotide of this disclosure comprises binding sites for miR-142-5p and miR-155 (miR-155-3p or miR-155- 5p), miR-142-5p and miR-146 (miR-146-3 or miR-146-5p), or miR-142-5p and miR-126 (miR-126-3p or miR-126-5p). In yet another embodiment, the polynucleotide of this disclosure comprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-155-5p. In various embodiments, the polynucleotide of this disclosure comprises binding sites for miR-155-5p and miR-142 (miR-142-3p or miR-142- 5p), miR-155-5p and miR-146 (miR-146-3 or miR-146-5p), or miR-155-5p and miR-126 (miR-126-3p or miR-126-5p). In some embodiments, a polynucleotide of this disclosure comprises a miRNA binding site, wherein the miRNA binding site comprises one or more nucleotide sequences selected from Table 6, including one or more copies of any one or more of the miRNA binding site sequences. In some embodiments, a polynucleotide of this disclosure further comprises at least one, two, three, four, five, six, seven, eight, nine, ten, or more of the same or different miRNA binding sites selected from Table 6, including any combination thereof. In some embodiments, the miRNA binding site binds to miR-142 or is complementary to miR-142. In some embodiments, the miR-142 comprises SEQ ID NO:172. In some embodiments, the miRNA binding site binds to miR-142-3p or miR-142-5p. In some embodiments, the miR-142-3p binding site comprises SEQ ID NO:174. In some embodiments, the miR-142-5p binding site comprises SEQ ID NO:210. In some embodiments, the miRNA binding site comprises a nucleotide sequence at least 80%, at least 85%, at least 90%, at least 95%, or 100% identical to SEQ ID NO:174 or SEQ ID NO:210. In some embodiments, the miRNA binding site binds to miR-126 or is complementary to miR-126. In some embodiments, the miR-126 comprises SEQ ID NO: 150. In some embodiments, the miRNA binding site binds to miR-126-3p or miR-126-5p. In some embodiments, the miR-126-3p binding site comprises SEQ ID NO: 152. In some embodiments, the miR-126-5p binding site comprises SEQ ID NO: 154. In some embodiments, the miRNA binding site comprises a nucleotide sequence at least 80%, at least 85%, at least 90%, at least 95%, or 100% identical to SEQ ID NO: 152 or SEQ ID NO: 154. In one embodiment, the 3′ UTR comprises two miRNA binding sites, wherein a first miRNA binding site binds to miR-142 and a second miRNA binding site binds to miR-126.
In an embodiment, the 3′ UTR comprises a micro RNA (miRNA) binding site, e.g., as described herein, which binds to a miR present in a human cell. In an embodiment, the 3′ UTR comprises a miRNA binding site of SEQ ID NO: 212, SEQ ID NO: 174, SEQ ID NO: 152 or a combination thereof. In an embodiment, the 3′ UTR comprises a plurality of miRNA binding sites, e.g., 2, 3, 4, 5, 6, 7 or 8 miRNA binding sites. In an embodiment, the plurality of miRNA binding sites comprises the same or different miRNA binding sites. miR122 bs = CAAACACCAUUGUCACACUCCA (SEQ ID NO: 212) miR-142-3p bs = UCCAUAAAGUAGGAAACACUACA (SEQ ID NO: 174) miR-126 bs = CGCAUUAUUACUCACGGUACGA (SEQ ID NO: 152) In an aspect, disclosed herein is a polynucleotide encoding a polypeptide, wherein the polynucleotide comprises: (a) a 5′-UTR, e.g., as described herein; (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3′-UTR (e.g., as described herein). In an aspect, an LNP composition comprising a polynucleotide comprising an open reading frame encoding an IL-22 polypeptide (e.g., SEQ ID NO:1) and comprising a 3′ UTR disclosed herein comprises: (i) an ionizable lipid, e.g., an amino lipid; (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; and (iv) a PEG-lipid. In another aspect, the LNP compositions of the disclosure are used in a method of treating metabolic disease in a subject. In an aspect, an LNP composition comprising a polynucleotide disclosed herein encoding an IL-22 polypeptide, e.g., as described herein, can be administered with an additional agent, e.g., as described herein. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUC UGAGUGGGCGGC (SEQ ID NO:139) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:139. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:139 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:139 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:139 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:139 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:139. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGUCUAAGCUGGAGCCUCCUGAGAGACCUGUGUGAACUAUUGAGAAGAUCG GAACAGCUCCUUACUCUGAGGAAGUUGGUACCCCCGUGGUCUUUGAAUAAAG UCUGAGUGGGCGGC (SEQ ID NO:140) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:140. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:140 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:140 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:140 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:140 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:140. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGCAAACACCAUUGUCACACUCCAGCCUCGGUGGCCUAGCU UCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCCAGCCCCUCC UCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUCCAGUGGUCU UUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:141) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:141. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:141 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:141 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:141 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:141 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:141. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGUCCAUAAAGUAGGAAACACUACAGCUGGAGCCUCGGUG GCCUAGCUUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCC AGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUCU GAGUGGGCGGC (SEQ ID NO:142) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:142. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:142 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:142 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:142 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:142 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:142. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAAGCUCCCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUC CAGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:143) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:143. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:143 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:143 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:143 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:143 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:143. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGCAAACACCAUUGUCACACUCCAGCCUCGGUGGCCUAGC UUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCCAGCCCCUC CUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUCCAGUGGUCU UUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:144) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:144. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:144 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:144 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:144 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:144 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:144. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGUCCAUAAAGUAGGAAACACUACAGCCUCGGUGGCCUAG CUUCUUGCCCCUUGGGCCUCCAUAAAGUAGGAAACACUACAUCCCCCCAGCCC CUCCUCCCCUUCCUGCACCCGUACCCCCCGCAUUAUUACUCACGGUACGAGUG GUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:145) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:145. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:145 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:145 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:145 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:145 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:145. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGUCCAUAAAGUAGGAAACACUACAGCUGGAGCCUCGGUG GCCUAGCUUCUUGCCCCUUGGGCCCAAACACCAUUGUCACACUCCAUCCCCCC AGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCGUGGUCUUUGAAUAAAGUCU GAGUGGGCGGC (SEQ ID NO:146) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:146. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:146 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:146 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:146 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:146 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:146. In an embodiment, the polynucleotide comprises a stop element and 3’-UTR, wherein the sequence is (stop element is italicized): UAAGCCCCUCCGGGGGCCUCGGUGGCCUAGCUUCUUGCCCCUUGGGCCUCCCCC CAGCCCCUCCUCCCCUUCCUGCACCCGUACCCCCCAAACACCAUUGUCACACUC CAGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGC (SEQ ID NO:147) or a variant or fragment thereof (e.g., a fragment that lacks the first one, two, three, four, five, six, or more nucleotides of nucleotides of SEQ ID NO:147. In an embodiment, the polynucleotide having a 3’ UTR sequence provided in SEQ ID NO:147 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3-fold or more. In an embodiment, the increase in half-life is about 4-fold or more. In an embodiment, the increase in half-life is about 5-fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more. In an embodiment, the polynucleotide having a 3′ UTR sequence provided in SEQ ID NO:147 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3′ UTR, has a different 3′ UTR, or does not have a 3′ UTR of SEQ ID NO:147 or a variant or fragment thereof. In an embodiment, the polynucleotide comprises a 3′ UTR sequence provided in SEQ ID NO:147 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3′ UTR sequence provided in SEQ ID NO:147. 13. MicroRNA (miRNA) Binding Sites Polynucleotides of this disclosure can include regulatory elements, for example, microRNA (miRNA) binding sites, transcription factor binding sites, structured mRNA sequences and/or motifs, artificial binding sites engineered to act as pseudo-receptors for endogenous nucleic acid binding molecules, and combinations thereof. In some embodiments, polynucleotides including such regulatory elements are referred to as including “sensor sequences”. In some embodiments, a polynucleotide (e.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)) of this disclosure comprises an open reading frame (ORF) encoding a polypeptide of interest and further comprises one or more miRNA binding site(s). Inclusion or incorporation of miRNA binding site(s) provides for regulation of polynucleotides of this disclosure, and in turn, of the polypeptides encoded therefrom, based on tissue-specific and/or cell-type specific expression of naturally-occurring miRNAs. The present invention also provides pharmaceutical compositions and Formulations that comprise any of the polynucleotides described above. In some embodiments, the composition or Formulation further comprises a delivery agent. In some embodiments, the composition or Formulation can contain a polynucleotide comprising a sequence optimized nucleic acid sequence disclosed herein which encodes a polypeptide. In some embodiments, the composition or Formulation can contain a polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a polynucleotide (e.g., an ORF) having significant sequence identity to a sequence optimized nucleic acid sequence disclosed herein which encodes a polypeptide. In some embodiments, the polynucleotide further comprises a miRNA binding site, e.g., a miRNA binding site that binds A miRNA, e.g., a natural-occurring miRNA, is a 19-25 nucleotide long noncoding RNA that binds to a polynucleotide and down-regulates gene expression either by reducing stability or by inhibiting translation of the polynucleotide. A miRNA sequence comprises a “seed” region, i.e., a sequence in the region of positions 2-8 of the mature miRNA. A miRNA seed can comprise positions 2-8 or 2-7 of the mature miRNA. microRNAs derive enzymatically from regions of RNA transcripts that fold back on themselves to form short hairpin structures often termed a pre-miRNA (precursor-miRNA). A pre-miRNA typically has a two-nucleotide overhang at its 3′ end, and has 3′ hydroxyl and 5′ phosphate groups. This precursor-mRNA is processed in the nucleus and subsequently transported to the cytoplasm where it is further processed by DICER (a RNase III enzyme), to form a mature microRNA of approximately 22 nucleotides. The mature microRNA is then incorporated into a ribonuclear particle to form the RNA-induced silencing complex, RISC, which mediates gene silencing. Art-recognized nomenclature for mature miRNAs typically designates the arm of the pre-miRNA from which the mature miRNA derives; "5p" means the microRNA is from the 5 prime arm of the pre-miRNA hairpin and "3p" means the microRNA is from the 3 prime end of the pre-miRNA hairpin. A miR referred to by number herein can refer to either of the two mature microRNAs originating from opposite arms of the same pre- miRNA (e.g., either the 3p or 5p microRNA). All miRs referred to herein are intended to include both the 3p and 5p arms/sequences, unless particularly specified by the 3p or 5p designation. As used herein, the term “microRNA (miRNA or miR) binding site” refers to a sequence within a polynucleotide, e.g., within a DNA or within an RNA transcript, including in the 5′UTR and/or 3′UTR, that has sufficient complementarity to all or a region of a miRNA to interact with, associate with or bind to the miRNA. In some embodiments, a polynucleotide of this disclosure comprising an ORF encoding a polypeptide of interest and further comprises one or more miRNA binding site(s). In exemplary embodiments, a 5′ UTR and/or 3′ UTR of the polynucleotide (e.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)) comprises the one or more miRNA binding site(s). A miRNA binding site having sufficient complementarity to a miRNA refers to a degree of complementarity sufficient to facilitate miRNA-mediated regulation of a polynucleotide, e.g., miRNA-mediated translational repression or degradation of the polynucleotide. In exemplary aspects of this disclosure, a miRNA binding site having sufficient complementarity to the miRNA refers to a degree of complementarity sufficient to facilitate miRNA-mediated degradation of the polynucleotide, e.g., miRNA-guided RNA- induced silencing complex (RISC)-mediated cleavage of mRNA. The miRNA binding site can have complementarity to, for example, a 19-25 nucleotide long miRNA sequence, to a 19-23 nucleotide long miRNA sequence, or to a 22 nucleotide long miRNA sequence. A miRNA binding site can be complementary to only a portion of a miRNA, e.g., to a portion less than 1, 2, 3, or 4 nucleotides of the full length of a naturally-occurring miRNA sequence, or to a portion less than 1, 2, 3, or 4 nucleotides shorter than a naturally-occurring miRNA sequence. Full or complete complementarity (e.g., full complementarity or complete complementarity over all or a significant portion of the length of a naturally-occurring miRNA) is preferred when the desired regulation is mRNA degradation. In some embodiments, a miRNA binding site includes a sequence that has complementarity (e.g., partial or complete complementarity) with an miRNA seed sequence. In some embodiments, the miRNA binding site includes a sequence that has complete complementarity with a miRNA seed sequence. In some embodiments, a miRNA binding site includes a sequence that has complementarity (e.g., partial or complete complementarity) with an miRNA sequence. In some embodiments, the miRNA binding site includes a sequence that has complete complementarity with a miRNA sequence. In other embodiments, the sequence is not completely complementary. In some embodiments, a miRNA binding site has complete complementarity with a miRNA sequence but for 1, 2, or 3 nucleotide substitutions, terminal additions, and/or truncations. In some embodiments, the miRNA binding site is the same length as the corresponding miRNA. In other embodiments, the miRNA binding site is one, two, three, four, five, six, seven, eight, nine, ten, eleven or twelve nucleotide(s) shorter than the corresponding miRNA at the 5′ terminus, the 3′ terminus, or both. In still other embodiments, the microRNA binding site is two nucleotides shorter than the corresponding microRNA at the 5′ terminus, the 3′ terminus, or both. The miRNA binding sites that are shorter than the corresponding miRNAs are still capable of degrading the mRNA incorporating one or more of the miRNA binding sites or preventing the mRNA from translation. In some embodiments, the miRNA binding site binds the corresponding mature miRNA that is part of an active RISC containing Dicer. In another embodiment, binding of the miRNA binding site to the corresponding miRNA in RISC degrades the mRNA containing the miRNA binding site or prevents the mRNA from being translated. In some embodiments, the miRNA binding site has sufficient complementarity to miRNA so that a RISC complex comprising the miRNA cleaves the polynucleotide comprising the miRNA binding site. In other embodiments, the miRNA binding site has imperfect complementarity so that a RISC complex comprising the miRNA induces instability in the polynucleotide comprising the miRNA binding site. In another embodiment, the miRNA binding site has imperfect complementarity so that a RISC complex comprising the miRNA represses transcription of the polynucleotide comprising the miRNA binding site. In some embodiments, the miRNA binding site has one, two, three, four, five, six, seven, eight, nine, ten, eleven or twelve mismatch(es) from the corresponding miRNA. In some embodiments, the miRNA binding site has at least about ten, at least about eleven, at least about twelve, at least about thirteen, at least about fourteen, at least about fifteen, at least about sixteen, at least about seventeen, at least about eighteen, at least about nineteen, at least about twenty, or at least about twenty-one contiguous nucleotides complementary to at least about ten, at least about eleven, at least about twelve, at least about thirteen, at least about fourteen, at least about fifteen, at least about sixteen, at least about seventeen, at least about eighteen, at least about nineteen, at least about twenty, or at least about twenty-one, respectively, contiguous nucleotides of the corresponding miRNA. By engineering one or more miRNA binding sites into a polynucleotide of this disclosure, the polynucleotide can be targeted for degradation or reduced translation, provided the miRNA in question is available. This can reduce off-target effects upon delivery of the polynucleotide. For example, if a polynucleotide of this disclosure is not intended to be delivered to a tissue or cell but ends up is said tissue or cell, then a miRNA abundant in the tissue or cell can inhibit the expression of the gene of interest if one or multiple binding sites of the miRNA are engineered into the 5′ UTR and/or 3′ UTR of the polynucleotide. Thus, in some embodiments, incorporation of one or more miRNA binding sites into an mRNA of the disclosure may reduce the hazard of off-target effects upon nucleic acid molecule delivery and/or enable tissue-specific regulation of expression of a polypeptide encoded by the mRNA. In yet other embodiments, incorporation of one or more miRNA binding sites into an mRNA of the disclosure can modulate immune responses upon nucleic acid delivery in vivo. In further embodiments, incorporation of one or more miRNA binding sites into an mRNA of the disclosure can modulate accelerated blood clearance (ABC) of lipid-comprising compounds and compositions described herein. Conversely, miRNA binding sites can be removed from polynucleotide sequences in which they naturally occur to increase protein expression in specific tissues. For example, a binding site for a specific miRNA can be removed from a polynucleotide to improve protein expression in tissues or cells containing the miRNA. Regulation of expression in multiple tissues can be accomplished through introduction or removal of one or more miRNA binding sites, e.g., one or more distinct miRNA binding sites. The decision whether to remove or insert a miRNA binding site can be made based on miRNA expression patterns and/or their profilings in tissues and/or cells in development and/or disease. Identification of miRNAs, miRNA binding sites, and their expression patterns and role in biology have been reported (e.g., Bonauer et al., Curr Drug Targets 2010 11:943-949; Anand and Cheresh Curr Opin Hematol 201118:171-176; Contreras and Rao Leukemia 201226:404-413 (2011 Dec 20. doi: 10.1038/leu.2011.356); Bartel Cell 2009 136:215-233; Landgraf et al, Cell, 2007129:1401-1414; Gentner and Naldini, Tissue Antigens.201280:393-403 and all references therein; each of which is incorporated herein by reference in its entirety). Examples of tissues where miRNA are known to regulate mRNA, and thereby protein expression, include, but are not limited to, liver (miR-122), muscle (miR-133, miR-206, miR- 208), endothelial cells (miR-17-92, miR-126), myeloid cells (miR-142-3p, miR-142-5p, miR- 16, miR-21, miR-223, miR-24, miR-27), adipose tissue (let-7, miR-30c), heart (miR-1d, miR- 149), kidney (miR-192, miR-194, miR-204), and lung epithelial cells (let-7, miR-133, miR- 126). Specifically, miRNAs are known to be differentially expressed in immune cells (also called hematopoietic cells), such as antigen presenting cells (APCs) (e.g., dendritic cells and macrophages), macrophages, monocytes, B lymphocytes, T lymphocytes, granulocytes, natural killer cells, etc. Immune cell specific miRNAs are involved in immunogenicity, autoimmunity, the immune-response to infection, inflammation, as well as unwanted immune response after gene therapy and tissue/organ transplantation. Immune cells specific miRNAs also regulate many aspects of development, proliferation, differentiation and apoptosis of hematopoietic cells (immune cells). For example, miR-142 and miR-146 are exclusively expressed in immune cells, particularly abundant in myeloid dendritic cells. It has been demonstrated that the immune response to a polynucleotide can be shut-off by adding miR- 142 binding sites to the 3′-UTR of the polynucleotide, enabling more stable gene transfer in tissues and cells. miR-142 efficiently degrades exogenous polynucleotides in antigen presenting cells and suppresses cytotoxic elimination of transduced cells (e.g., Annoni A et al., blood, 2009, 114, 5152-5161; Brown BD, et al., Nat med.2006, 12(5), 585-591; Brown BD, et al., blood, 2007, 110(13): 4144-4152, each of which is incorporated herein by reference in its entirety). An antigen-mediated immune response can refer to an immune response triggered by foreign antigens, which, when entering an organism, are processed by the antigen presenting cells and displayed on the surface of the antigen presenting cells. T cells can recognize the presented antigen and induce a cytotoxic elimination of cells that express the antigen. Introducing one or more (e.g., one, two, or three) miR-142 binding sites into the 5′ UTR and/or 3′UTR of a polynucleotide of this disclosure can selectively repress gene expression in antigen presenting cells through miR-142 mediated degradation, limiting antigen presentation in antigen presenting cells (e.g., dendritic cells) and thereby preventing antigen-mediated immune response after the delivery of the polynucleotide. The polynucleotide is then stably expressed in target tissues or cells without triggering cytotoxic elimination. In some embodiments, it may be beneficial to target the same cell type with multiple miRs and to incorporate binding sites to each of the 3p and 5p arm if both are abundant (e.g., both miR-142-3p and miR142-5p are abundant in hematopoietic stem cells). Thus, in certain embodiments, polynucleotides of this disclosure contain two or more (e.g., two, three, four or more) miR bindings sites from: (i) the group consisting of miR-142, miR-144, miR-150, miR-155 and miR-223 (which are expressed in many hematopoietic cells); or (ii) the group consisting of miR-142, miR150, miR-16 and miR-223 (which are expressed in B cells); or the group consisting of miR-223, miR-451, miR-26a, miR-16 (which are expressed in progenitor hematopoietic cells). In some embodiments, it may also be beneficial to combine various miRs such that multiple cell types of interest are targeted at the same time (e.g., miR-142 and miR-126 to target many cells of the hematopoietic lineage and endothelial cells). Thus, for example, in certain embodiments, polynucleotides of this disclosure comprise two or more (e.g., two, three, four or more) miRNA bindings sites, wherein: (i) at least one of the miRs targets cells of the hematopoietic lineage (e.g., miR-142, miR-144, miR-150, miR-155 or miR-223) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or (ii) at least one of the miRs targets B cells (e.g., miR-142, miR150, miR-16 or miR-223) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or (iii) at least one of the miRs targets progenitor hematopoietic cells (e.g., miR-223, miR-451, miR-26a or miR-16) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or (iv) at least one of the miRs targets cells of the hematopoietic lineage (e.g., miR-142, miR- 144, miR-150, miR-155 or miR-223), at least one of the miRs targets B cells (e.g., miR-142, miR150, miR-16 or miR-223) and at least one of the miRs targets plasmacytoid dendritic cells, platelets or endothelial cells (e.g., miR-126); or any other possible combination of the foregoing four classes of miR binding sites (i.e., those targeting the hematopoietic lineage, those targeting B cells, those targeting progenitor hematopoietic cells and/or those targeting plasmacytoid dendritic cells/platelets/endothelial cells). In one embodiment, to modulate immune responses, polynucleotides of the present invention can comprise one or more miRNA binding sequences that bind to one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro-inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells). It has now been discovered that incorporation into an mRNA of one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro- inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells) reduces or inhibits immune cell activation (e.g., B cell activation, as measured by frequency of activated B cells) and/or cytokine production (e.g., production of IL-6, IFN-γ and/or TNFα). Furthermore, it has now been discovered that incorporation into an mRNA of one or more miRs that are expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro- inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells) can reduce or inhibit an anti-drug antibody (ADA) response against a protein of interest encoded by the mRNA. In another embodiment, to modulate accelerated blood clearance of a polynucleotide delivered in a lipid-comprising compound or composition, polynucleotides of this disclosure can comprise one or more miR binding sequences that bind to one or more miRNAs expressed in conventional immune cells or any cell that expresses TLR7 and/or TLR8 and secrete pro-inflammatory cytokines and/or chemokines (e.g., in immune cells of peripheral lymphoid organs and/or splenocytes and/or endothelial cells). It has now been discovered that incorporation into an mRNA of one or more miR binding sites reduces or inhibits accelerated blood clearance (ABC) of the lipid-comprising compound or composition for use in delivering the mRNA. Furthermore, it has now been discovered that incorporation of one or more miR binding sites into an mRNA reduces serum levels of anti-PEG anti-IgM (e.g, reduces or inhibits the acute production of IgMs that recognize polyethylene glycol (PEG) by B cells) and/or reduces or inhibits proliferation and/or activation of plasmacytoid dendritic cells following administration of a lipid-comprising compound or composition comprising the mRNA. In some embodiments, miR sequences may correspond to any known microRNA expressed in immune cells, including but not limited to those taught in US Publication US2005/0261218 and US Publication US2005/0059005, the contents of which are incorporated herein by reference in their entirety. Non-limiting examples of miRs expressed in immune cells include those expressed in spleen cells, myeloid cells, dendritic cells, plasmacytoid dendritic cells, B cells, T cells and/or macrophages. For example, miR-142-3p, miR-142-5p, miR-16, miR-21, miR-223, miR-24 and miR-27 are expressed in myeloid cells, miR-155 is expressed in dendritic cells, B cells and T cells, miR-146 is upregulated in macrophages upon TLR stimulation and miR-126 is expressed in plasmacytoid dendritic cells. In certain embodiments, the miR(s) is expressed abundantly or preferentially in immune cells. For example, miR-142 (miR-142-3p and/or miR-142-5p), miR-126 (miR-126- 3p and/or miR-126-5p), miR-146 (miR-146-3p and/or miR-146-5p) and miR-155 (miR-155- 3p and/or miR155-5p) are expressed abundantly in immune cells. These microRNA sequences are known in the art and, thus, one of ordinary skill in the art can readily design binding sequences or target sequences to which these microRNAs will bind based upon Watson-Crick complementarity. In one embodiment, the polynucleotide of this disclosure comprises three copies of the same miRNA binding site. In certain embodiments, use of three copies of the same miR binding site can exhibit beneficial properties as compared to use of a single miRNA binding site. In another embodiment, the polynucleotide of this disclosure comprises two or more (e.g., two, three, four) copies of at least two different miR binding sites expressed in immune cells. In another embodiment, the polynucleotide of this disclosure comprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-142-3p. In various embodiments, the polynucleotide of this disclosure comprises binding sites for miR-142-3p and miR-155 (miR-155-3p or miR-155- 5p), miR-142-3p and miR-146 (miR-146-3 or miR-146-5p), or miR-142-3p and miR-126 (miR-126-3p or miR-126-5p). In another embodiment, the polynucleotide of this disclosure comprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-126-3p. In various embodiments, the polynucleotide of this disclosure comprises binding sites for miR-126-3p and miR-155 (miR-155-3p or miR-155- 5p), miR-126-3p and miR-146 (miR-146-3p or miR-146-5p), or miR-126-3p and miR-142 (miR-142-3p or miR-142-5p). In another embodiment, the polynucleotide of this disclosure comprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-142-5p. In various embodiments, the polynucleotide of this disclosure comprises binding sites for miR-142-5p and miR-155 (miR-155-3p or miR-155- 5p), miR-142-5p and miR-146 (miR-146-3 or miR-146-5p), or miR-142-5p and miR-126 (miR-126-3p or miR-126-5p). In yet another embodiment, the polynucleotide of this disclosure comprises at least two miR binding sites for microRNAs expressed in immune cells, wherein one of the miR binding sites is for miR-155-5p. In various embodiments, the polynucleotide of this disclosure comprises binding sites for miR-155-5p and miR-142 (miR-142-3p or miR-142- 5p), miR-155-5p and miR-146 (miR-146-3 or miR-146-5p), or miR-155-5p and miR-126 (miR-126-3p or miR-126-5p). In some embodiments, a polynucleotide of this disclosure comprises a miRNA binding site, wherein the miRNA binding site comprises one or more nucleotide sequences selected from Table 6, including one or more copies of any one or more of the miRNA binding site sequences. In some embodiments, a polynucleotide of this disclosure further comprises at least one, two, three, four, five, six, seven, eight, nine, ten, or more of the same or different miRNA binding sites selected from Table 6, including any combination thereof. In some embodiments, the miRNA binding site binds to miR-142 or is complementary to miR-142. In some embodiments, the miR-142 comprises SEQ ID NO:172. In some embodiments, the miRNA binding site binds to miR-142-3p or miR-142-5p. In some embodiments, the miR-142-3p binding site comprises SEQ ID NO:174. In some embodiments, the miR-142-5p binding site comprises SEQ ID NO:210. In some embodiments, the miRNA binding site comprises a nucleotide sequence at least 80%, at least 85%, at least 90%, at least 95%, or 100% identical to SEQ ID NO:174 or SEQ ID NO:210. In some embodiments, the miRNA binding site binds to miR-126 or is complementary to miR-126. In some embodiments, the miR-126 comprises SEQ ID NO: 150. In some embodiments, the miRNA binding site binds to miR-126-3p or miR-126-5p. In some embodiments, the miR-126-3p binding site comprises SEQ ID NO: 152. In some embodiments, the miR-126-5p binding site comprises SEQ ID NO: 154. In some embodiments, the miRNA binding site comprises a nucleotide sequence at least 80%, at least 85%, at least 90%, at least 95%, or 100% identical to SEQ ID NO: 152 or SEQ ID NO: 154. In one embodiment, the 3′ UTR comprises two miRNA binding sites, wherein a first miRNA binding site binds to miR-142 and a second miRNA binding site binds to miR-126.









Table 6. miR-142, miR-126, and miR-142 and miR-126 binding sites In some embodiments, a miRNA binding site is inserted in the polynucleotide of this disclosure in any position of the polynucleotide (e.g., the 5′ UTR and/or 3′ UTR). In some embodiments, the 5′ UTR comprises a miRNA binding site. In some embodiments, the 3′ UTR comprises a miRNA binding site. In some embodiments, the 5′ UTR and the 3′ UTR comprise a miRNA binding site. The insertion site in the polynucleotide can be anywhere in the polynucleotide as long as the insertion of the miRNA binding site in the polynucleotide does not interfere with the translation of a functional polypeptide in the absence of the corresponding miRNA; and in the presence of the miRNA, the insertion of the miRNA binding site in the polynucleotide and the binding of the miRNA binding site to the corresponding miRNA are capable of degrading the polynucleotide or preventing the translation of the polynucleotide. In some embodiments, a miRNA binding site is inserted in at least about 30 nucleotides downstream from the stop codon of an ORF in a polynucleotide of this disclosure comprising the ORF. In some embodiments, a miRNA binding site is inserted in at least about 10 nucleotides, at least about 15 nucleotides, at least about 20 nucleotides, at least about 25 nucleotides, at least about 30 nucleotides, at least about 35 nucleotides, at least about 40 nucleotides, at least about 45 nucleotides, at least about 50 nucleotides, at least about 55 nucleotides, at least about 60 nucleotides, at least about 65 nucleotides, at least about 70 nucleotides, at least about 75 nucleotides, at least about 80 nucleotides, at least about 85 nucleotides, at least about 90 nucleotides, at least about 95 nucleotides, or at least about 100 nucleotides downstream from the stop codon of an ORF in a polynucleotide of this disclosure. In some embodiments, a miRNA binding site is inserted in about 10 nucleotides to about 100 nucleotides, about 20 nucleotides to about 90 nucleotides, about 30 nucleotides to about 80 nucleotides, about 40 nucleotides to about 70 nucleotides, about 50 nucleotides to about 60 nucleotides, about 45 nucleotides to about 65 nucleotides downstream from the stop codon of an ORF in a polynucleotide of this disclosure. In some embodiments, a miRNA binding site is inserted within the 3′ UTR immediately following the stop codon of the coding region within the polynucleotide of this disclosure, e.g., mRNA. In some embodiments, if there are multiple copies of a stop codon in the construct, a miRNA binding site is inserted immediately following the final stop codon. In some embodiments, a miRNA binding site is inserted further downstream of the stop codon, in which case there are 3′ UTR bases between the stop codon and the miR binding site(s). In some embodiments, one or more miRNA binding sites can be positioned within the 5′ UTR at one or more possible insertion sites. In one embodiment, a codon optimized open reading frame encoding a polypeptide of interest comprises a stop codon and the at least one microRNA binding site is located within the 3′ UTR 1-100 nucleotides after the stop codon. In one embodiment, the codon optimized open reading frame encoding the polypeptide of interest comprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR 30-50 nucleotides after the stop codon. In another embodiment, the codon optimized open reading frame encoding the polypeptide of interest comprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR at least 50 nucleotides after the stop codon. In other embodiments, the codon optimized open reading frame encoding the polypeptide of interest comprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR immediately after the stop codon, or within the 3′ UTR 15-20 nucleotides after the stop codon or within the 3′ UTR 70-80 nucleotides after the stop codon. In other embodiments, the 3′ UTR comprises more than one miRNA binding site (e.g., 2-4 miRNA binding sites), wherein there can be a spacer region (e.g., of 10-100, 20-70 or 30-50 nucleotides in length) between each miRNA binding site. In another embodiment, the 3′ UTR comprises a spacer region between the end of the miRNA binding site(s) and the poly A tail nucleotides. For example, a spacer region of 10-100, 20-70 or 30-50 nucleotides in length can be situated between the end of the miRNA binding site(s) and the beginning of the poly A tail. In one embodiment, a codon optimized open reading frame encoding a polypeptide of interest comprises a start codon and the at least one microRNA binding site is located within the 5′ UTR 1-100 nucleotides before (upstream of) the start codon. In one embodiment, the codon optimized open reading frame encoding the polypeptide of interest comprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR 10-50 nucleotides before (upstream of) the start codon. In another embodiment, the codon optimized open reading frame encoding the polypeptide of interest comprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR at least 25 nucleotides before (upstream of) the start codon. In other embodiments, the codon optimized open reading frame encoding the polypeptide of interest comprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR immediately before the start codon, or within the 5′ UTR 15-20 nucleotides before the start codon or within the 5′ UTR 70-80 nucleotides before the start codon. In other embodiments, the 5′ UTR comprises more than one miRNA binding site (e.g., 2-4 miRNA binding sites), wherein there can be a spacer region (e.g., of 10-100, 20-70 or 30-50 nucleotides in length) between each miRNA binding site. In one embodiment, the 3′ UTR comprises more than one stop codon, wherein at least one miRNA binding site is positioned downstream of the stop codons. For example, a 3′ UTR can comprise 1, 2 or 3 stop codons. Non-limiting examples of triple stop codons that can be used include: UGAUAAUAG (SEQ ID NO:182), UGAUAGUAA (SEQ ID NO:183), UAAUGAUAG (SEQ ID NO:184), UGAUAAUAA (SEQ ID NO:185), UGAUAGUAG (SEQ ID NO:186), UAAUGAUGA (SEQ ID NO:187), UAAUAGUAG (SEQ ID NO:188), UGAUGAUGA (SEQ ID NO:179), UAAUAAUAA (SEQ ID NO:180), and UAGUAGUAG (SEQ ID NO:181). Within a 3′ UTR, for example, 1, 2, 3 or 4 miRNA binding sites, e.g., miR-142-3p binding sites, can be positioned immediately adjacent to the stop codon(s) or at any number of nucleotides downstream of the final stop codon. When the 3′ UTR comprises multiple miRNA binding sites, these binding sites can be positioned directly next to each other in the construct (i.e., one after the other) or, alternatively, spacer nucleotides can be positioned between each binding site. In one embodiment, the 3′ UTR comprises three stop codons with a single miR-142- 3p binding site located downstream of the 3rd stop codon. In one embodiment, the polynucleotide of this disclosure comprises a 5′ UTR, a codon optimized open reading frame encoding a polypeptide of interest, a 3′ UTR comprising the at least one miRNA binding site for a miR expressed in immune cells, and a 3′ tailing region of linked nucleosides. In various embodiments, the 3′ UTR comprises 1-4, at least two, one, two, three or four miRNA binding sites for miRs expressed in immune cells, preferably abundantly or preferentially expressed in immune cells. In one embodiment, the at least one miRNA expressed in immune cells is a miR-142- 3p microRNA binding site. In one embodiment, the miR-142-3p microRNA binding site comprises the sequence shown in SEQ ID NO: 174. In one embodiment, the at least one miRNA expressed in immune cells is a miR-126 microRNA binding site. In one embodiment, the miR-126 binding site is a miR-126-3p binding site. In one embodiment, the miR-126-3p microRNA binding site comprises the sequence shown in SEQ ID NO: 152. Non-limiting exemplary sequences for miRs to which a microRNA binding site(s) of the disclosure can bind include the following: miR-142-3p (SEQ ID NO: 173), miR-142-5p (SEQ ID NO: 175), miR-146-3p (SEQ ID NO: 155), miR-146-5p (SEQ ID NO: 156), miR- 155-3p (SEQ ID NO: 157), miR-155-5p (SEQ ID NO: 158), miR-126-3p (SEQ ID NO: 151), miR-126-5p (SEQ ID NO: 153), miR-16-3p (SEQ ID NO: 159), miR-16-5p (SEQ ID NO: 160), miR-21-3p (SEQ ID NO: 161), miR-21-5p (SEQ ID NO: 162), miR-223-3p (SEQ ID NO: 163), miR-223-5p (SEQ ID NO: 164), miR-24-3p (SEQ ID NO: 165), miR-24-5p (SEQ ID NO: 166), miR-27-3p (SEQ ID NO: 167) and miR-27-5p (SEQ ID NO: 168). Other suitable miR sequences expressed in immune cells (e.g., abundantly or preferentially expressed in immune cells) are known and available in the art, for example at the University of Manchester’s microRNA database, miRBase. Sites that bind any of the aforementioned miRs can be designed based on Watson-Crick complementarity to the miR, typically 100% complementarity to the miR, and inserted into an mRNA construct of the disclosure as described herein. In another embodiment, a polynucleotide of the present invention (e.g., and mRNA, e.g., the 3′ UTR thereof) can comprise at least one miRNA bindingsite to thereby reduce or inhibit accelerated blood clearance, for example by reducing or inhibiting production of IgMs, e.g., against PEG, by B cells and/or reducing or inhibiting proliferation and/or activation of pDCs, and can comprise at least one miRNA bindingsite for modulating tissue expression of an encoded protein of interest. miRNA gene regulation can be influenced by the sequence surrounding the miRNA such as, but not limited to, the species of the surrounding sequence, the type of sequence (e.g., heterologous, homologous, exogenous, endogenous, or artificial), regulatory elements in the surrounding sequence and/or structural elements in the surrounding sequence. The miRNA can be influenced by the 5′UTR and/or 3′UTR. As a non-limiting example, a non- human 3′UTR can increase the regulatory effect of the miRNA sequence on the expression of a polypeptide of interest compared to a human 3′ UTR of the same sequence type. In one embodiment, other regulatory elements and/or structural elements of the 5′ UTR can influence miRNA mediated gene regulation. One example of a regulatory element and/or structural element is a structured IRES (Internal Ribosome Entry Site) in the 5′ UTR, which is necessary for the binding of translational elongation factors to initiate protein translation. EIF4A2 binding to this secondarily structured element in the 5′-UTR is necessary for miRNA mediated gene expression (Meijer HA et al., Science, 2013, 340, 82-85, herein incorporated by reference in its entirety). The polynucleotides of this disclosure can further include this structured 5′ UTR in order to enhance microRNA mediated gene regulation. At least one miRNA binding site can be engineered into the 3′ UTR of a polynucleotide of this disclosure. In this context, at least two, at least three, at least four, at least five, at least six, at least seven, at least eight, at least nine, at least ten, or more miRNA binding sites can be engineered into a 3′ UTR of a polynucleotide of this disclosure. For example, 1 to 10, 1 to 9, 1 to 8, 1 to 7, 1 to 6, 1 to 5, 1 to 4, 1 to 3, 2, or 1 miRNA binding sites can be engineered into the 3′UTR of a polynucleotide of this disclosure. In one embodiment, miRNA binding sites incorporated into a polynucleotide of this disclosure can be the same or can be different miRNA sites. A combination of different miRNA binding sites incorporated into a polynucleotide of this disclosure can include combinations in which more than one copy of any of the different miRNA sites are incorporated. In another embodiment, miRNA binding sites incorporated into a polynucleotide of this disclosure can target the same or different tissues in the body. As a non-limiting example, through the introduction of tissue-, cell-type-, or disease-specific miRNA binding sites in the 3′-UTR of a polynucleotide of this disclosure, the degree of expression in specific cell types (e.g., myeloid cells, endothelial cells, etc.) can be reduced. In one embodiment, a miRNA binding site can be engineered near the 5′ terminus of the 3′UTR, about halfway between the 5′ terminus and 3′ terminus of the 3′UTR and/or near the 3′ terminus of the 3′ UTR in a polynucleotide of this disclosure. As a non-limiting example, a miRNA binding site can be engineered near the 5′ terminus of the 3′UTR and about halfway between the 5′ terminus and 3′ terminus of the 3′UTR. As another non- limiting example, a miRNA binding site can be engineered near the 3′ terminus of the 3′UTR and about halfway between the 5′ terminus and 3′ terminus of the 3′ UTR. As yet another non-limiting example, a miRNA binding site can be engineered near the 5′ terminus of the 3′ UTR and near the 3′ terminus of the 3′ UTR. In another embodiment, a 3′UTR can comprise 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 miRNA binding sites. The miRNA binding sites can be complementary to a miRNA, miRNA seed sequence, and/or miRNA sequences flanking the seed sequence. In some embodiments, the expression of a polynucleotide of this disclosure can be controlled by incorporating at least one sensor sequence in the polynucleotide and Formulating the polynucleotide for administration. As a non-limiting example, a polynucleotide of this disclosure can be targeted to a tissue or cell by incorporating a miRNA binding site and Formulating the polynucleotide in a lipid nanoparticle comprising an ionizable amino lipid, including any of the lipids described herein. A polynucleotide of this disclosure can be engineered for more targeted expression in specific tissues, cell types, or biological conditions based on the expression patterns of miRNAs in the different tissues, cell types, or biological conditions. Through introduction of tissue-specific miRNA binding sites, a polynucleotide of this disclosure can be designed for optimal protein expression in a tissue or cell, or in the context of a biological condition. In some embodiments, a polynucleotide of this disclosure can be designed to incorporate miRNA binding sites that either have 100% identity to known miRNA seed sequences or have less than 100% identity to miRNA seed sequences. In some embodiments, a polynucleotide of this disclosure can be designed to incorporate miRNA binding sites that have at least: 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to known miRNA seed sequences. The miRNA seed sequence can be partially mutated to decrease miRNA binding affinity and as such result in reduced downmodulation of the polynucleotide. In essence, the degree of match or mis-match between the miRNA binding site and the miRNA seed can act as a rheostat to more finely tune the ability of the miRNA to modulate protein expression. In addition, mutation in the non-seed region of a miRNA binding site can also impact the ability of a miRNA to modulate protein expression. In one embodiment, a miRNA sequence can be incorporated into the loop of a stem loop. In another embodiment, a miRNA seed sequence can be incorporated in the loop of a stem loop and a miRNA binding site can be incorporated into the 5′ or 3′ stem of the stem loop. In one embodiment the miRNA sequence in the 5′ UTR can be used to stabilize a polynucleotide of this disclosure described herein. In another embodiment, a miRNA sequence in the 5′ UTR of a polynucleotide of this disclosure can be used to decrease the accessibility of the site of translation initiation such as, but not limited to a start codon. See, e.g., Matsuda et al., PLoS One.201011(5):e15057; incorporated herein by reference in its entirety, which used antisense locked nucleic acid (LNA) oligonucleotides and exon-junction complexes (EJCs) around a start codon (-4 to +37 where the A of the AUG codons is +1) in order to decrease the accessibility to the first start codon (AUG). Matsuda showed that altering the sequence around the start codon with an LNA or EJC affected the efficiency, length and structural stability of a polynucleotide. A polynucleotide of this disclosure can comprise a miRNA sequence, instead of the LNA or EJC sequence described by Matsuda et al, near the site of translation initiation in order to decrease the accessibility to the site of translation initiation. The site of translation initiation can be prior to, after or within the miRNA sequence. As a non-limiting example, the site of translation initiation can be located within a miRNA sequence such as a seed sequence or binding site. In some embodiments, a polynucleotide of this disclosure can include at least one miRNA in order to dampen the antigen presentation by antigen presenting cells. The miRNA can be the complete miRNA sequence, the miRNA seed sequence, the miRNA sequence without the seed, or a combination thereof. As a non-limiting example, a miRNA incorporated into a polynucleotide of this disclosure can be specific to the hematopoietic system. As another non-limiting example, a miRNA incorporated into a polynucleotide of this disclosure to dampen antigen presentation is miR-142-3p. In some embodiments, a polynucleotide of this disclosure can include at least one miRNA in order to dampen expression of the encoded polypeptide in a tissue or cell of interest. As a non-limiting example a polynucleotide of this disclosure can include at least one miR-142-3p binding site, miR-142-3p seed sequence, miR-142-3p binding site without the seed, miR-142-5p binding site, miR-142-5p seed sequence, miR-142-5p binding site without the seed, miR-146 binding site, miR-146 seed sequence and/or miR-146 binding site without the seed sequence. In some embodiments, a polynucleotide of this disclosure can comprise at least one miRNA binding site in the 3′UTR in order to selectively degrade mRNA therapeutics in the immune cells to subdue unwanted immunogenic reactions caused by therapeutic delivery. As a non-limiting example, the miRNA binding site can make a polynucleotide of this disclosure more unstable in antigen presenting cells. Non-limiting examples of these miRNAs include miR-142-5p, miR-142-3p, miR-146a-5p, and miR-146-3p. In one embodiment, a polynucleotide of this disclosure comprises at least one miRNA sequence in a region of the polynucleotide that can interact with a RNA binding protein. In some embodiments, the polynucleotide of this disclosure (e.g., a RNA, e.g., an mRNA) comprising (i) a sequence-optimized nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, variant thereof, or a fusion protein comprising the same) and (ii) a miRNA binding site (e.g., a miRNA binding site that binds to miR-142) and/or a miRNA binding site that binds to miR-126. 14. Regions having a 5′ Cap The disclosure also includes a polynucleotide that comprises both a 5′ Cap and a polynucleotide of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide to be expressed). The 5′ cap structure of a natural mRNA is involved in nuclear export, increasing mRNA stability and binds the mRNA Cap Binding Protein (CBP), which is responsible for mRNA stability in the cell and translation competency through the association of CBP with poly(A) binding protein to form the mature cyclic mRNA species. The cap further assists the removal of 5′ proximal introns during mRNA splicing. Endogenous mRNA molecules can be 5′-end capped generating a 5′-ppp-5′- triphosphate linkage between a terminal guanosine cap residue and the 5′-terminal transcribed sense nucleotide of the mRNA molecule. This 5′-guanylate cap can then be methylated to generate an N7-methyl-guanylate residue. The ribose sugars of the terminal and/or anteterminal transcribed nucleotides of the 5′ end of the mRNA can optionally also be 2′-O- methylated.5′-decapping through hydrolysis and cleavage of the guanylate cap structure can target a nucleic acid molecule, such as an mRNA molecule, for degradation. In some embodiments, the polynucleotides of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide) incorporate a cap moiety. In some embodiments, polynucleotides of the present invention comprise a non- hydrolyzable cap structure preventing decapping and thus increasing mRNA half-life. Because cap structure hydrolysis requires cleavage of 5′-ppp-5′ phosphorodiester linkages, modified nucleotides can be used during the capping reaction. For example, a Vaccinia Capping Enzyme from New England Biolabs (Ipswich, MA) can be used with α-thio- guanosine nucleotides according to the manufacturer's instructions to create a phosphorothioate linkage in the 5′-ppp-5′ cap. Additional modified guanosine nucleotides can be used such as α-methyl-phosphonate and seleno-phosphate nucleotides. Additional modifications include, but are not limited to, 2′-O-methylation of the ribose sugars of 5′-terminal and/or 5′-anteterminal nucleotides of the polynucleotide (as mentioned above) on the 2′-hydroxyl group of the sugar ring. Multiple distinct 5′-cap structures can be used to generate the 5′-cap of a nucleic acid molecule, such as a polynucleotide that functions as an mRNA molecule. Cap analogs, which herein are also referred to as synthetic cap analogs, chemical caps, chemical cap analogs, or structural or functional cap analogs, differ from natural (i.e., endogenous, wild-type or physiological) 5′- caps in their chemical structure, while retaining cap function. Cap analogs can be chemically (i.e., non-enzymatically) or enzymatically synthesized and/or linked to the polynucleotides of this disclosure. For example, the Anti-Reverse Cap Analog (ARCA) cap contains two guanines linked by a 5′-5′-triphosphate group, wherein one guanine contains an N7 methyl group as well as a 3′-O-methyl group (i.e., N7,3′-O-dimethyl-guanosine-5′-triphosphate-5′-guanosine (m7G- 3′mppp-G; which can equivalently be designated 3′ O-Me-m7G(5′)ppp(5′)G). The 3′-O atom of the other, unmodified, guanine becomes linked to the 5′-terminal nucleotide of the capped polynucleotide. The N7- and 3′-O-methlyated guanine provides the terminal moiety of the capped polynucleotide. Another exemplary cap is mCAP, which is similar to ARCA but has a 2′-O-methyl group on guanosine (i.e., N7,2′-O-dimethyl-guanosine-5′-triphosphate-5′-guanosine, m7Gm- ppp-G). Another exemplary cap is m7G-ppp-Gm-A (i.e., N7,guanosine-5′-triphosphate-2′-O- dimethyl-guanosine-adenosine). In some embodiments, the cap is a dinucleotide cap analog. As a non-limiting example, the dinucleotide cap analog can be modified at different phosphate positions with a boranophosphate group or a phosphoroselenoate group such as the dinucleotide cap analogs described in U.S. Patent No. US 8,519,110, the contents of which are herein incorporated by reference in its entirety. In another embodiment, the cap is a cap analog is a N7-(4-chlorophenoxyethyl) substituted dinucleotide form of a cap analog known in the art and/or described herein. Non- limiting examples of a N7-(4-chlorophenoxyethyl) substituted dinucleotide form of a cap analog include a N7-(4-chlorophenoxyethyl)-G(5′)ppp(5′)G and a N7-(4- chlorophenoxyethyl)-m3′-OG(5′)ppp(5′)G cap analog (See, e.g., the various cap analogs and the methods of synthesizing cap analogs described in Kore et al. Bioorganic & Medicinal Chemistry 201321:4570-4574; the contents of which are herein incorporated by reference in its entirety). In another embodiment, a cap analog of the present invention is a 4- chloro/bromophenoxyethyl analog. Polynucleotides of this disclosure can also be capped post-manufacture (whether IVT or chemical synthesis), using enzymes, in order to generate more authentic 5′-cap structures. As used herein, the phrase "more authentic" refers to a feature that closely mirrors or mimics, either structurally or functionally, an endogenous or wild type feature. That is, a "more authentic" feature is better representative of an endogenous, wild-type, natural or physiological cellular function and/or structure as compared to synthetic features or analogs, etc., of the prior art, or which outperforms the corresponding endogenous, wild-type, natural or physiological feature in one or more respects. Non-limiting examples of more authentic 5′cap structures of the present invention are those that, among other things, have enhanced binding of cap binding proteins, increased half-life, reduced susceptibility to 5′ endonucleases and/or reduced 5′decapping, as compared to synthetic 5′cap structures known in the art (or to a wild-type, natural or physiological 5′cap structure). For example, recombinant Vaccinia Virus Capping Enzyme and recombinant 2′-O-methyltransferase enzyme can create a canonical 5′-5′-triphosphate linkage between the 5′-terminal nucleotide of a polynucleotide and a guanine cap nucleotide wherein the cap guanine contains an N7 methylation and the 5′- terminal nucleotide of the mRNA contains a 2′-O-methyl. Such a structure is termed the Cap1 structure. This cap results in a higher translational-competency and cellular stability and a reduced activation of cellular pro-inflammatory cytokines, as compared, e.g., to other 5′cap analog structures known in the art. Cap structures include, but are not limited to, 7mG(5′)ppp(5′)N1pN2p (cap 0), 7mG(5′)ppp(5′)N1mpNp (cap 1), and 7mG(5′)- ppp(5′)N1mpN2mp (cap 2). As a non-limiting example, capping chimeric polynucleotides post-manufacture can be more efficient as nearly 100% of the chimeric polynucleotides can be capped. This is in contrast to ~80% when a cap analog is linked to a chimeric polynucleotide in the course of an in vitro transcription reaction. According to the present invention, 5′ terminal caps can include endogenous caps or cap analogs. According to the present invention, a 5′ terminal cap can comprise a guanine analog. Useful guanine analogs include, but are not limited to, inosine, N1-methyl-guanosine, 2′fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino-guanosine, LNA- guanosine, and 2-azido-guanosine. Also provided herein are exemplary caps including those that can be used in co- transcriptional capping methods for ribonucleic acid (RNA) synthesis, using RNA polymerase, e.g., wild type RNA polymerase or variants thereof, e.g., such as those variants described herein. In one embodiment, caps can be added when RNA is produced in a “one- pot” reaction, without the need for a separate capping reaction. Thus, the methods, in some embodiments, comprise reacting a polynucleotide template with an RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript. As used here the term “cap” includes the inverted G nucleotide and can comprise one or more additional nucleotides 3’ of the inverted G nucleotide, e.g., 1, 2, 3, or more nucleotides 3’ of the inverted G nucleotide and 5’ to the 5’ UTR, e.g., a 5’ UTR described herein. Exemplary caps comprise a sequence of GG, GA, or GGA, wherein the underlined, italicized G is an in inverted G nucleotide followed by a 5’-5’-triphosphate group. In one embodiment, a cap comprises a compound of formula (C-I)
In some embodiments, a miRNA binding site is inserted in the polynucleotide of this disclosure in any position of the polynucleotide (e.g., the 5′ UTR and/or 3′ UTR). In some embodiments, the 5′ UTR comprises a miRNA binding site. In some embodiments, the 3′ UTR comprises a miRNA binding site. In some embodiments, the 5′ UTR and the 3′ UTR comprise a miRNA binding site. The insertion site in the polynucleotide can be anywhere in the polynucleotide as long as the insertion of the miRNA binding site in the polynucleotide does not interfere with the translation of a functional polypeptide in the absence of the corresponding miRNA; and in the presence of the miRNA, the insertion of the miRNA binding site in the polynucleotide and the binding of the miRNA binding site to the corresponding miRNA are capable of degrading the polynucleotide or preventing the translation of the polynucleotide. In some embodiments, a miRNA binding site is inserted in at least about 30 nucleotides downstream from the stop codon of an ORF in a polynucleotide of this disclosure comprising the ORF. In some embodiments, a miRNA binding site is inserted in at least about 10 nucleotides, at least about 15 nucleotides, at least about 20 nucleotides, at least about 25 nucleotides, at least about 30 nucleotides, at least about 35 nucleotides, at least about 40 nucleotides, at least about 45 nucleotides, at least about 50 nucleotides, at least about 55 nucleotides, at least about 60 nucleotides, at least about 65 nucleotides, at least about 70 nucleotides, at least about 75 nucleotides, at least about 80 nucleotides, at least about 85 nucleotides, at least about 90 nucleotides, at least about 95 nucleotides, or at least about 100 nucleotides downstream from the stop codon of an ORF in a polynucleotide of this disclosure. In some embodiments, a miRNA binding site is inserted in about 10 nucleotides to about 100 nucleotides, about 20 nucleotides to about 90 nucleotides, about 30 nucleotides to about 80 nucleotides, about 40 nucleotides to about 70 nucleotides, about 50 nucleotides to about 60 nucleotides, about 45 nucleotides to about 65 nucleotides downstream from the stop codon of an ORF in a polynucleotide of this disclosure. In some embodiments, a miRNA binding site is inserted within the 3′ UTR immediately following the stop codon of the coding region within the polynucleotide of this disclosure, e.g., mRNA. In some embodiments, if there are multiple copies of a stop codon in the construct, a miRNA binding site is inserted immediately following the final stop codon. In some embodiments, a miRNA binding site is inserted further downstream of the stop codon, in which case there are 3′ UTR bases between the stop codon and the miR binding site(s). In some embodiments, one or more miRNA binding sites can be positioned within the 5′ UTR at one or more possible insertion sites. In one embodiment, a codon optimized open reading frame encoding a polypeptide of interest comprises a stop codon and the at least one microRNA binding site is located within the 3′ UTR 1-100 nucleotides after the stop codon. In one embodiment, the codon optimized open reading frame encoding the polypeptide of interest comprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR 30-50 nucleotides after the stop codon. In another embodiment, the codon optimized open reading frame encoding the polypeptide of interest comprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR at least 50 nucleotides after the stop codon. In other embodiments, the codon optimized open reading frame encoding the polypeptide of interest comprises a stop codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 3′ UTR immediately after the stop codon, or within the 3′ UTR 15-20 nucleotides after the stop codon or within the 3′ UTR 70-80 nucleotides after the stop codon. In other embodiments, the 3′ UTR comprises more than one miRNA binding site (e.g., 2-4 miRNA binding sites), wherein there can be a spacer region (e.g., of 10-100, 20-70 or 30-50 nucleotides in length) between each miRNA binding site. In another embodiment, the 3′ UTR comprises a spacer region between the end of the miRNA binding site(s) and the poly A tail nucleotides. For example, a spacer region of 10-100, 20-70 or 30-50 nucleotides in length can be situated between the end of the miRNA binding site(s) and the beginning of the poly A tail. In one embodiment, a codon optimized open reading frame encoding a polypeptide of interest comprises a start codon and the at least one microRNA binding site is located within the 5′ UTR 1-100 nucleotides before (upstream of) the start codon. In one embodiment, the codon optimized open reading frame encoding the polypeptide of interest comprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR 10-50 nucleotides before (upstream of) the start codon. In another embodiment, the codon optimized open reading frame encoding the polypeptide of interest comprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR at least 25 nucleotides before (upstream of) the start codon. In other embodiments, the codon optimized open reading frame encoding the polypeptide of interest comprises a start codon and the at least one microRNA binding site for a miR expressed in immune cells is located within the 5′ UTR immediately before the start codon, or within the 5′ UTR 15-20 nucleotides before the start codon or within the 5′ UTR 70-80 nucleotides before the start codon. In other embodiments, the 5′ UTR comprises more than one miRNA binding site (e.g., 2-4 miRNA binding sites), wherein there can be a spacer region (e.g., of 10-100, 20-70 or 30-50 nucleotides in length) between each miRNA binding site. In one embodiment, the 3′ UTR comprises more than one stop codon, wherein at least one miRNA binding site is positioned downstream of the stop codons. For example, a 3′ UTR can comprise 1, 2 or 3 stop codons. Non-limiting examples of triple stop codons that can be used include: UGAUAAUAG (SEQ ID NO:182), UGAUAGUAA (SEQ ID NO:183), UAAUGAUAG (SEQ ID NO:184), UGAUAAUAA (SEQ ID NO:185), UGAUAGUAG (SEQ ID NO:186), UAAUGAUGA (SEQ ID NO:187), UAAUAGUAG (SEQ ID NO:188), UGAUGAUGA (SEQ ID NO:179), UAAUAAUAA (SEQ ID NO:180), and UAGUAGUAG (SEQ ID NO:181). Within a 3′ UTR, for example, 1, 2, 3 or 4 miRNA binding sites, e.g., miR-142-3p binding sites, can be positioned immediately adjacent to the stop codon(s) or at any number of nucleotides downstream of the final stop codon. When the 3′ UTR comprises multiple miRNA binding sites, these binding sites can be positioned directly next to each other in the construct (i.e., one after the other) or, alternatively, spacer nucleotides can be positioned between each binding site. In one embodiment, the 3′ UTR comprises three stop codons with a single miR-142- 3p binding site located downstream of the 3rd stop codon. In one embodiment, the polynucleotide of this disclosure comprises a 5′ UTR, a codon optimized open reading frame encoding a polypeptide of interest, a 3′ UTR comprising the at least one miRNA binding site for a miR expressed in immune cells, and a 3′ tailing region of linked nucleosides. In various embodiments, the 3′ UTR comprises 1-4, at least two, one, two, three or four miRNA binding sites for miRs expressed in immune cells, preferably abundantly or preferentially expressed in immune cells. In one embodiment, the at least one miRNA expressed in immune cells is a miR-142- 3p microRNA binding site. In one embodiment, the miR-142-3p microRNA binding site comprises the sequence shown in SEQ ID NO: 174. In one embodiment, the at least one miRNA expressed in immune cells is a miR-126 microRNA binding site. In one embodiment, the miR-126 binding site is a miR-126-3p binding site. In one embodiment, the miR-126-3p microRNA binding site comprises the sequence shown in SEQ ID NO: 152. Non-limiting exemplary sequences for miRs to which a microRNA binding site(s) of the disclosure can bind include the following: miR-142-3p (SEQ ID NO: 173), miR-142-5p (SEQ ID NO: 175), miR-146-3p (SEQ ID NO: 155), miR-146-5p (SEQ ID NO: 156), miR- 155-3p (SEQ ID NO: 157), miR-155-5p (SEQ ID NO: 158), miR-126-3p (SEQ ID NO: 151), miR-126-5p (SEQ ID NO: 153), miR-16-3p (SEQ ID NO: 159), miR-16-5p (SEQ ID NO: 160), miR-21-3p (SEQ ID NO: 161), miR-21-5p (SEQ ID NO: 162), miR-223-3p (SEQ ID NO: 163), miR-223-5p (SEQ ID NO: 164), miR-24-3p (SEQ ID NO: 165), miR-24-5p (SEQ ID NO: 166), miR-27-3p (SEQ ID NO: 167) and miR-27-5p (SEQ ID NO: 168). Other suitable miR sequences expressed in immune cells (e.g., abundantly or preferentially expressed in immune cells) are known and available in the art, for example at the University of Manchester’s microRNA database, miRBase. Sites that bind any of the aforementioned miRs can be designed based on Watson-Crick complementarity to the miR, typically 100% complementarity to the miR, and inserted into an mRNA construct of the disclosure as described herein. In another embodiment, a polynucleotide of the present invention (e.g., and mRNA, e.g., the 3′ UTR thereof) can comprise at least one miRNA bindingsite to thereby reduce or inhibit accelerated blood clearance, for example by reducing or inhibiting production of IgMs, e.g., against PEG, by B cells and/or reducing or inhibiting proliferation and/or activation of pDCs, and can comprise at least one miRNA bindingsite for modulating tissue expression of an encoded protein of interest. miRNA gene regulation can be influenced by the sequence surrounding the miRNA such as, but not limited to, the species of the surrounding sequence, the type of sequence (e.g., heterologous, homologous, exogenous, endogenous, or artificial), regulatory elements in the surrounding sequence and/or structural elements in the surrounding sequence. The miRNA can be influenced by the 5′UTR and/or 3′UTR. As a non-limiting example, a non- human 3′UTR can increase the regulatory effect of the miRNA sequence on the expression of a polypeptide of interest compared to a human 3′ UTR of the same sequence type. In one embodiment, other regulatory elements and/or structural elements of the 5′ UTR can influence miRNA mediated gene regulation. One example of a regulatory element and/or structural element is a structured IRES (Internal Ribosome Entry Site) in the 5′ UTR, which is necessary for the binding of translational elongation factors to initiate protein translation. EIF4A2 binding to this secondarily structured element in the 5′-UTR is necessary for miRNA mediated gene expression (Meijer HA et al., Science, 2013, 340, 82-85, herein incorporated by reference in its entirety). The polynucleotides of this disclosure can further include this structured 5′ UTR in order to enhance microRNA mediated gene regulation. At least one miRNA binding site can be engineered into the 3′ UTR of a polynucleotide of this disclosure. In this context, at least two, at least three, at least four, at least five, at least six, at least seven, at least eight, at least nine, at least ten, or more miRNA binding sites can be engineered into a 3′ UTR of a polynucleotide of this disclosure. For example, 1 to 10, 1 to 9, 1 to 8, 1 to 7, 1 to 6, 1 to 5, 1 to 4, 1 to 3, 2, or 1 miRNA binding sites can be engineered into the 3′UTR of a polynucleotide of this disclosure. In one embodiment, miRNA binding sites incorporated into a polynucleotide of this disclosure can be the same or can be different miRNA sites. A combination of different miRNA binding sites incorporated into a polynucleotide of this disclosure can include combinations in which more than one copy of any of the different miRNA sites are incorporated. In another embodiment, miRNA binding sites incorporated into a polynucleotide of this disclosure can target the same or different tissues in the body. As a non-limiting example, through the introduction of tissue-, cell-type-, or disease-specific miRNA binding sites in the 3′-UTR of a polynucleotide of this disclosure, the degree of expression in specific cell types (e.g., myeloid cells, endothelial cells, etc.) can be reduced. In one embodiment, a miRNA binding site can be engineered near the 5′ terminus of the 3′UTR, about halfway between the 5′ terminus and 3′ terminus of the 3′UTR and/or near the 3′ terminus of the 3′ UTR in a polynucleotide of this disclosure. As a non-limiting example, a miRNA binding site can be engineered near the 5′ terminus of the 3′UTR and about halfway between the 5′ terminus and 3′ terminus of the 3′UTR. As another non- limiting example, a miRNA binding site can be engineered near the 3′ terminus of the 3′UTR and about halfway between the 5′ terminus and 3′ terminus of the 3′ UTR. As yet another non-limiting example, a miRNA binding site can be engineered near the 5′ terminus of the 3′ UTR and near the 3′ terminus of the 3′ UTR. In another embodiment, a 3′UTR can comprise 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 miRNA binding sites. The miRNA binding sites can be complementary to a miRNA, miRNA seed sequence, and/or miRNA sequences flanking the seed sequence. In some embodiments, the expression of a polynucleotide of this disclosure can be controlled by incorporating at least one sensor sequence in the polynucleotide and Formulating the polynucleotide for administration. As a non-limiting example, a polynucleotide of this disclosure can be targeted to a tissue or cell by incorporating a miRNA binding site and Formulating the polynucleotide in a lipid nanoparticle comprising an ionizable amino lipid, including any of the lipids described herein. A polynucleotide of this disclosure can be engineered for more targeted expression in specific tissues, cell types, or biological conditions based on the expression patterns of miRNAs in the different tissues, cell types, or biological conditions. Through introduction of tissue-specific miRNA binding sites, a polynucleotide of this disclosure can be designed for optimal protein expression in a tissue or cell, or in the context of a biological condition. In some embodiments, a polynucleotide of this disclosure can be designed to incorporate miRNA binding sites that either have 100% identity to known miRNA seed sequences or have less than 100% identity to miRNA seed sequences. In some embodiments, a polynucleotide of this disclosure can be designed to incorporate miRNA binding sites that have at least: 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to known miRNA seed sequences. The miRNA seed sequence can be partially mutated to decrease miRNA binding affinity and as such result in reduced downmodulation of the polynucleotide. In essence, the degree of match or mis-match between the miRNA binding site and the miRNA seed can act as a rheostat to more finely tune the ability of the miRNA to modulate protein expression. In addition, mutation in the non-seed region of a miRNA binding site can also impact the ability of a miRNA to modulate protein expression. In one embodiment, a miRNA sequence can be incorporated into the loop of a stem loop. In another embodiment, a miRNA seed sequence can be incorporated in the loop of a stem loop and a miRNA binding site can be incorporated into the 5′ or 3′ stem of the stem loop. In one embodiment the miRNA sequence in the 5′ UTR can be used to stabilize a polynucleotide of this disclosure described herein. In another embodiment, a miRNA sequence in the 5′ UTR of a polynucleotide of this disclosure can be used to decrease the accessibility of the site of translation initiation such as, but not limited to a start codon. See, e.g., Matsuda et al., PLoS One.201011(5):e15057; incorporated herein by reference in its entirety, which used antisense locked nucleic acid (LNA) oligonucleotides and exon-junction complexes (EJCs) around a start codon (-4 to +37 where the A of the AUG codons is +1) in order to decrease the accessibility to the first start codon (AUG). Matsuda showed that altering the sequence around the start codon with an LNA or EJC affected the efficiency, length and structural stability of a polynucleotide. A polynucleotide of this disclosure can comprise a miRNA sequence, instead of the LNA or EJC sequence described by Matsuda et al, near the site of translation initiation in order to decrease the accessibility to the site of translation initiation. The site of translation initiation can be prior to, after or within the miRNA sequence. As a non-limiting example, the site of translation initiation can be located within a miRNA sequence such as a seed sequence or binding site. In some embodiments, a polynucleotide of this disclosure can include at least one miRNA in order to dampen the antigen presentation by antigen presenting cells. The miRNA can be the complete miRNA sequence, the miRNA seed sequence, the miRNA sequence without the seed, or a combination thereof. As a non-limiting example, a miRNA incorporated into a polynucleotide of this disclosure can be specific to the hematopoietic system. As another non-limiting example, a miRNA incorporated into a polynucleotide of this disclosure to dampen antigen presentation is miR-142-3p. In some embodiments, a polynucleotide of this disclosure can include at least one miRNA in order to dampen expression of the encoded polypeptide in a tissue or cell of interest. As a non-limiting example a polynucleotide of this disclosure can include at least one miR-142-3p binding site, miR-142-3p seed sequence, miR-142-3p binding site without the seed, miR-142-5p binding site, miR-142-5p seed sequence, miR-142-5p binding site without the seed, miR-146 binding site, miR-146 seed sequence and/or miR-146 binding site without the seed sequence. In some embodiments, a polynucleotide of this disclosure can comprise at least one miRNA binding site in the 3′UTR in order to selectively degrade mRNA therapeutics in the immune cells to subdue unwanted immunogenic reactions caused by therapeutic delivery. As a non-limiting example, the miRNA binding site can make a polynucleotide of this disclosure more unstable in antigen presenting cells. Non-limiting examples of these miRNAs include miR-142-5p, miR-142-3p, miR-146a-5p, and miR-146-3p. In one embodiment, a polynucleotide of this disclosure comprises at least one miRNA sequence in a region of the polynucleotide that can interact with a RNA binding protein. In some embodiments, the polynucleotide of this disclosure (e.g., a RNA, e.g., an mRNA) comprising (i) a sequence-optimized nucleotide sequence (e.g., an ORF) encoding an IL-22 polypeptide (e.g., the wild-type sequence, functional fragment, variant thereof, or a fusion protein comprising the same) and (ii) a miRNA binding site (e.g., a miRNA binding site that binds to miR-142) and/or a miRNA binding site that binds to miR-126. 14. Regions having a 5′ Cap The disclosure also includes a polynucleotide that comprises both a 5′ Cap and a polynucleotide of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide to be expressed). The 5′ cap structure of a natural mRNA is involved in nuclear export, increasing mRNA stability and binds the mRNA Cap Binding Protein (CBP), which is responsible for mRNA stability in the cell and translation competency through the association of CBP with poly(A) binding protein to form the mature cyclic mRNA species. The cap further assists the removal of 5′ proximal introns during mRNA splicing. Endogenous mRNA molecules can be 5′-end capped generating a 5′-ppp-5′- triphosphate linkage between a terminal guanosine cap residue and the 5′-terminal transcribed sense nucleotide of the mRNA molecule. This 5′-guanylate cap can then be methylated to generate an N7-methyl-guanylate residue. The ribose sugars of the terminal and/or anteterminal transcribed nucleotides of the 5′ end of the mRNA can optionally also be 2′-O- methylated.5′-decapping through hydrolysis and cleavage of the guanylate cap structure can target a nucleic acid molecule, such as an mRNA molecule, for degradation. In some embodiments, the polynucleotides of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding an IL-22 polypeptide) incorporate a cap moiety. In some embodiments, polynucleotides of the present invention comprise a non- hydrolyzable cap structure preventing decapping and thus increasing mRNA half-life. Because cap structure hydrolysis requires cleavage of 5′-ppp-5′ phosphorodiester linkages, modified nucleotides can be used during the capping reaction. For example, a Vaccinia Capping Enzyme from New England Biolabs (Ipswich, MA) can be used with α-thio- guanosine nucleotides according to the manufacturer's instructions to create a phosphorothioate linkage in the 5′-ppp-5′ cap. Additional modified guanosine nucleotides can be used such as α-methyl-phosphonate and seleno-phosphate nucleotides. Additional modifications include, but are not limited to, 2′-O-methylation of the ribose sugars of 5′-terminal and/or 5′-anteterminal nucleotides of the polynucleotide (as mentioned above) on the 2′-hydroxyl group of the sugar ring. Multiple distinct 5′-cap structures can be used to generate the 5′-cap of a nucleic acid molecule, such as a polynucleotide that functions as an mRNA molecule. Cap analogs, which herein are also referred to as synthetic cap analogs, chemical caps, chemical cap analogs, or structural or functional cap analogs, differ from natural (i.e., endogenous, wild-type or physiological) 5′- caps in their chemical structure, while retaining cap function. Cap analogs can be chemically (i.e., non-enzymatically) or enzymatically synthesized and/or linked to the polynucleotides of this disclosure. For example, the Anti-Reverse Cap Analog (ARCA) cap contains two guanines linked by a 5′-5′-triphosphate group, wherein one guanine contains an N7 methyl group as well as a 3′-O-methyl group (i.e., N7,3′-O-dimethyl-guanosine-5′-triphosphate-5′-guanosine (m7G- 3′mppp-G; which can equivalently be designated 3′ O-Me-m7G(5′)ppp(5′)G). The 3′-O atom of the other, unmodified, guanine becomes linked to the 5′-terminal nucleotide of the capped polynucleotide. The N7- and 3′-O-methlyated guanine provides the terminal moiety of the capped polynucleotide. Another exemplary cap is mCAP, which is similar to ARCA but has a 2′-O-methyl group on guanosine (i.e., N7,2′-O-dimethyl-guanosine-5′-triphosphate-5′-guanosine, m7Gm- ppp-G). Another exemplary cap is m7G-ppp-Gm-A (i.e., N7,guanosine-5′-triphosphate-2′-O- dimethyl-guanosine-adenosine). In some embodiments, the cap is a dinucleotide cap analog. As a non-limiting example, the dinucleotide cap analog can be modified at different phosphate positions with a boranophosphate group or a phosphoroselenoate group such as the dinucleotide cap analogs described in U.S. Patent No. US 8,519,110, the contents of which are herein incorporated by reference in its entirety. In another embodiment, the cap is a cap analog is a N7-(4-chlorophenoxyethyl) substituted dinucleotide form of a cap analog known in the art and/or described herein. Non- limiting examples of a N7-(4-chlorophenoxyethyl) substituted dinucleotide form of a cap analog include a N7-(4-chlorophenoxyethyl)-G(5′)ppp(5′)G and a N7-(4- chlorophenoxyethyl)-m3′-OG(5′)ppp(5′)G cap analog (See, e.g., the various cap analogs and the methods of synthesizing cap analogs described in Kore et al. Bioorganic & Medicinal Chemistry 201321:4570-4574; the contents of which are herein incorporated by reference in its entirety). In another embodiment, a cap analog of the present invention is a 4- chloro/bromophenoxyethyl analog. Polynucleotides of this disclosure can also be capped post-manufacture (whether IVT or chemical synthesis), using enzymes, in order to generate more authentic 5′-cap structures. As used herein, the phrase "more authentic" refers to a feature that closely mirrors or mimics, either structurally or functionally, an endogenous or wild type feature. That is, a "more authentic" feature is better representative of an endogenous, wild-type, natural or physiological cellular function and/or structure as compared to synthetic features or analogs, etc., of the prior art, or which outperforms the corresponding endogenous, wild-type, natural or physiological feature in one or more respects. Non-limiting examples of more authentic 5′cap structures of the present invention are those that, among other things, have enhanced binding of cap binding proteins, increased half-life, reduced susceptibility to 5′ endonucleases and/or reduced 5′decapping, as compared to synthetic 5′cap structures known in the art (or to a wild-type, natural or physiological 5′cap structure). For example, recombinant Vaccinia Virus Capping Enzyme and recombinant 2′-O-methyltransferase enzyme can create a canonical 5′-5′-triphosphate linkage between the 5′-terminal nucleotide of a polynucleotide and a guanine cap nucleotide wherein the cap guanine contains an N7 methylation and the 5′- terminal nucleotide of the mRNA contains a 2′-O-methyl. Such a structure is termed the Cap1 structure. This cap results in a higher translational-competency and cellular stability and a reduced activation of cellular pro-inflammatory cytokines, as compared, e.g., to other 5′cap analog structures known in the art. Cap structures include, but are not limited to, 7mG(5′)ppp(5′)N1pN2p (cap 0), 7mG(5′)ppp(5′)N1mpNp (cap 1), and 7mG(5′)- ppp(5′)N1mpN2mp (cap 2). As a non-limiting example, capping chimeric polynucleotides post-manufacture can be more efficient as nearly 100% of the chimeric polynucleotides can be capped. This is in contrast to ~80% when a cap analog is linked to a chimeric polynucleotide in the course of an in vitro transcription reaction. According to the present invention, 5′ terminal caps can include endogenous caps or cap analogs. According to the present invention, a 5′ terminal cap can comprise a guanine analog. Useful guanine analogs include, but are not limited to, inosine, N1-methyl-guanosine, 2′fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino-guanosine, LNA- guanosine, and 2-azido-guanosine. Also provided herein are exemplary caps including those that can be used in co- transcriptional capping methods for ribonucleic acid (RNA) synthesis, using RNA polymerase, e.g., wild type RNA polymerase or variants thereof, e.g., such as those variants described herein. In one embodiment, caps can be added when RNA is produced in a “one- pot” reaction, without the need for a separate capping reaction. Thus, the methods, in some embodiments, comprise reacting a polynucleotide template with an RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript. As used here the term “cap” includes the inverted G nucleotide and can comprise one or more additional nucleotides 3’ of the inverted G nucleotide, e.g., 1, 2, 3, or more nucleotides 3’ of the inverted G nucleotide and 5’ to the 5’ UTR, e.g., a 5’ UTR described herein. Exemplary caps comprise a sequence of GG, GA, or GGA, wherein the underlined, italicized G is an in inverted G nucleotide followed by a 5’-5’-triphosphate group. In one embodiment, a cap comprises a compound of formula (C-I)









































Table 13. Tested mRNA constructs EXAMPLE 10: In vitro validation of expression and bioactivity of human IL-22 mRNA constructs The present Example illustrates that human IL-22 mRNA constructs express and are bioactive in vitro. To assay the expression of various human IL-22 mRNA constructs, HeLa cells were transfected with 500 ng of one of the IL-22 mRNA constructs listed below in Table 14. HeLa cell culture medium was changed 6 hours after the transfection. The conditioned media was collected 24 hours after the transfection, and IL22 concentrations were measured by ELISA. Each human IL-22 construct tested expressed in HeLa cells (Fig.15). To assay the bioactivity of human IL-22 mRNA constructs, HEK-Blue™ IL-22 cells plated at 50,000 cells/well in 96 well plates, and were treated with serially diluted samples of conditioned media from HeLa cells transfected with the mRNA constructs.24 hours after treatment, 20 µL of conditioned media was added to 180 uL QUANTI-Blue solution and incubated at 37ºC for 30 minutes, before absorbance reading at 655 nm on a Synergy HT2 plate reader. All human IL-22 constructs exhibited activation of the IL-22 signaling pathway as measured by the sample OD at 655 nm at the lowest concentrations of IL-22 construct protein tested (Fig.16). Human IL-22 constructs in which IL-22 was linked to the albumin domain antibody (adab) exhibited enhanced bioactivity compared to those in which IL-22 was linked to human serum albumin (HSA). Construct identities are summarized in the Table 14 below: Table 14. Tested mRNA construct identities
EXAMPLE 10: In vitro validation of expression and bioactivity of human IL-22 mRNA constructs The present Example illustrates that human IL-22 mRNA constructs express and are bioactive in vitro. To assay the expression of various human IL-22 mRNA constructs, HeLa cells were transfected with 500 ng of one of the IL-22 mRNA constructs listed below in Table 14. HeLa cell culture medium was changed 6 hours after the transfection. The conditioned media was collected 24 hours after the transfection, and IL22 concentrations were measured by ELISA. Each human IL-22 construct tested expressed in HeLa cells (Fig.15). To assay the bioactivity of human IL-22 mRNA constructs, HEK-Blue™ IL-22 cells plated at 50,000 cells/well in 96 well plates, and were treated with serially diluted samples of conditioned media from HeLa cells transfected with the mRNA constructs.24 hours after treatment, 20 µL of conditioned media was added to 180 uL QUANTI-Blue solution and incubated at 37ºC for 30 minutes, before absorbance reading at 655 nm on a Synergy HT2 plate reader. All human IL-22 constructs exhibited activation of the IL-22 signaling pathway as measured by the sample OD at 655 nm at the lowest concentrations of IL-22 construct protein tested (Fig.16). Human IL-22 constructs in which IL-22 was linked to the albumin domain antibody (adab) exhibited enhanced bioactivity compared to those in which IL-22 was linked to human serum albumin (HSA). Construct identities are summarized in the Table 14 below: Table 14. Tested mRNA construct identities EXAMPLE 11: In vivo probing of efficacy of IL-22 mRNA constructs metabolic disease signatures in aged mice The present Example illustrates experiments designed to test the efficiacy of IL-22 mRNA constructs on signatures of metabolic disease in aged mice. To further study the effects of IL-22 mRNA constructs on fibrosis regression, an IL- 22 mRNA construct is administered in older animals. C57BL/6NTac male mice are fed an AMLN diet or a chow diet for more than 40 weeks. Previous studies have shown that such a duration of ALMN diet results in robust fibrosis in mice. Due to the high inter-animal variability of aged mice, these studies carry the potential risk of inadvertently including animals that do not display late stage NASH/fibrosis, as there are currently no reliable biomarkers available. Accordingly, a survivable liver biopsy strategy is employed to randomize animals into treatment groups based on a molecular analysis of their NASH/fibrosis stage before treatment. Employing this approach allows for the tracking of the liver phenotype of the same mouse over time. This approach closely mimics human trial designs for NASH intervention, in which liver biopsies are obtained pre- and post- treatment. EXAMPLE 12: In vivo probing of efficacy of IL-22 mRNA constructs following IV or IM administration in aged mice The present Example illustrates the efficiacy of IL-22 mRNA constructs on signatures of metabolic disease in aged mice, following IV and IM administration of IL-22 mRNA constructs. Briefly, male C57BL/6Tac mice were fed an AMLN diet starting from 6 weeks of age through 40 weeks of age, for a total of 34 weeks of AMLN feeding. LNP-encapsulated MSA mRNA (n=10) or Construct #1 mRNA was administered at 0.5 mg/kg weekly IV (n=10) or 0.1 mg/kg IM (n=10) to mice fed the AMLN diet, with an untreated group of mice (n=8) and mice fed in normal chow diet (n=10) maintained in parallel. Body weight was determined at the indicated time points over the course of the study (Fig.17). Liver weight and epididymal white adipose tissue (eWAT) weight were measured 72 hours after the last dose in a 7-week treatment period. eWAT and eWAT (%) were analyzed together, and liver weight and liver weight (%) were also analyzed together with group and measures as fixed effects. Metabolic efficiency was measured as weight gain (in grams (g)) over MJ caloric intake. Alanine aminotransferase (ALT) levels (U/L), serum cholesterol levels, HDL cholesterol levels, LDL cholesterol levels, and triglyceride levels were determined at the end of the study in the serum collection. As shown in Fig.18, aged mice administered Construct #1 mRNA displayed a consistent and increasingly pronounced reduction in body weight starting immediately after the initial IV injection of 0.5 mpk of Construct #1 mRNA. This reduction persisted throughout the seven-week study. A significant decrease in body weight was also observed following IM administration of Construct #1 mRNA at a dose of 0.1 mpk. As shown in Fig. 19A-B, daily caloric intake was transiently reduced by approximately 50% in the IV- administered group, and was mildly reduced in the early weeks for the IM group. By the end of the study, caloric intake was identical across all groups. These data suggest that Construct #1 mRNA-dependent changes in bodyweight can be observed following administration by other routes besides IV or SC, including IM. As shown in Fig.19C, the total cumulative energy intake was lowest in lean mice, followed by the IV group, and then the IM group. As shown in Fig.20A-B, metabolic efficiency (weight gain per caloric intake over the course of the study) was significantly reduced in the IV group and 0.1 mpk IM group, with the IV group showing the most pronounced effect. These data suggest that, in addition to lowering food intake, Construct #1 mRNA increases energy expenditure. Weight gain was significantly reduced in the IV group and 0.1 mpk IM group compared to controls. As shown in Fig.21A-D, a reduction in liver weight following Construct #1 administration (by either IV or IM administration) was observed at the conclusion of the study. In addition, a decrease in epididymal white adipose tissue (eWAT) weight following both IV and IM treatments was observed. The liver weight/body weight ratio was unaffected by Construct #1, suggesting that the loss of liver weight was driven by the overall body weight loss and not due to the liver being preferentially targeted. Conversely, the eWAT weight/body weight ratio was significantly lower in the IV group, possibly indicating increased lipolysis. As shown in Fig.22A-D, all lipid readings such as cholesterol, HDL, LDL, and triglycerides showed reduced levels following the IV treatment via Construct #1 mRNA when compared to MSA mRNA and the untreated mice on a high cholesterol diet. As shown in Fig.22E, alanine aminotransferase (ALT) levels in these aged animals were notably elevated in those fed with the specified diet. However, they remarkably reached the same levels as the lean control following IV Construct #1 mRNA treatment. Intriguingly, even though less potent, the IM treatment also significantly reduced the ALT levels. Taken together, these results strongly suggest that even in the more severe disease state of aged animals, Construct #1 mRNA treatment by multiple administration routes can improve the condition of the animals. The effects observed with Construct #1 mRNA administered intramuscularly suggest the potential for further investigation with lower dosages and alternative routes of administration. * * * * It is to be appreciated that the Detailed Description section, and not the Summary and Abstract sections, is intended to be used to interpret the claims. The Summary and Abstract sections can set forth one or more but not all exemplary embodiments of the present invention as contemplated by the inventor(s), and thus, are not intended to limit the present invention and the appended claims in any way. The present invention has been described above with the aid of functional building blocks illustrating the implementation of specified functions and relationships thereof. The boundaries of these functional building blocks have been arbitrarily defined herein for the convenience of the description. Alternate boundaries can be defined so long as the specified functions and relationships thereof are appropriately performed. The foregoing description of the specific embodiments will so fully reveal the general nature of this disclosure that others can, by applying knowledge within the skill of the art, readily modify and/or adapt for various applications such specific embodiments, without undue experimentation, without departing from the general concept of the present invention. Therefore, such adaptations and modifications are intended to be within the meaning and range of equivalents of the disclosed embodiments, based on the teaching and guidance presented herein. It is to be understood that the phraseology or terminology herein is for the purpose of description and not of limitation, such that the terminology or phraseology of the present specification is to be interpreted by the skilled artisan in light of the teachings and guidance. The breadth and scope of the present invention should not be limited by any of the above-described exemplary embodiments, but should be defined only in accordance with the following claims and their equivalents. All publications, patent applications, patents, and other references mentioned herein are incorporated by reference in their entirety. In case of conflict, the present specification, including definitions, will control. ENUMERATED EMBODIMENTS 1. A lipid nanoparticle comprising: (a) an ionizable lipid of Formula (I): a
EXAMPLE 11: In vivo probing of efficacy of IL-22 mRNA constructs metabolic disease signatures in aged mice The present Example illustrates experiments designed to test the efficiacy of IL-22 mRNA constructs on signatures of metabolic disease in aged mice. To further study the effects of IL-22 mRNA constructs on fibrosis regression, an IL- 22 mRNA construct is administered in older animals. C57BL/6NTac male mice are fed an AMLN diet or a chow diet for more than 40 weeks. Previous studies have shown that such a duration of ALMN diet results in robust fibrosis in mice. Due to the high inter-animal variability of aged mice, these studies carry the potential risk of inadvertently including animals that do not display late stage NASH/fibrosis, as there are currently no reliable biomarkers available. Accordingly, a survivable liver biopsy strategy is employed to randomize animals into treatment groups based on a molecular analysis of their NASH/fibrosis stage before treatment. Employing this approach allows for the tracking of the liver phenotype of the same mouse over time. This approach closely mimics human trial designs for NASH intervention, in which liver biopsies are obtained pre- and post- treatment. EXAMPLE 12: In vivo probing of efficacy of IL-22 mRNA constructs following IV or IM administration in aged mice The present Example illustrates the efficiacy of IL-22 mRNA constructs on signatures of metabolic disease in aged mice, following IV and IM administration of IL-22 mRNA constructs. Briefly, male C57BL/6Tac mice were fed an AMLN diet starting from 6 weeks of age through 40 weeks of age, for a total of 34 weeks of AMLN feeding. LNP-encapsulated MSA mRNA (n=10) or Construct #1 mRNA was administered at 0.5 mg/kg weekly IV (n=10) or 0.1 mg/kg IM (n=10) to mice fed the AMLN diet, with an untreated group of mice (n=8) and mice fed in normal chow diet (n=10) maintained in parallel. Body weight was determined at the indicated time points over the course of the study (Fig.17). Liver weight and epididymal white adipose tissue (eWAT) weight were measured 72 hours after the last dose in a 7-week treatment period. eWAT and eWAT (%) were analyzed together, and liver weight and liver weight (%) were also analyzed together with group and measures as fixed effects. Metabolic efficiency was measured as weight gain (in grams (g)) over MJ caloric intake. Alanine aminotransferase (ALT) levels (U/L), serum cholesterol levels, HDL cholesterol levels, LDL cholesterol levels, and triglyceride levels were determined at the end of the study in the serum collection. As shown in Fig.18, aged mice administered Construct #1 mRNA displayed a consistent and increasingly pronounced reduction in body weight starting immediately after the initial IV injection of 0.5 mpk of Construct #1 mRNA. This reduction persisted throughout the seven-week study. A significant decrease in body weight was also observed following IM administration of Construct #1 mRNA at a dose of 0.1 mpk. As shown in Fig. 19A-B, daily caloric intake was transiently reduced by approximately 50% in the IV- administered group, and was mildly reduced in the early weeks for the IM group. By the end of the study, caloric intake was identical across all groups. These data suggest that Construct #1 mRNA-dependent changes in bodyweight can be observed following administration by other routes besides IV or SC, including IM. As shown in Fig.19C, the total cumulative energy intake was lowest in lean mice, followed by the IV group, and then the IM group. As shown in Fig.20A-B, metabolic efficiency (weight gain per caloric intake over the course of the study) was significantly reduced in the IV group and 0.1 mpk IM group, with the IV group showing the most pronounced effect. These data suggest that, in addition to lowering food intake, Construct #1 mRNA increases energy expenditure. Weight gain was significantly reduced in the IV group and 0.1 mpk IM group compared to controls. As shown in Fig.21A-D, a reduction in liver weight following Construct #1 administration (by either IV or IM administration) was observed at the conclusion of the study. In addition, a decrease in epididymal white adipose tissue (eWAT) weight following both IV and IM treatments was observed. The liver weight/body weight ratio was unaffected by Construct #1, suggesting that the loss of liver weight was driven by the overall body weight loss and not due to the liver being preferentially targeted. Conversely, the eWAT weight/body weight ratio was significantly lower in the IV group, possibly indicating increased lipolysis. As shown in Fig.22A-D, all lipid readings such as cholesterol, HDL, LDL, and triglycerides showed reduced levels following the IV treatment via Construct #1 mRNA when compared to MSA mRNA and the untreated mice on a high cholesterol diet. As shown in Fig.22E, alanine aminotransferase (ALT) levels in these aged animals were notably elevated in those fed with the specified diet. However, they remarkably reached the same levels as the lean control following IV Construct #1 mRNA treatment. Intriguingly, even though less potent, the IM treatment also significantly reduced the ALT levels. Taken together, these results strongly suggest that even in the more severe disease state of aged animals, Construct #1 mRNA treatment by multiple administration routes can improve the condition of the animals. The effects observed with Construct #1 mRNA administered intramuscularly suggest the potential for further investigation with lower dosages and alternative routes of administration. * * * * It is to be appreciated that the Detailed Description section, and not the Summary and Abstract sections, is intended to be used to interpret the claims. The Summary and Abstract sections can set forth one or more but not all exemplary embodiments of the present invention as contemplated by the inventor(s), and thus, are not intended to limit the present invention and the appended claims in any way. The present invention has been described above with the aid of functional building blocks illustrating the implementation of specified functions and relationships thereof. The boundaries of these functional building blocks have been arbitrarily defined herein for the convenience of the description. Alternate boundaries can be defined so long as the specified functions and relationships thereof are appropriately performed. The foregoing description of the specific embodiments will so fully reveal the general nature of this disclosure that others can, by applying knowledge within the skill of the art, readily modify and/or adapt for various applications such specific embodiments, without undue experimentation, without departing from the general concept of the present invention. Therefore, such adaptations and modifications are intended to be within the meaning and range of equivalents of the disclosed embodiments, based on the teaching and guidance presented herein. It is to be understood that the phraseology or terminology herein is for the purpose of description and not of limitation, such that the terminology or phraseology of the present specification is to be interpreted by the skilled artisan in light of the teachings and guidance. The breadth and scope of the present invention should not be limited by any of the above-described exemplary embodiments, but should be defined only in accordance with the following claims and their equivalents. All publications, patent applications, patents, and other references mentioned herein are incorporated by reference in their entirety. In case of conflict, the present specification, including definitions, will control. ENUMERATED EMBODIMENTS 1. A lipid nanoparticle comprising: (a) an ionizable lipid of Formula (I): a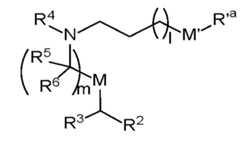 (I) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is:
(I) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is: ; wherein
; wherein denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; wherein n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13; and (b) an mRNA molecule comprising an open reading frame (ORF) encoding an Interleukin-22 (IL-22) polypeptide or an IL-22 polypeptide-containing fusion protein. 2. The lipid nanoparticle of embodiment 1, wherein in the compound of Formula (I): R’a is R’branched; R’branched is
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; wherein n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13; and (b) an mRNA molecule comprising an open reading frame (ORF) encoding an Interleukin-22 (IL-22) polypeptide or an IL-22 polypeptide-containing fusion protein. 2. The lipid nanoparticle of embodiment 1, wherein in the compound of Formula (I): R’a is R’branched; R’branched is ;
; denotes a point of attachment; and Raα is C2-12 alkyl; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is
denotes a point of attachment; and Raα is C2-12 alkyl; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is R10 is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. 3. The lipid nanoparticle of embodiment 1, wherein in the compound of Formula (I): R’a is R’branched; R’branched is
R10 is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. 3. The lipid nanoparticle of embodiment 1, wherein in the compound of Formula (I): R’a is R’branched; R’branched is ;
; denotes a point of attachment; Raα, Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. 4. The lipid nanoparticle of embodiment 1, wherein in the compound of Formula (I): R’a is R’branched; R’branched is
denotes a point of attachment; Raα, Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. 4. The lipid nanoparticle of embodiment 1, wherein in the compound of Formula (I): R’a is R’branched; R’branched is ;
; denotes a point of attachment; Raβ and Raδ are each H; Raγ is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. 5. The lipid nanoparticle of embodiment 1, wherein the ionizable lipid is:
denotes a point of attachment; Raβ and Raδ are each H; Raγ is C2-12 alkyl; R2 and R3 are each C5-14 alkyl; R4 is -(CH2)nOH; n is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7. 5. The lipid nanoparticle of embodiment 1, wherein the ionizable lipid is: ,
,



















Claims
WHAT IS CLAIMED IS: 1. A lipid nanoparticle comprising: (a) an ionizable lipid of Formula (I): a (I) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is:
(I) or its N-oxide, or a salt or isomer thereof, wherein R’a is R’branched; wherein R’branched is: ; wherein
; wherein denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and
denotes a point of attachment; wherein Raα, Raβ, Raγ, and Raδ are each independently selected from the group consisting of H, C2-12 alkyl, and C2-12 alkenyl; R2 and R3 are each independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl; R4 is selected from the group consisting of -(CH2)nOH, wherein n is selected from the group consisting of 1, 2, 3, 4, and 5, and wherein
wherein denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; wherein n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13; and (b) an mRNA molecule comprising an open reading frame (ORF) encoding an Interleukin-22 (IL-22) polypeptide or an IL-22 polypeptide-containing fusion protein.
denotes a point of attachment; wherein R10 is N(R)2; each R is independently selected from the group consisting of C1-6 alkyl, C2-3 alkenyl, and H; wherein n2 is selected from the group consisting of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10; each R5 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R6 is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; M and M’ are each independently selected from the group consisting of -C(O)O- and -OC(O)-; R’ is a C1-12 alkyl or C2-12 alkenyl; l is selected from the group consisting of 1, 2, 3, 4, and 5; and m is selected from the group consisting of 5, 6, 7, 8, 9, 10, 11, 12, and 13; and (b) an mRNA molecule comprising an open reading frame (ORF) encoding an Interleukin-22 (IL-22) polypeptide or an IL-22 polypeptide-containing fusion protein.





2. The lipid nanoparticle of claim 1, wherein in the compound of Formula (I): R’a is R’branched; R’branched is
 denotes a point of attachment; and Raα is C2-12 alkyl; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is
denotes a point of attachment; and Raα is C2-12 alkyl; Raβ, Raγ, and Raδ are each H; R2 and R3 are each C5-14 alkyl; R4 is R10 is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7.
R10 is NH(C1-6 alkyl); n2 is 2; each R5 is H; each R6 is H; M and M’ are each -C(O)O-; R’ is a C1-12 alkyl; l is 5; and m is 7.



3. The lipid nanoparticle of claim 1, wherein the ionizable lipid is: , or N-oxides, salts, or isomers thereof. 4. The lipid nanoparticle of any one of claims 1-3, wherein the mRNA molecule, when administered as a single dose to a human subject, is sufficient to increase serum IL-22 levels in the human subject to a level at least 2-fold, 3-fold,
, or N-oxides, salts, or isomers thereof. 4. The lipid nanoparticle of any one of claims 1-3, wherein the mRNA molecule, when administered as a single dose to a human subject, is sufficient to increase serum IL-22 levels in the human subject to a level at least 2-fold, 3-fold,

4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20-fold, 30-fold, 40-fold, or at least 50-fold higher than the level observed prior to the administration, for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120 hours, or at least one week post-administration. 5. The lipid nanoparticle of any one of claims 1-4, wherein the mRNA molecule, when administered as a single dose to a human subject, is sufficient to: (i) increase serum IL-22 levels in the human subject by at least about 1.1-fold, at least about 1.2-fold, at least about 1.5-fold, at least about 2.0-fold, at least about 5.0-fold, or at least about 10.0-fold compared to the human subject’s baseline serum IL-22 levels for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120 hours, or at least one week post-administration; or (ii) increase IL-22 activity in the human subject by at least about 1.5-fold, at least bout 2.0-fold, or at least about 2.
5-fold, compared to the human subject’s baseline IL-22 activity for at least 12 hours, at least 24 hours, at least 48 hours, at least 72 hours, at least 96 hours, at least 120, or at least one week post-administration, and/or wherein the mRNA molecule, when administered as a single dose to a human subject, is sufficient to: (i) reduce body weight of the human subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, or at least about 50%; (ii) reduce total cholesterol levels in the human subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%; (iii) reduce total HDL cholesterol levels in the human subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%; (iv) reduce total LDL cholesterol levels in the human subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%; (v) reduce total triglyceride levels in the human subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%; (vi) reduce liver steatosis in the human subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%; or (vii) reduce blood glucose levels in the human subject by at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 20%, at least about 50%, or 100%.
6. The lipid nanoparticle of any one of claims 1-5, wherein the IL-22 polypeptide has an amino acid sequence identical to any one of SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, or SEQ ID NO:4.
7. The lipid nanoparticle of any one of claims 1-5, wherein the IL-22 polypeptide- containing fusion protein has an amino acid sequence identical to SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25, SEQ ID NO:26, SEQ ID NO:27, SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38.
8. The lipid nanoparticle of any one of claims 1-7, wherein the ORF is at least 65% identical to SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328.
9. The lipid nanoparticle of any one of claims 1-8, wherein the mRNA molecule further comprises a 5’UTR having a nucleic acid sequence according to SEQ ID NO:50, SEQ ID NO:55, or SEQ ID NO:56; and/or the mRNA molecule further comprises a 3’UTR having a nucleic acid sequence according to SEQ ID NO:108, SEQ ID NO:125, or SEQ ID NO:139.
10. A pharmaceutical composition comprising: the lipid nanoparticle of any one of claims 1-9; and a pharmaceutically acceptable carrier.
11. A pharmaceutical composition according to claim 10 for use in treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject.
12. A lipid nanoparticle according to any one of claims 1-9 for use in the manufacture of a medicament or pharmaceutical composition for use in treating or delaying the onset and/or progression of metabolic disease, increasing serum IL-22 levels, or increasing IL-22 activity in a subject.
13. A method of treating or delaying the onset and/or progression of metabolic disease in a human subject, or increasing serum IL-22 levels in a human subject, or increasing IL-22 activity in a human subject, or reducing body weight of a human subject, or reducing total cholesterol levels in a human subject, reducing total LDL cholesterol levels in a human subject, or reducing total HDL cholesterol levels in a human subject, or reducing total triglyceride levels in a human subject, or reducing liver steatosis in a human subject, or reducing blood glucose levels in a human subject, comprising: administering to the human subject an effective amount of the pharmaceutical composition according to claim 10.
14. The method of claim 13, wherein (i) the administration is a single administration; (ii) the administration is a repeated administration; or (iii) the administration is about once per day, about once per week, about once per two weeks, or about once per month.
15. The method of claim 13 or 14, wherein the pharmaceutical composition is administered intravenously, intramuscularly, or subcutaneously.
16. An IL-22 polypeptide-containing fusion protein having an amino acid sequence identical to SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25, SEQ ID NO:26, SEQ ID NO:27, SEQ ID NO:28, SEQ ID NO:29, SEQ ID NO:30, SEQ ID NO:31, SEQ ID NO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35, SEQ ID NO:36, SEQ ID NO:37, or SEQ ID NO:38.
17. A pharmaceutical composition comprising: the IL-22 polypeptide of claim 16; and a pharmaceutically acceptable carrier.
18. A codon-optimized mRNA molecule encoding the IL-22 polypeptide according to claim 16.
19. The codon-optimized mRNA molecule of claim 18, having an open reading frame (ORF) having at least about 60% identity to the nucleic acid sequence of SEQ ID NO:310, SEQ ID NO:311, SEQ ID NO:312, SEQ ID NO:313, SEQ ID NO:314, SEQ ID NO:315, SEQ ID NO:316, SEQ ID NO:317, SEQ ID NO:318, SEQ ID NO:319, SEQ ID NO:320, SEQ ID NO:321, SEQ ID NO:322, SEQ ID NO:323, SEQ ID NO:324, SEQ ID NO:325, SEQ ID NO:326, SEQ ID NO:327, or SEQ ID NO:328.
20. A cell comprising the IL-22 polypeptide-containing fusion protein according to claim 16 or the codon-optimized mRNA molecule of claim 18 or 19.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202263433292P | 2022-12-16 | 2022-12-16 | |
| US63/433,292 | 2022-12-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024130158A1true WO2024130158A1 (en) | 2024-06-20 |
Family
ID=89767349
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2023/084348CeasedWO2024130158A1 (en) | 2022-12-16 | 2023-12-15 | Lipid nanoparticles and polynucleotides encoding extended serum half-life interleukin-22 for the treatment of metabolic disease |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024130158A1 (en) |
Citations (83)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0307434A1 (en) | 1987-03-18 | 1989-03-22 | Medical Res Council | Altered antibodies. |
| EP0367166A1 (en) | 1988-10-31 | 1990-05-09 | Takeda Chemical Industries, Ltd. | Modified interleukin-2 and production thereof |
| EP0394827A1 (en) | 1989-04-26 | 1990-10-31 | F. Hoffmann-La Roche Ag | Chimaeric CD4-immunoglobulin polypeptides |
| EP0413622A1 (en) | 1989-08-03 | 1991-02-20 | Rhone-Poulenc Sante | Albumin derivatives with therapeutic functions |
| WO1991006570A1 (en) | 1989-10-25 | 1991-05-16 | The University Of Melbourne | HYBRID Fc RECEPTOR MOLECULES |
| US5112946A (en) | 1989-07-06 | 1992-05-12 | Repligen Corporation | Modified pf4 compositions and methods of use |
| WO1993015200A1 (en) | 1992-01-31 | 1993-08-05 | Rhone-Poulenc Rorer S.A. | Antithrombotic polypeptides as antagonists of the binding of vwf to platelets or to subendothelium |
| WO1993015199A1 (en) | 1992-01-31 | 1993-08-05 | Rhone-Poulenc Rorer S.A. | Novel biologically active polypeptides, preparation thereof and pharmaceutical composition containing said polypeptides |
| US5336603A (en) | 1987-10-02 | 1994-08-09 | Genentech, Inc. | CD4 adheson variants |
| US5349053A (en) | 1990-06-01 | 1994-09-20 | Protein Design Labs, Inc. | Chimeric ligand/immunoglobulin molecules and their uses |
| US5359046A (en) | 1990-12-14 | 1994-10-25 | Cell Genesys, Inc. | Chimeric chains for receptor-associated signal transduction pathways |
| US5447851A (en) | 1992-04-02 | 1995-09-05 | Board Of Regents, The University Of Texas System | DNA encoding a chimeric polypeptide comprising the extracellular domain of TNF receptor fused to IgG, vectors, and host cells |
| WO1996004388A1 (en) | 1994-07-29 | 1996-02-15 | Smithkline Beecham Plc | Novel compounds |
| WO1996022024A1 (en) | 1995-01-17 | 1996-07-25 | Brigham And Women's Hospital, Inc. | Receptor specific transepithelial transport of immunogens |
| US5622929A (en) | 1992-01-23 | 1997-04-22 | Bristol-Myers Squibb Company | Thioether conjugates |
| WO1997034631A1 (en) | 1996-03-18 | 1997-09-25 | Board Of Regents, The University Of Texas System | Immunoglobin-like domains with increased half lives |
| US5681702A (en) | 1994-08-30 | 1997-10-28 | Chiron Corporation | Reduction of nonspecific hybridization by using novel base-pairing schemes |
| US5723125A (en) | 1995-12-28 | 1998-03-03 | Tanox Biosystems, Inc. | Hybrid with interferon-alpha and an immunoglobulin Fc linked through a non-immunogenic peptide |
| WO1998023289A1 (en) | 1996-11-27 | 1998-06-04 | The General Hospital Corporation | MODULATION OF IgG BINDING TO FcRn |
| US5783181A (en) | 1994-07-29 | 1998-07-21 | Smithkline Beecham Corporation | Therapeutic uses of fusion proteins between mutant IL 4/IL13 antagonists and immunoglobulins |
| US5844095A (en) | 1991-06-27 | 1998-12-01 | Bristol-Myers Squibb Company | CTLA4 Ig fusion proteins |
| WO1999004813A1 (en) | 1997-07-24 | 1999-02-04 | Brigham & Women's Hospital, Inc. | Receptor specific transepithelial transport of therapeutics |
| US5977307A (en) | 1989-09-07 | 1999-11-02 | Alkermes, Inc. | Transferrin receptor specific ligand-neuropharmaceutical agent fusion proteins |
| US6277375B1 (en) | 1997-03-03 | 2001-08-21 | Board Of Regents, The University Of Texas System | Immunoglobulin-like domains with increased half-lives |
| WO2001077137A1 (en) | 2000-04-12 | 2001-10-18 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| WO2003006007A1 (en) | 2001-07-11 | 2003-01-23 | Research & Innovation Soc.Coop. A R.L. | Use of compounds as functional antagonists to the central cannabinoid receptors |
| US6586524B2 (en) | 2001-07-19 | 2003-07-01 | Expression Genetics, Inc. | Cellular targeting poly(ethylene glycol)-grafted polymeric gene carrier |
| WO2003059934A2 (en)* | 2001-12-21 | 2003-07-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US6835393B2 (en) | 1998-01-05 | 2004-12-28 | University Of Washington | Enhanced transport using membrane disruptive agents |
| US20040262223A1 (en) | 2001-07-27 | 2004-12-30 | President And Fellows Of Harvard College | Laminar mixing apparatus and methods |
| US20050059005A1 (en) | 2001-09-28 | 2005-03-17 | Thomas Tuschl | Microrna molecules |
| US20050222064A1 (en) | 2002-02-20 | 2005-10-06 | Sirna Therapeutics, Inc. | Polycationic compositions for cellular delivery of polynucleotides |
| US20050261218A1 (en) | 2003-07-31 | 2005-11-24 | Christine Esau | Oligomeric compounds and compositions for use in modulation small non-coding RNAs |
| US7404969B2 (en) | 2005-02-14 | 2008-07-29 | Sirna Therapeutics, Inc. | Lipid nanoparticle based compositions and methods for the delivery of biologically active molecules |
| WO2008121949A1 (en) | 2007-03-30 | 2008-10-09 | Bind Biosciences, Inc. | Cancer cell targeting using nanoparticles |
| WO2010005740A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences, Inc. | Methods for the preparation of targeting agent functionalized diblock copolymers for use in fabrication of therapeutic targeted nanoparticles |
| WO2010005721A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences, Inc. | Drug loaded polymeric nanoparticles and methods of making and using same |
| WO2010005725A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences, Inc. | Therapeutic polymeric nanoparticles comprising vinca alkaloids and methods of making and using same |
| WO2010005726A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences Inc. | Therapeutic polymeric nanoparticles with mtor inhibitors and methods of making and using same |
| WO2010030763A2 (en) | 2008-09-10 | 2010-03-18 | Bind Biosciences, Inc. | High throughput fabrication of nanoparticles |
| US20100129877A1 (en) | 2005-09-28 | 2010-05-27 | Ugur Sahin | Modification of RNA, Producing an Increased Transcript Stability and Translation Efficiency |
| US7737108B1 (en) | 2000-01-07 | 2010-06-15 | University Of Washington | Enhanced transport using membrane disruptive agents |
| WO2010075072A2 (en) | 2008-12-15 | 2010-07-01 | Bind Biosciences | Long circulating nanoparticles for sustained release of therapeutic agents |
| US20100293625A1 (en) | 2007-09-26 | 2010-11-18 | Interexon Corporation | Synthetic 5'UTRs, Expression Vectors, and Methods for Increasing Transgene Expression |
| WO2011062965A2 (en) | 2009-11-18 | 2011-05-26 | University Of Washington Through Its Center For Commercialization | Targeting monomers and polymers having targeting blocks |
| WO2011084518A2 (en) | 2009-12-15 | 2011-07-14 | Bind Biosciences, Inc. | Therapeutic polymeric nanoparticles comprising corticosteroids and methods of making and using same |
| WO2011084521A2 (en) | 2009-12-15 | 2011-07-14 | Bind Biosciences, Inc. | Therapeutic polymeric nanoparticles comprising epothilone and methods of making and using same |
| US20110262491A1 (en) | 2010-04-12 | 2011-10-27 | Selecta Biosciences, Inc. | Emulsions and methods of making nanocarriers |
| WO2012040524A1 (en) | 2010-09-24 | 2012-03-29 | Mallinckrodt Llc | Aptamer conjugates for targeting of therapeutic and/or diagnostic nanocarriers |
| US8158601B2 (en) | 2009-06-10 | 2012-04-17 | Alnylam Pharmaceuticals, Inc. | Lipid formulation |
| WO2012054923A2 (en) | 2010-10-22 | 2012-04-26 | Bind Biosciences, Inc. | Therapeutic nanoparticles with high molecular weight copolymers |
| US20120140790A1 (en) | 2009-12-15 | 2012-06-07 | Ali Mir M | Therapeutic Polymeric Nanoparticle Compositions with High Glass Transition Termperature or High Molecular Weight Copolymers |
| WO2012099755A1 (en) | 2011-01-11 | 2012-07-26 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| US20120201859A1 (en) | 2002-05-02 | 2012-08-09 | Carrasquillo Karen G | Drug Delivery Systems and Use Thereof |
| US8241670B2 (en) | 2004-04-15 | 2012-08-14 | Chiasma Inc. | Compositions capable of facilitating penetration across a biological barrier |
| WO2012109121A1 (en) | 2011-02-07 | 2012-08-16 | Purdue Research Foundation | Carbohydrate nanoparticles for prolonged efficacy of antimicrobial peptide |
| WO2012135805A2 (en) | 2011-03-31 | 2012-10-04 | modeRNA Therapeutics | Delivery and formulation of engineered nucleic acids |
| US20120276209A1 (en) | 2009-11-04 | 2012-11-01 | The University Of British Columbia | Nucleic acid-containing lipid particles and related methods |
| WO2012170889A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc. | Cleavable lipids |
| WO2013039857A1 (en) | 2011-09-12 | 2013-03-21 | modeRNA Therapeutics | Engineered nucleic acids and methods of use thereof |
| WO2013052523A1 (en) | 2011-10-03 | 2013-04-11 | modeRNA Therapeutics | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| WO2013082111A2 (en) | 2011-11-29 | 2013-06-06 | The University Of North Carolina At Chapel Hill | Geometrically engineered particles and methods for modulating macrophage or immune responses |
| WO2013086354A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20130150295A1 (en) | 2006-12-21 | 2013-06-13 | Stryker Corporation | Sustained-Release Formulations Comprising Crystals, Macromolecular Gels, and Particulate Suspensions of Biologic Agents |
| US20130183244A1 (en) | 2010-09-10 | 2013-07-18 | The Johns Hopkins University | Rapid Diffusion of Large Polymeric Nanoparticles in the Mammalian Brain |
| WO2013105101A1 (en) | 2012-01-13 | 2013-07-18 | Department Of Biotechnology | Solid lipid nanoparticles entrapping hydrophilic/ amphiphilic drug and a process for preparing the same |
| WO2013110028A1 (en) | 2012-01-19 | 2013-07-25 | The Johns Hopkins University | Nanoparticle formulations with enhanced mucosal penetration |
| US20130195799A1 (en) | 2010-08-19 | 2013-08-01 | Peg Biosciences, Inc. | Synergistic biomolecule-polymer conjugates |
| WO2013116126A1 (en) | 2012-02-01 | 2013-08-08 | Merck Sharp & Dohme Corp. | Novel low molecular weight, biodegradable cationic lipids for oligonucleotide delivery |
| US8507653B2 (en) | 2006-12-27 | 2013-08-13 | Nektar Therapeutics | Factor IX moiety-polymer conjugates having a releasable linkage |
| US20130211249A1 (en) | 2010-07-22 | 2013-08-15 | The Johns Hopkins University | Drug eluting hydrogels for catheter delivery |
| US20130216610A1 (en) | 2007-03-30 | 2013-08-22 | Helix Biopharma Corp. | Biphasic lipid-vesicle composition and method for treating cervical dysplasia by intravaginal delivery |
| US8519110B2 (en) | 2008-06-06 | 2013-08-27 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | mRNA cap analogs |
| WO2013151671A1 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of cosmetic proteins and peptides |
| WO2014093924A1 (en) | 2012-12-13 | 2014-06-19 | Moderna Therapeutics, Inc. | Modified nucleic acid molecules and uses thereof |
| WO2014164253A1 (en) | 2013-03-09 | 2014-10-09 | Moderna Therapeutics, Inc. | Heterologous untranslated regions for mrna |
| WO2015130584A2 (en) | 2014-02-25 | 2015-09-03 | Merck Sharp & Dohme Corp. | Lipid nanoparticle vaccine adjuvants and antigen delivery systems |
| WO2017051367A1 (en) | 2015-09-24 | 2017-03-30 | Medidata Sp. Z O.O. | Cryoapplicator for minimally invasive surgical cardiac ablation |
| WO2017066797A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Trinucleotide mrna cap analogs |
| WO2017201325A1 (en) | 2016-05-18 | 2017-11-23 | Modernatx, Inc. | Combinations of mrnas encoding immune modulating polypeptides and uses thereof |
| WO2017201333A1 (en)* | 2016-05-18 | 2017-11-23 | Modernatx, Inc. | Polynucleotides encoding lipoprotein lipase for the treatment of hyperlipidemia |
| WO2019148026A1 (en)* | 2018-01-26 | 2019-08-01 | Genentech, Inc. | Il-22 fc fusion proteins and methods of use |
| WO2022026643A1 (en)* | 2020-07-30 | 2022-02-03 | Anwita Biosciences, Inc. | Interleukin-22 fusion proteins, and their pharmaceutical compositions and therapeutic applications |
- 2023
- 2023-12-15WOPCT/US2023/084348patent/WO2024130158A1/ennot_activeCeased
Patent Citations (110)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0307434A1 (en) | 1987-03-18 | 1989-03-22 | Medical Res Council | Altered antibodies. |
| US5336603A (en) | 1987-10-02 | 1994-08-09 | Genentech, Inc. | CD4 adheson variants |
| EP0367166A1 (en) | 1988-10-31 | 1990-05-09 | Takeda Chemical Industries, Ltd. | Modified interleukin-2 and production thereof |
| EP0394827A1 (en) | 1989-04-26 | 1990-10-31 | F. Hoffmann-La Roche Ag | Chimaeric CD4-immunoglobulin polypeptides |
| US5112946A (en) | 1989-07-06 | 1992-05-12 | Repligen Corporation | Modified pf4 compositions and methods of use |
| EP0413622A1 (en) | 1989-08-03 | 1991-02-20 | Rhone-Poulenc Sante | Albumin derivatives with therapeutic functions |
| US5977307A (en) | 1989-09-07 | 1999-11-02 | Alkermes, Inc. | Transferrin receptor specific ligand-neuropharmaceutical agent fusion proteins |
| WO1991006570A1 (en) | 1989-10-25 | 1991-05-16 | The University Of Melbourne | HYBRID Fc RECEPTOR MOLECULES |
| US5349053A (en) | 1990-06-01 | 1994-09-20 | Protein Design Labs, Inc. | Chimeric ligand/immunoglobulin molecules and their uses |
| US5359046A (en) | 1990-12-14 | 1994-10-25 | Cell Genesys, Inc. | Chimeric chains for receptor-associated signal transduction pathways |
| US5844095A (en) | 1991-06-27 | 1998-12-01 | Bristol-Myers Squibb Company | CTLA4 Ig fusion proteins |
| US5622929A (en) | 1992-01-23 | 1997-04-22 | Bristol-Myers Squibb Company | Thioether conjugates |
| WO1993015200A1 (en) | 1992-01-31 | 1993-08-05 | Rhone-Poulenc Rorer S.A. | Antithrombotic polypeptides as antagonists of the binding of vwf to platelets or to subendothelium |
| WO1993015199A1 (en) | 1992-01-31 | 1993-08-05 | Rhone-Poulenc Rorer S.A. | Novel biologically active polypeptides, preparation thereof and pharmaceutical composition containing said polypeptides |
| US5447851A (en) | 1992-04-02 | 1995-09-05 | Board Of Regents, The University Of Texas System | DNA encoding a chimeric polypeptide comprising the extracellular domain of TNF receptor fused to IgG, vectors, and host cells |
| US5447851B1 (en) | 1992-04-02 | 1999-07-06 | Univ Texas System Board Of | Dna encoding a chimeric polypeptide comprising the extracellular domain of tnf receptor fused to igg vectors and host cells |
| WO1996004388A1 (en) | 1994-07-29 | 1996-02-15 | Smithkline Beecham Plc | Novel compounds |
| US5783181A (en) | 1994-07-29 | 1998-07-21 | Smithkline Beecham Corporation | Therapeutic uses of fusion proteins between mutant IL 4/IL13 antagonists and immunoglobulins |
| US5681702A (en) | 1994-08-30 | 1997-10-28 | Chiron Corporation | Reduction of nonspecific hybridization by using novel base-pairing schemes |
| US5780610A (en) | 1994-08-30 | 1998-07-14 | Collins; Mark L. | Reduction of nonspecific hybridization by using novel base-pairing schemes |
| WO1996022024A1 (en) | 1995-01-17 | 1996-07-25 | Brigham And Women's Hospital, Inc. | Receptor specific transepithelial transport of immunogens |
| US5723125A (en) | 1995-12-28 | 1998-03-03 | Tanox Biosystems, Inc. | Hybrid with interferon-alpha and an immunoglobulin Fc linked through a non-immunogenic peptide |
| US5908626A (en) | 1995-12-28 | 1999-06-01 | Tanox, Inc. | Hybrid with interferon-β and an immunoglobulin Fc joined by a peptide linker |
| WO1997034631A1 (en) | 1996-03-18 | 1997-09-25 | Board Of Regents, The University Of Texas System | Immunoglobin-like domains with increased half lives |
| WO1998023289A1 (en) | 1996-11-27 | 1998-06-04 | The General Hospital Corporation | MODULATION OF IgG BINDING TO FcRn |
| US6277375B1 (en) | 1997-03-03 | 2001-08-21 | Board Of Regents, The University Of Texas System | Immunoglobulin-like domains with increased half-lives |
| WO1999004813A1 (en) | 1997-07-24 | 1999-02-04 | Brigham & Women's Hospital, Inc. | Receptor specific transepithelial transport of therapeutics |
| US6835393B2 (en) | 1998-01-05 | 2004-12-28 | University Of Washington | Enhanced transport using membrane disruptive agents |
| US8003129B2 (en) | 1998-01-05 | 2011-08-23 | University Of Washington | Enhanced transport using membrane disruptive agents |
| US7374778B2 (en) | 1998-01-05 | 2008-05-20 | University Of Washington | Enhanced transport using membrane disruptive agents |
| US7737108B1 (en) | 2000-01-07 | 2010-06-15 | University Of Washington | Enhanced transport using membrane disruptive agents |
| WO2001077137A1 (en) | 2000-04-12 | 2001-10-18 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| WO2003006007A1 (en) | 2001-07-11 | 2003-01-23 | Research & Innovation Soc.Coop. A R.L. | Use of compounds as functional antagonists to the central cannabinoid receptors |
| US6586524B2 (en) | 2001-07-19 | 2003-07-01 | Expression Genetics, Inc. | Cellular targeting poly(ethylene glycol)-grafted polymeric gene carrier |
| US20040262223A1 (en) | 2001-07-27 | 2004-12-30 | President And Fellows Of Harvard College | Laminar mixing apparatus and methods |
| US20050059005A1 (en) | 2001-09-28 | 2005-03-17 | Thomas Tuschl | Microrna molecules |
| WO2003059934A2 (en)* | 2001-12-21 | 2003-07-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US20050222064A1 (en) | 2002-02-20 | 2005-10-06 | Sirna Therapeutics, Inc. | Polycationic compositions for cellular delivery of polynucleotides |
| US20120201859A1 (en) | 2002-05-02 | 2012-08-09 | Carrasquillo Karen G | Drug Delivery Systems and Use Thereof |
| US20050261218A1 (en) | 2003-07-31 | 2005-11-24 | Christine Esau | Oligomeric compounds and compositions for use in modulation small non-coding RNAs |
| US8241670B2 (en) | 2004-04-15 | 2012-08-14 | Chiasma Inc. | Compositions capable of facilitating penetration across a biological barrier |
| US7404969B2 (en) | 2005-02-14 | 2008-07-29 | Sirna Therapeutics, Inc. | Lipid nanoparticle based compositions and methods for the delivery of biologically active molecules |
| US20100129877A1 (en) | 2005-09-28 | 2010-05-27 | Ugur Sahin | Modification of RNA, Producing an Increased Transcript Stability and Translation Efficiency |
| US20130150295A1 (en) | 2006-12-21 | 2013-06-13 | Stryker Corporation | Sustained-Release Formulations Comprising Crystals, Macromolecular Gels, and Particulate Suspensions of Biologic Agents |
| US8507653B2 (en) | 2006-12-27 | 2013-08-13 | Nektar Therapeutics | Factor IX moiety-polymer conjugates having a releasable linkage |
| US20130216610A1 (en) | 2007-03-30 | 2013-08-22 | Helix Biopharma Corp. | Biphasic lipid-vesicle composition and method for treating cervical dysplasia by intravaginal delivery |
| WO2008121949A1 (en) | 2007-03-30 | 2008-10-09 | Bind Biosciences, Inc. | Cancer cell targeting using nanoparticles |
| US20100293625A1 (en) | 2007-09-26 | 2010-11-18 | Interexon Corporation | Synthetic 5'UTRs, Expression Vectors, and Methods for Increasing Transgene Expression |
| US20130172406A1 (en) | 2007-09-28 | 2013-07-04 | Bind Biosciences, Inc. | Cancer Cell Targeting Using Nanoparticles |
| US20120004293A1 (en) | 2007-09-28 | 2012-01-05 | Zale Stephen E | Cancer Cell Targeting Using Nanoparticles |
| US8519110B2 (en) | 2008-06-06 | 2013-08-27 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | mRNA cap analogs |
| US20130230567A1 (en) | 2008-06-16 | 2013-09-05 | Bind Therapeutics, Inc. | Drug Loaded Polymeric Nanoparticles and Methods of Making and Using Same |
| WO2010005725A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences, Inc. | Therapeutic polymeric nanoparticles comprising vinca alkaloids and methods of making and using same |
| US20100104645A1 (en) | 2008-06-16 | 2010-04-29 | Bind Biosciences, Inc. | Methods for the preparation of targeting agent functionalized diblock copolymers for use in fabrication of therapeutic targeted nanoparticles |
| WO2010005740A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences, Inc. | Methods for the preparation of targeting agent functionalized diblock copolymers for use in fabrication of therapeutic targeted nanoparticles |
| US8293276B2 (en) | 2008-06-16 | 2012-10-23 | Bind Biosciences, Inc. | Drug loaded polymeric nanoparticles and methods of making and using same |
| US20100068285A1 (en) | 2008-06-16 | 2010-03-18 | Zale Stephen E | Drug Loaded Polymeric Nanoparticles and Methods of Making and Using Same |
| US20100068286A1 (en) | 2008-06-16 | 2010-03-18 | Greg Troiano | Drug Loaded Polymeric Nanoparticles and Methods of Making and Using Same |
| US20100069426A1 (en) | 2008-06-16 | 2010-03-18 | Zale Stephen E | Therapeutic polymeric nanoparticles with mTor inhibitors and methods of making and using same |
| WO2010005726A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences Inc. | Therapeutic polymeric nanoparticles with mtor inhibitors and methods of making and using same |
| US20100104655A1 (en) | 2008-06-16 | 2010-04-29 | Zale Stephen E | Therapeutic Polymeric Nanoparticles Comprising Vinca Alkaloids and Methods of Making and Using Same |
| WO2010005723A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences, Inc. | Drug loaded polymeric nanoparticles and methods of making and using same |
| US20110274759A1 (en) | 2008-06-16 | 2011-11-10 | Greg Troiano | Drug Loaded Polymeric Nanoparticles and Methods of Making and Using Same |
| WO2010005721A2 (en) | 2008-06-16 | 2010-01-14 | Bind Biosciences, Inc. | Drug loaded polymeric nanoparticles and methods of making and using same |
| US8206747B2 (en) | 2008-06-16 | 2012-06-26 | Bind Biosciences, Inc. | Drug loaded polymeric nanoparticles and methods of making and using same |
| US8318211B2 (en) | 2008-06-16 | 2012-11-27 | Bind Biosciences, Inc. | Therapeutic polymeric nanoparticles comprising vinca alkaloids and methods of making and using same |
| US8318208B1 (en) | 2008-06-16 | 2012-11-27 | Bind Biosciences, Inc. | Drug loaded polymeric nanoparticles and methods of making and using same |
| US20120288541A1 (en) | 2008-06-16 | 2012-11-15 | Zale Stephen E | Drug Loaded Polymeric Nanoparticles and Methods of Making and Using Same |
| US20130123351A1 (en) | 2008-09-10 | 2013-05-16 | Bind Biosciences, Inc. | High throughput fabrication of nanoparticles |
| WO2010030763A2 (en) | 2008-09-10 | 2010-03-18 | Bind Biosciences, Inc. | High throughput fabrication of nanoparticles |
| US20100087337A1 (en) | 2008-09-10 | 2010-04-08 | Bind Biosciences, Inc. | High Throughput Fabrication of Nanoparticles |
| WO2010075072A2 (en) | 2008-12-15 | 2010-07-01 | Bind Biosciences | Long circulating nanoparticles for sustained release of therapeutic agents |
| US20110217377A1 (en) | 2008-12-15 | 2011-09-08 | Zale Stephen E | Long Circulating Nanoparticles for Sustained Release of Therapeutic Agents |
| US20100216804A1 (en) | 2008-12-15 | 2010-08-26 | Zale Stephen E | Long Circulating Nanoparticles for Sustained Release of Therapeutic Agents |
| US8158601B2 (en) | 2009-06-10 | 2012-04-17 | Alnylam Pharmaceuticals, Inc. | Lipid formulation |
| US20120276209A1 (en) | 2009-11-04 | 2012-11-01 | The University Of British Columbia | Nucleic acid-containing lipid particles and related methods |
| WO2011062965A2 (en) | 2009-11-18 | 2011-05-26 | University Of Washington Through Its Center For Commercialization | Targeting monomers and polymers having targeting blocks |
| US20120140790A1 (en) | 2009-12-15 | 2012-06-07 | Ali Mir M | Therapeutic Polymeric Nanoparticle Compositions with High Glass Transition Termperature or High Molecular Weight Copolymers |
| WO2011084521A2 (en) | 2009-12-15 | 2011-07-14 | Bind Biosciences, Inc. | Therapeutic polymeric nanoparticles comprising epothilone and methods of making and using same |
| WO2011084518A2 (en) | 2009-12-15 | 2011-07-14 | Bind Biosciences, Inc. | Therapeutic polymeric nanoparticles comprising corticosteroids and methods of making and using same |
| US20110262491A1 (en) | 2010-04-12 | 2011-10-27 | Selecta Biosciences, Inc. | Emulsions and methods of making nanocarriers |
| US20130211249A1 (en) | 2010-07-22 | 2013-08-15 | The Johns Hopkins University | Drug eluting hydrogels for catheter delivery |
| US20130195799A1 (en) | 2010-08-19 | 2013-08-01 | Peg Biosciences, Inc. | Synergistic biomolecule-polymer conjugates |
| US20130183244A1 (en) | 2010-09-10 | 2013-07-18 | The Johns Hopkins University | Rapid Diffusion of Large Polymeric Nanoparticles in the Mammalian Brain |
| WO2012040524A1 (en) | 2010-09-24 | 2012-03-29 | Mallinckrodt Llc | Aptamer conjugates for targeting of therapeutic and/or diagnostic nanocarriers |
| WO2012054923A2 (en) | 2010-10-22 | 2012-04-26 | Bind Biosciences, Inc. | Therapeutic nanoparticles with high molecular weight copolymers |
| WO2012099755A1 (en) | 2011-01-11 | 2012-07-26 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| WO2012109121A1 (en) | 2011-02-07 | 2012-08-16 | Purdue Research Foundation | Carbohydrate nanoparticles for prolonged efficacy of antimicrobial peptide |
| US8710200B2 (en) | 2011-03-31 | 2014-04-29 | Moderna Therapeutics, Inc. | Engineered nucleic acids encoding a modified erythropoietin and their expression |
| WO2012135805A2 (en) | 2011-03-31 | 2012-10-04 | modeRNA Therapeutics | Delivery and formulation of engineered nucleic acids |
| WO2012170889A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc. | Cleavable lipids |
| WO2013039857A1 (en) | 2011-09-12 | 2013-03-21 | modeRNA Therapeutics | Engineered nucleic acids and methods of use thereof |
| WO2013052523A1 (en) | 2011-10-03 | 2013-04-11 | modeRNA Therapeutics | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US20130115272A1 (en) | 2011-10-03 | 2013-05-09 | modeRNA Therapeutics | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| WO2013082111A2 (en) | 2011-11-29 | 2013-06-06 | The University Of North Carolina At Chapel Hill | Geometrically engineered particles and methods for modulating macrophage or immune responses |
| WO2013086354A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| WO2013105101A1 (en) | 2012-01-13 | 2013-07-18 | Department Of Biotechnology | Solid lipid nanoparticles entrapping hydrophilic/ amphiphilic drug and a process for preparing the same |
| WO2013110028A1 (en) | 2012-01-19 | 2013-07-25 | The Johns Hopkins University | Nanoparticle formulations with enhanced mucosal penetration |
| WO2013116126A1 (en) | 2012-02-01 | 2013-08-08 | Merck Sharp & Dohme Corp. | Novel low molecular weight, biodegradable cationic lipids for oligonucleotide delivery |
| US8999380B2 (en) | 2012-04-02 | 2015-04-07 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| WO2013151671A1 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of cosmetic proteins and peptides |
| WO2014093924A1 (en) | 2012-12-13 | 2014-06-19 | Moderna Therapeutics, Inc. | Modified nucleic acid molecules and uses thereof |
| WO2014164253A1 (en) | 2013-03-09 | 2014-10-09 | Moderna Therapeutics, Inc. | Heterologous untranslated regions for mrna |
| WO2015130584A2 (en) | 2014-02-25 | 2015-09-03 | Merck Sharp & Dohme Corp. | Lipid nanoparticle vaccine adjuvants and antigen delivery systems |
| WO2017051367A1 (en) | 2015-09-24 | 2017-03-30 | Medidata Sp. Z O.O. | Cryoapplicator for minimally invasive surgical cardiac ablation |
| WO2017066797A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Trinucleotide mrna cap analogs |
| WO2017201325A1 (en) | 2016-05-18 | 2017-11-23 | Modernatx, Inc. | Combinations of mrnas encoding immune modulating polypeptides and uses thereof |
| WO2017201333A1 (en)* | 2016-05-18 | 2017-11-23 | Modernatx, Inc. | Polynucleotides encoding lipoprotein lipase for the treatment of hyperlipidemia |
| WO2019148026A1 (en)* | 2018-01-26 | 2019-08-01 | Genentech, Inc. | Il-22 fc fusion proteins and methods of use |
| WO2022026643A1 (en)* | 2020-07-30 | 2022-02-03 | Anwita Biosciences, Inc. | Interleukin-22 fusion proteins, and their pharmaceutical compositions and therapeutic applications |
Non-Patent Citations (66)
| Title |
|---|
| "NCBI", Database accession no. NM 020525.5 |
| "Oxford Dictionary Of Biochemistry And Molecular Biology", 2000, OXFORD UNIVERSITY PRESS |
| "Pharmaceutical Salts: Properties, Selection, and Use", 2008, WILEY-VCH |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY, pages: 1418 |
| "The Dictionary of Cell and Molecular Biology", 1999, ACADEMIC PRESS |
| A. R. GENNARO: "Remington: The Science and Practice of Pharmacy", 2006, LIPPINCOTT, WILLIAMS & WILKINS |
| ABRAHAM ET AL.: "Chaotic Mixer for Microchannels", SCIENCE, vol. 295, 2002, pages 647 - 651 |
| ALEKU ET AL., CANCER RES, vol. 68, 2008, pages 9788 - 9798 |
| ANANDCHERESH, CURR OPIN HEMATOL, vol. 18, 2011, pages 171 - 176 |
| ANNONI A ET AL., BLOOD, vol. 114, 2009, pages 5152 - 5161 |
| ASHKENAZI ET AL., PROC. NATL. ACAD. SCI. USA, vol. 88, 1991, pages 10535 - 10539 |
| BARTEL, CELL, vol. 136, 2009, pages 215 - 233 |
| BELLIVEAU ET AL.: "Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA", MOLECULAR THERAPY-NUCLEIC ACIDS, vol. 1, 2012, pages e37, XP002715253, DOI: 10.1038/mtna.2012.28 |
| BERGE ET AL., JOURNAL OF PHARMACEUTICAL SCIENCE, vol. 66, 1977, pages 1 - 19 |
| BONAUER ET AL., CURR DRUG TARGETS, vol. 11, 2010, pages 943 - 949 |
| BROWN BD ET AL., BLOOD, vol. 110, no. 13, 2007, pages 4144 - 4152 |
| BROWN BD ET AL., NAT MED, vol. 12, no. 5, 2006, pages 585 - 591 |
| CHEN ET AL.: "Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation", J. AM. CHEM. SOC., vol. 134, no. 16, 2012, pages 6948 - 51, XP002715254, DOI: 10.1021/ja301621z |
| CONTRERASRAO, LEUKEMIA, vol. 26, 20 December 2011 (2011-12-20), pages 404 - 413 |
| DEFOUGEROLLES, HUM GENE THER., vol. 19, 2008, pages 125 - 132 |
| FOTIN-MLECZEK ET AL., J. IMMUNOTHER., vol. 34, 2011, pages 1 - 15 |
| GAO MINSONG ET AL: "Synthetic modified messenger RNA for therapeutic applications", ACTA BIOMATERIALIA, ELSEVIER, AMSTERDAM, NL, vol. 131, 13 June 2021 (2021-06-13), pages 1 - 15, XP086746515, ISSN: 1742-7061, [retrieved on 20210613], DOI: 10.1016/J.ACTBIO.2021.06.020* |
| GENTNERNALDINI, TISSUE ANTIGENS, vol. 80, 2012, pages 393 - 403 |
| GUTBIER ET AL., PULM PHARMACOL. THER., vol. 23, 2010, pages 334 - 344 |
| HINNEBUSCH A ET AL., SCIENCE, vol. 352, no. 6292, 2016, pages 1413 - 6 |
| HOLLIGER ET AL., NAT BIOTECHNOL, vol. 15, no. 7, 1997, pages 632 - 6 |
| IMMUNOL, vol. 165, no. 1, 2000, pages 263 - 70 |
| JUNJIE LI ET AL., CURRENT BIOLOGY, vol. 15, 23 August 2005 (2005-08-23), pages 1501 - 1507 |
| JUO, PEI-SHOW: "Concise Dictionary of Biomedicine and Molecular Biology", 2002, CRC PRESS |
| KAUFMANN ET AL., MICROVASC RES, vol. 80, 2010, pages 286 - 293 |
| KORE ET AL., BIOORGANIC & MEDICINAL CHEMISTRY, vol. 21, 2013, pages 4570 - 4574 |
| LANDGRAF ET AL., CELL, vol. 129, 2007, pages 1401 - 1414 |
| LEACH ET AL., J. AM. CHEM. SOC., vol. 114, 1992, pages 3675 - 3683 |
| MANDALROSSI, NAT. PROTOC., vol. 8, no. 3, 2013, pages 568 - 82 |
| MANTSCH ET AL., BIOCHEM., vol. 14, 1993, pages 5593 - 5601 |
| MATSUDA ET AL., PLOS ONE, vol. 11, no. 5, 2010, pages e15057 |
| MATSUDAMAURO, PLOS ONE, vol. 5, 2010, pages 11 |
| MAYR C., COLD SPRING HARB PERSP BIOL, vol. 11, no. 10, 1 October 2019 (2019-10-01), pages a034728 |
| MEIJER HA ET AL., SCIENCE, vol. 340, 2013, pages 82 - 85 |
| MICHAEL E. ROTHENBERG ET AL: "-22Fc): A Potential Therapy for Epithelial Injury", CLINICAL PHARMACOLOGY AND THERAPEUTICS, vol. 105, no. 1, 9 January 2019 (2019-01-09), US, pages 177 - 189, XP055744981, ISSN: 0009-9236, DOI: 10.1002/cpt.1164* |
| NASERI ET AL.: "Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: Structure, Preparation and Application", ADV. PHARM. BULL., vol. 5, 2015, pages 305 - 13 |
| NATURE, vol. 402, 1999, pages 304 - 309 |
| NORBURY: "Nature Reviews Molecular Cell Biology", 29 August 2013, AOP, article "Cytoplasmic RNA: a case of the tail wagging the dog" |
| PASCOLO, EXPERT OPIN. BIOL. THER., vol. 4, pages 1285 - 1294 |
| PEER ET AL., PROC NATL ACAD SCI USA, vol. 104, 2007, pages 4095 - 4100 |
| PICCIRILLI ET AL., NATURE, vol. 343, 1990, pages 33 - 37 |
| SANTEL ET AL., GENE THER, vol. 13, 2006, pages 1360 - 1370 |
| SILVA ET AL.: "Lipid nanoparticles for the delivery of biopharmaceuticals", CURR. PHARM. BIOTECHNOL., vol. 16, 2015, pages 291 - 302, XP055602369 |
| SILVA: "Delivery Systems for Biopharmaceuticals. Part I: Nanoparticles and Microparticles", CURR. PHARM. TECHNOL., vol. 16, 2015, pages 940 - 954 |
| SONG ET AL., NATURE BIOTECHNOL, vol. 23, 2005, pages 709 - 717 |
| SSU-HSUEH TSENG: "Albumin and interferon-[beta] fusion protein serves as an effective vaccine adjuvant to enhance antigen-specific CD8+ T cell-mediated antitumor immunity", JOURNAL FOR IMMUNOTHERAPY OF CANCER, vol. 10, no. 4, 1 April 2022 (2022-04-01), GB, pages e004342, XP093157633, ISSN: 2051-1426, DOI: 10.1136/jitc-2021-004342* |
| STRUMBERG ET AL., INT J CLIN PHARMACOL THER, vol. 50, 2012, pages 76 - 78 |
| SUNG JUNSIK ET AL: "Oral delivery of IL-22 mRNA-loaded lipid nanoparticles targeting the injured intestinal mucosa: A novel therapeutic solution to treat ulcerative colitis", BIOMATERIALS, ELSEVIER, AMSTERDAM, NL, vol. 288, 3 August 2022 (2022-08-03), XP087169223, ISSN: 0142-9612, [retrieved on 20220803], DOI: 10.1016/J.BIOMATERIALS.2022.121707* |
| SWITZER ET AL., BIOCHEMISTRY, vol. 32, 1993, pages 10489 - 10496 |
| TOR ET AL., J. AM. CHEM. SOC., vol. 115, 1993, pages 4461 - 4467 |
| TOURIOL ET AL., BIOLOGY OF THE CELL, vol. 95, 2003, pages 169 - 178 |
| TRAUNECKER ET AL., NATURE, vol. 331, 1988, pages 84 - 86 |
| VIL ET AL., PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 11337 - 11341 |
| WANG ET AL.: "Delivery of oligonucleotides with lipid nanoparticles", ADV. DRUG DELIV. REV., vol. 87, 2015, pages 68 - 80 |
| WARDGHETIE, THER. IMMUNOL., vol. 2, 1995, pages 77 - 94 |
| WEIDE ET AL., J IMMUNOTHER., vol. 31, 2008, pages 180 - 188 |
| WEIDE ET AL., J IMMUNOTHER., vol. 32, 2009, pages 498 - 507 |
| WHITESIDES, GEORGE M.: "The Origins and the Future of Microfluidics", NATURE, vol. 442, 2006, pages 368 - 373, XP055123139, DOI: 10.1038/nature05058 |
| YAKUBOV ET AL., BIOCHEM. BIOPHYS. RES. COMMUN., vol. 394, no. 1, 2010, pages 189 - 193 |
| ZHENG ET AL., J. IMMUNOL., vol. 154, 1995, pages 5590 - 5600 |
| ZHIGALTSEV ET AL.: "Bottom-up design and synthesis of limit size lipid nanoparticle systems with aqueous and triglyceride cores using millisecond microfluidic mixing", LANGMUIR, vol. 28, 2012, pages 3633 - 40, XP055150435, DOI: 10.1021/la204833h |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12239742B2 (en) | Polynucleotides encoding Citrin for the treatment of Citrullinemia type 2 | |
| US20230323371A1 (en) | Polynucleotides encoding alpha-galactosidase a for the treatment of fabry disease | |
| US12128113B2 (en) | Polynucleotides encoding JAGGED1 for the treatment of Alagille syndrome | |
| US20240247239A1 (en) | Polynucleotides encoding ornithine transcarbamylase for the treatment of urea cycle disorders | |
| US20200131498A1 (en) | Polynucleotides encoding methylmalonyl-coa mutase | |
| US11939601B2 (en) | Polynucleotides encoding phenylalanine hydroxylase for the treatment of phenylketonuria | |
| US20240024506A1 (en) | Polynucleotides encoding propionyl-coa carboxylase alpha and beta subunits for the treatment of propionic acidemia | |
| US20250170070A1 (en) | Polynucleotides encoding glucose-6-phosphatase for the treatment of glycogen storage disease | |
| US20250017867A1 (en) | Polynucleotides encoding relaxin for the treatment of fibrosis and/or cardiovascular disease | |
| US20230406895A1 (en) | Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis | |
| US20240207444A1 (en) | Lipid nanoparticles containing polynucleotides encoding phenylalanine hydroxylase and uses thereof | |
| US20250262320A1 (en) | Polynucleotides encoding uridine diphosphate glycosyltransferase 1 family, polypeptide a1 for the treatment of crigler-najjar syndrome | |
| US20240207374A1 (en) | Lipid nanoparticles containing polynucleotides encoding glucose-6-phosphatase and uses thereof | |
| US20220110966A1 (en) | Polynucleotides encoding very long-chain acyl-coa dehydrogenase for the treatment of very long-chain acyl-coa dehydrogenase deficiency | |
| US20220243182A1 (en) | Polynucleotides encoding branched-chain alpha-ketoacid dehydrogenase complex e1-alpha, e1-beta, and e2 subunits for the treatment of maple syrup urine disease | |
| US20240226025A1 (en) | Polynucleotides encoding methylmalonyl-coa mutase for the treatment of methylmalonic acidemia | |
| WO2024130158A1 (en) | Lipid nanoparticles and polynucleotides encoding extended serum half-life interleukin-22 for the treatment of metabolic disease | |
| US20250221931A1 (en) | Polynucleotides encoding fanconi anemia, complementation group proteins for the treatment of fanconi anemia | |
| US20240376445A1 (en) | Polynucleotides encoding uridine diphosphate glycosyltransferase 1 family, polypeptide a1 for the treatment of crigler-najjar syndrome | |
| US20240216288A1 (en) | Lipid nanoparticles containing polynucleotides encoding propionyl-coa carboxylase alpha and beta subunits and uses thereof | |
| WO2023196399A1 (en) | Lipid nanoparticles and polynucleotides encoding argininosuccinate lyase for the treatment of argininosuccinic aciduria | |
| WO2024182301A2 (en) | Lipid nanoparticles and polynucleotides encoding galactose-1-phosphate uridylyltransferase (galt) for the treatment of galactosemia | |
| WO2024137953A2 (en) | Small nuclear ribonucleoprotein 13 polypeptides, polynucleotides, and uses thereof | |
| EP4314260A1 (en) | Lipid nanoparticles and polynucleotides encoding ornithine transcarbamylase for the treatment of ornithine transcarbamylase deficiency |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:23847811 Country of ref document:EP Kind code of ref document:A1 | |
| NENP | Non-entry into the national phase | Ref country code:DE |