WO2024115353A1 - Analytical composition for monitoring disinfection - Google Patents
Analytical composition for monitoring disinfectionDownload PDFInfo
- Publication number
- WO2024115353A1 WO2024115353A1PCT/EP2023/083109EP2023083109WWO2024115353A1WO 2024115353 A1WO2024115353 A1WO 2024115353A1EP 2023083109 WEP2023083109 WEP 2023083109WWO 2024115353 A1WO2024115353 A1WO 2024115353A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lipid
- nucleic acid
- mol
- test sample
- lipid nanoparticles
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
Definitions
- Analytical composition for monitoring disinfectionTechnical Field
- the present inventionrelates to analytical compositions and kits, their use for monitoring disinfection procedures, and a method for monitoring a disinfection procedure.
- the analytical compositioncomprises lipid nanoparticles encapsulating a nucleic acid.
- microbial surrogate markersare used for monitoring the transmission of microorganisms.
- examples of traditional surrogate markersare live non-pathogenic viruses, bacteriophages, such as bacteriophage MS2, or cauliflower mosaic virus DNA.
- SPEDsilica nanoparticles with encapsulated DNA
- the term “monitoring a disinfection procedure”relates to an evaluation whether a successful disinfection of a surface was achieved. “Monitoring a disinfection procedure” thus includes validation of a disinfection procedure.
- the termfurther includes “monitoring transmission pathways of microorganisms between surfaces”, i.e. providing information whether microorganisms (the LNPs encapsulating a nucleic acid as described herein being a surrogate for microorganisms) have been transferred from a first surface to a second surface or a third surface, etc.
- Nucleic acidrefers to a deoxyribonucleotide or ribonucleotide polymer, in linear or circular conformation, and in either single- or double-stranded form (ss or ds, respectively).
- the termscan encompass known analogs of natural nucleotides, as well as nucleotides that are modified in the base, sugar and/or phosphate moieties (e.g. phosphorothioate or phosphorodithioate backbones).
- NucleotideThe term “nucleotide” refers to deoxyribonucleotides or ribonucleotides.
- the nucleotidesmay be standard nucleotides (i.e., adenosine, guanosine, cytidine, thymidine, and uridine) or nucleotide analogs.
- a nucleotide analogrefers to a nucleotide having a modified purine or pyrimidine base or a modified ribose moiety.
- a nucleotide analogmay be a naturally occurring nucleotide (e.g. inosine) or a non-naturally occurring nucleotide.
- Non-naturally occurring nucleotidesinclude nucleotides that are modified at the 2’-O position of the sugar such as 2’-OMe, 2’-MOE, 2’-F modified nucleotides, as well as locked nucleic acids (LNAs), peptide nucleic acids (PNA), and morpholinos.
- LNAslocked nucleic acids
- PNApeptide nucleic acids
- morpholinosmorpholinos.
- the skilled personunderstands that this list of modified nucleotides is not exhaustive and is able to select suitable modified nucleo- tides based on the present disclosure.
- LipidThe term “lipid” is known in the field and advantageously relates to biomolecules that are soluble in non-polar solvents such as benzene, chloroform, acetone, etc.
- lipidincludes for example fatty acids, waxes, sterols (e.g. cholesterol), fat-soluble vitamins (such as vitamins A, D, E, and K), monoglycerides, diglycerides, triglycerides, and phospholipids.
- amphiphilic lipidse.g. ionisable lipids and phospholipids
- LNPlipid nanoparticle
- LNPsare typically spherical with an average diameter between 20 nm and 1500 nm, e.g. between 50 nm to 1500 nm, such as between 50 nm to 300 nm.
- components of the LNPsare typically selected from four types of lipids: an ionisable lipid (positive charge supports binding to negatively charged nucleic acid), a phospholipid (improves / stabilizes the structure of the LNPs), cholesterol (improves / stabilizes the structure of the LNPs), and a lipid anchored PEG (improves the stability of the LNP, e.g. by preventing aggregation).
- LNPs suitable for the present inventionare non-toxic.
- Dispersionis known in the field. In general, the term “dispersion” relates to a system in which distributed particles of one material are dispersed in a continuous phase of another material. In an aqueous dispersion, the continuous phase thus contains or consists of water.
- the term “dispersion”includes the terms “suspension” such as a “colloidal suspension”, “emulsion” and “solution”.
- a dispersion containing lipid nanoparticlesrelates to a suspension, usually a colloidal suspension.
- lipid nanoparticle dispersionstypically are aqueous suspensions, usually aqueous colloidal suspensions.
- Dispersed phaseThe term “dispersed phase” is known in the field. In general, the term “dispersed phase” relates to colloids of a substance dispersed in another substance. For example, the dispersed phase of a lipid nanoparticle dispersion comprises of the lipid nanoparticles.
- Continuous phaseThe term “continuous phase” is known in the field. In general, the term “continuous phase” relates to the fluid phase in a dispersion within which solid or fluid particles are distributed. For example, the continuous phase of a lipid nanoparticle dispersion is the aqueous liquid the lipid nanoparticles are dispersed in.
- Nnitrogen
- Pnegatively-charged nucleic acid phosphate
- Ppositively-chargeable polymer amine groups
- Ptypically present in ionisable lipids.
- PolyethylenglycolThe term “polyethylenglycol” (PEG; also referred to as polyethylene oxide) is known in the field and relates to a polymer, specifically a polyether, composed of ethylene oxide monomeric units.
- the structure of PEGis commonly expressed as H ⁇ (O ⁇ CH 2 ⁇ CH 2 ) n ⁇ OH, wherein n is the number of ethylene oxide monomeric units.
- a PEG polymeris defined by its molecular weight, e.g. PEG1000 refers to PEG having an average molecular weight of 1000 Da.
- PEG1000refers to PEG having an average molecular weight of 1000 Da.
- the molecular weight of PEG, such as PEG1000is an average molecular weight based on the polydispersity of PEG.
- Ionisable lipidThe term “ionisable lipid” is known in the field and relates to lipid molecules containing a chargeable group, typically an amine group, such as 306Oi10 [e.g. Hou et al., 2021, Nat Rev Mater 6, 1078–1094 and references therein].
- PhospholipidThe term “phospholipid” is known in the field and relates to amphiphilic lipid molecules containing a hydrophilic "head” containing a phosphate group and two hydrophobic "tails" derived from fatty acids, joined by an alcohol residue, typically a glycerol.
- the phosphate groupmay be further modified with organic molecules, typically serine (e.g. phosphatidylserine), choline (e.g.
- Lipid anchored PEGThe term “lipid anchored PEG” is known in the field and relates to a PEG polymer covalently linked to a lipid (also referred to as PEG-derivatized lipids, PEG-lipids or PEGylated lipids).
- Lipid-anchored PEGsare generally described in, for example, U.S. 5,672,662 (also see e.g. Hou et al., 2021, Nat. Rev. Mater. 6, 1078–1094 and references therein).
- a lipid anchored PEGis referred to in the following way: C x PEG y , wherein x is the number of carbon atoms in the fatty acid component of the lipid and y is the average molecular weight of the PEG component.
- xis the number of carbon atoms in the fatty acid component of the lipid
- yis the average molecular weight of the PEG component.
- the molecular weight of a lipid anchored PEG such as C14PEG1000is an average molecular weight based on the polydispersity of PEG.
- Lipid anchored PEGsgenerally increase the stability of LNPs, e.g. by preventing aggregation.
- C14PEG1000also referred to as 1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)-1000] is shown below (ammonium salt):
- DisaccharideThe term “disaccharide” is known in the field and particularly describes carbohydrates composed of two monosaccharide units bound together by glycosidic linkages. It is understood that a disaccharide may comprise two identical monosaccharide units or may comprise two different monosaccharide units, which may also be chemically modified (e.g.
- polysaccharideis known in the field and particularly describes polymeric carbohydrates composed of monosaccharide units bound together by glycosidic linkages, either linear or branched. Such polysaccharides are characterized by their repeating units, each repeating unit described with their respective monosaccharide composition. Said repeating units include one or more monosaccharides which can also be chemically modified (e.g. amidated, sulphonated, acetylated, phos- phorylated, etc).
- Typical found monosaccharides in said repeating unitsare cyclic or linear monosaccharides containing three to seven carbon atoms.
- a number of abbreviationsare used, including: Delta Ct Difference between Ct values DLS Dynamic light scattering DNA Deoxyribonucleic acid DOPE 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine ds double-stranded DSPC 1,2-distearoyl-sn-glycero-3-phosphocholin GC-MS gas chromatography mass spectrometry HPLC high performance liquid chromatography LNP lipid nanoparticle MALDI-TOF Matrix-assisted laser desorption ionisation- time-of- flight mass spectrometry MMLV Moloney Murine Leukemia Virus PEG polyethylenglycol RNA ribonucleic acid SEM Scanning electron microscopy SPED silica nanoparticles with encapsulated DNA ss single-stranded
- the analytical compositioncomprises lipid nanoparticles (LNPs) encapsulating a nucleic acid.
- the analytical compositionfurther complies with the following characteristics:
- the LNPsdisintegrate in 70% V/V ethanol.
- the term “disintegration of lipid nanoparticles”relates to an increase in concentration of any one of the lipid components of the LNP in the continuous phase of at least 100% compared the continuous phase of a reference dispersion of the same lipid nanoparticles in water. Disintegration can be measured by determining in the continuous phase the concentration of any one of the lipids that are present in the respective LNP in signification amounts, i.e.
- any one of the lipid components being present in the LNP in an amount of at least 1 mol%(also referred to as “constituent lipid”), e.g. in an amount of between 1 mol% to 100 mol%, such as 1 mol% to 50 mol%.
- the lipid component to be analysedmay be cholesterol or an ionisable lipid, such as oleylamine, dodecylamine, hexadecylamine or 306Oi10.
- the concentration of any one of the lipids in the continuous phasecan be measured by standard methods for lipid quantification, such as GC-MS, HPLC, MALDI-TOF.
- disintegrationis measured by GC-MS, e.g.
- the thereby determined concentration valueis compared to the concentration of the same lipid in the continuous phase of a reference dispersion of the same LNPs in water.
- the dispersed phaseis separated from the continuous phase by standard methods, usually employing centrifugation and filtration. Measuring the concentration of any one of the lipids in the continuous phase is within the ordinary skill.
- the term “disintegration”relates to an increase in concentration by 100% of a lipid component of the LNPs (i.e. any lipid being present in the LNP in an amount of at least 1 mol%, e.g.
- disintegrationmay be measured according to the following protocol: • Diluting a sample of the LNP (containing min.
- the LNPsprotect the nucleic acid encapsulated therein from nuclease degradation as measured by qPCR.
- the encapsulated nucleic acidhas a length of between 60 – 200 nucleotides, advantageously of between 60 – 120 nucleotides.
- the wt. ratio between the nucleic acid and the LNPs in the analytical compositionis in a range of between 0.01% to 15% nucleic acid, advantageously in a range of between 0.1% to 2% nucleic acid, such as 1% nucleic acid.
- the use of the analytical composition described abovehas the advantage that the encapsulated nucleic acid is protected from nuclease degradation as long as the LNPs are intact.
- the LNPsdisintegrate and the nucleic acid becomes susceptible to degradation, in particular nuclease degradation. It is considered beneficial that the nucleic acid can be quantified by well-established and sensitive detection methods, e.g. by quantitative polymerase chain reaction (qPCR).
- qPCRquantitative polymerase chain reaction
- the LNPs encapsulating the nucleic acid as described abovehave the advantage that they more closely mimic common pathogens than the tools provided in the prior art. Based on the present disclosure, the skilled person understands that the chemical composition of the LNPs can be adapted to particularly mimic certain pathogens, e.g.
- multi-drug resistant bacteriasuch as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci.
- MRSAmethicillin-resistant Staphylococcus aureus
- vancomycin-resistant enterococciSuch pathogens are of particular relevance in certain environments, e.g. in the intensive care unit of a hospital.
- the LNPs encapsulating the nucleic acidare thus improved surrogate markers for pathogens compared to the prior art.
- analytical compositions as described abovehave the advantage that the LNPs are susceptible to disinfection procedures.
- the use of the analytical compositionenables both monitoring the disinfection procedure of a single surface and monitoring the transmission of pathogens from a first surface to a further surface (e.g.
- the use of the analytical composition as described abovehas the advantage that the LNPs are non- infectious and thus well suited for use in a hospital, e.g. in an intensive care unit.
- the use of the analytical compositionhas the advantage that the nucleic acid can be detected with high sensitivity by established nucleic acid amplification techniques such as qPCR.
- the use of the analytical compositionhas the advantage that results can be obtained in a relatively short time period, typically results can be obtained within 2 hours.
- the encapsulated nucleic acidis deoxyribonucleic acid (DNA).
- the encapsulated DNAmay have the nucleotide sequence according to SEQ ID NO: 1 shown below: SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTT GAACACGAACAAACCTCTTT
- the encapsulated nucleic acidis ribonucleic acid.
- the nucleic acidcomprises or consists of modified nucleotides. Based on the present disclosure, the skilled person understands that the nucleic acid may be either single- stranded (ss) or double-stranded (ds).
- the use of multiple analytical compositions, each containing LNPs encapsulating a nucleic acid, e.g. DNA, having a different sequenceenables monitoring of transmission pathways from multiple surfaces while providing information on the efficiency of the disinfection procedure.
- the lipid nanoparticles and the DNA encapsulated thereinhave a N/P ratio of between 1:1 and 10:1, e.g. between 5:1 and 10:1, such as 8:1 or 10:1.
- “protection from nuclease degradation”, such as protection from DNAse degradationrelates to a decrease of max. 75% of intact nucleic acid present in the analytical composition after treatment with a nuclease, e.g.
- benzonase(protection test sample E) compared to a reference analytical composition that was not treated with the nuclease (protection negative control F), as measured by quantitative polymerase chain reaction (qPCR).
- Decrease2 ⁇ (Delta Ct)
- a difference between Ct-values measured for (protection test sample E) – (protection negative control F) of 2corresponds to a 4-fold decrease in intact nucleic acid.
- a decrease of max. 75% intact nucleic acidcorresponds to a delta Ct between protection test sample E and protection negative control F of max. 2.
- nucleaseis adapted to the nucleic acid to be analysed.
- DNase degradationmay be measured by subjecting the analytical composition to digestion with benzonase.
- protection from nuclease degradationmay be measured according to the following protocol (cf.
- protection test sample E2.5 units Benzonase in 10 ⁇ l sample (1.25 ng/ml) diluted in 90 ⁇ l Tris buffer (10 mM, pH 7) with MgCl 2 (20 mM)
- Protection negative control F10 ⁇ l sample (1.25 ng/ml) diluted in 90 ⁇ l miliQ water • Incubating protection test sample E for 1 h at 37°C • Inactivating enzyme by incubation for 10 min at 75 °C • Measuring DNA concentration of protection test sample E and protection negative control F via qPCR (see Example 6, Step 5) • Calculating the concentration difference between protection test sample E and protection negative control F based on the difference between Ct values (delta Ct).
- RNAis to be analysed, i.e. the encapsulated nucleic acid being RNA
- a suitable RNasee.g. RNase A
- qPCR measurement of RNArequires a reverse transcription step, e.g. using a reverse transcriptase such as Moloney Murine Leukemia Virus (MMLV) Reverse Transcriptase, prior to the PCR step (referred to as RT-qPCR).
- MMLVMoloney Murine Leukemia Virus
- RT-qPCRPerforming qPCR or RT-qPCR is well known in the art and all required components are commercially available.
- the qPCR step, and optionally the RT stepneed to be adapted to these modified nucleotides, e.g. by varying the enzymes, buffer or temperature or by using an alternative qPCR / RT-qPCR technique such as e.g. chemical-ligation qPCR (e.g. Boos et al., 2013, Nucleic Acids Res 41(15):e145; Shivalingam et al., 2020, Angew. Chem. Int. Ed., 59:11416–11422).
- “stability upon drying on a surface”relates to a decrease of max.
- a difference between Ct-values measured for (stability test sample G) – (stability negative control H) of 2corresponds to a 4-fold decrease in intact nucleic acid. Accordingly, a decrease of max. 75% intact nucleic acid corresponds to a delta Ct between stability test sample G and stability negative control H of max. 2.
- “stability upon drying on a surface”may be measured according to the following protocol (cf.
- example 5Air drying 50 ⁇ l of the analytical composition containing 0.005 ng/ ⁇ l of the LNPs encapsulating the nucleic acid on a glass surface, • rehydrating the dried composition with 200 ⁇ l water, • Stability test sample G: 2.5 units Benzonase in 10 ⁇ l rehydrated sample (1.25 ng/ml) diluted in 90 ⁇ l Tris buffer (10 mM, pH 7) with MgCl 2 (20 mM) • Stability negative control H: 10 ⁇ l rehydrated sample (1.25 ng/ml) diluted in 90 ⁇ l miliQ water • Incubating stability test sample G for 1 h at 37°C • Inactivating benzonase by incubation for 10 min at 75 °C • Measuring DNA concentration of stability test sample G and stability negative control H via qPCR (see Example 6, Step 5) • Calculating the concentration difference between stability test sample G and stability negative control H based on the difference between Ct values (delta Ct).
- the lipid nanoparticleshave a particle diameter in the range of between 50 nm to 1500 nm, advantageously in the range of between 50 nm to 300 nm, as measured by dynamic light scattering.
- the LNPs encapsulating the nucleic acidare lyophilized prior to use.
- the lyophilized LNPsare dissolved in a liquid prior to application on a surface.
- the lyophilized LNPsare dissolved in water or buffer, optionally containing additives as described herein.
- additives as described hereinmay also be lyophilized together with the LNPs.
- lipid nanoparticlescontain at least one lipid component.
- the at least one lipid componentis selected from the group consisting of ionisable lipids, phospholipids, cholesterol, lipid anchored PEGs, and mixtures thereof.
- the lipid nanoparticlescontain an ionisable lipid, advantageously in an amount of between 35 mol% and 100 mol%, e.g. 35 mol%, e.g. 50 mol% to 60 mol% such as 55 mol%, 56 mol%, 57 mol%, and 58 mol%, e.g. 100 mol%.
- the lipid nanoparticlesconsist of, or essentially consist of, ionisable lipids.
- the lipid nanoparticlescontain a phospholipid, advantageously in an amount of between 10 mol% and 50 mol%, e.g. 16 mol%, e.g. in a range of between 20 mol% to 35 mol% such as 26 mol%, 27 mol%, 28 mol%, 29 mol% and 30 mol%.
- the lipid nanoparticlescontain cholesterol, advantageously in an amount of between 10 mol% and 50 mol%, e.g.
- the lipid nanoparticlescontain a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%, advantageously in an amount of between 2 mol% and 8 mol% such as 2 mol%, 2.5 mol%, 5 mol% and 7 mol%.
- the lipid nanoparticlescontain an ionisable lipid, advantageously in an amount as described above and further contain at least one of the following: • a phospholipid, advantageously in an amount of between 10 mol% and 50 mol%, e.g. 16 mol%, e.g. in a range of between 20 mol% to 35 mol% such as 26 mol%, 28 mol% and 29 mol%; • cholesterol, advantageously in an amount of between 10 mol% and 50 mol%, e.g. in a range of between 10 mol% to 15 mol% such as 11 mol% and 12 mol%, e.g.
- the lipid nanoparticlescontain or consist of an ionisable lipid, advantageously in an amount as described above, and a phospholipid, advantageously in an amount as described above.
- the lipid nanoparticlescontain or consist of an ionisable lipid, advantageously in an amount as described above, and cholesterol, advantageously in an amount as described above.
- the lipid nanoparticlescontain or consist of an ionisable lipid, advantageously in an amount as described above, and a lipid anchored PEG, advantageously in an amount as described above.
- the lipid nanoparticlescontain or consist of an ionisable lipid, advantageously in an amount as described above, a phospholipid, advantageously in an amount as described above, and cholesterol, advantageously in an amount as described above.
- the lipid nanoparticlescontain or consist of an ionisable lipid, advantageously in an amount as described above, cholesterol, advantageously in an amount as described above and a lipid anchored PEG, advantageously in an amount as described above.
- the lipid nanoparticlescontain or consist of an ionisable lipid, advantageously in an amount as described above, a phospholipid, advantageously in an amount as described above, cholesterol, advantageously in an amount as described above, and a lipid anchored PEG, advantageously in an amount as described above.
- the ionisable lipidis selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more advantageously selected from the group consisting of oleylamine and 306Oi10, most advantageously 306Oi10.
- 306Oi10is also referred to as tetrakis(8-methylnonyl) 3,3',3'',3'''-((methylazanediyl)bis(propane-3,1- diyl))bis(azanetriyl))-tetrapropionate (cf. e.g.
- the phospholipidis selected from the group consisting of 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3- phosphocholin (DSPC), and mixtures thereof.
- DOPE1,2-dioleoyl-sn-glycero-3- phosphoethanolamine
- DSPC1,2-distearoyl-sn-glycero-3- phosphocholin
- the lipid anchored PEGis C14PEG1000, also referred to as 1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000].
- the lipid nanoparticlesconsist of, or essentially consist of, oleylamine.
- the lipid nanoparticlesconsist of, or essentially consist of, oleylamine, advantageously in an amount of between 55 mol% to 60 mol% such as 55 mol% or 58 mol%, DSPC, advantageously in an amount of between 25 mol% to 35 mol% such as 28 mol%, 29 mol% or 30 mol%, and cholesterol, advantageously in an amount of between 10 mol% to 15 mol% such as 12 mol% or 13 mol%.
- the lipid nanoparticlesconsist of, or essentially consist of, 58 mol% oleylamine, 29 mol% DSPC, and 13 mol% cholesterol.
- the lipid nanoparticlesconsist of, or essentially consist of, 58 mol% oleylamine, 30 mol% DSPC, and 12 mol% cholesterol.
- Non-lipid components of the lipid nanoparticlesin addition to the at least one lipid component, the lipid nanoparticles further contain an inert solid component, advantageously in an amount of 5 wt% to 80 wt%.
- inert solid componentrelates to an organic polymer, a metal oxide or semiconductor oxide that complies with the following characteristics: • Being solid at 22 °C; • having a primary particle size in the range of between 50 nm and 1500 nm; • being insoluble in water, ethanol and mixtures thereof, such as 70% V/V ethanol; i.e. max. 10 mg of the inert solid component can be dissolved in either of 100 ml of water, ethanol or a mixture thereof; • being unreactive with any of the lipid components described herein when being present in water at 22°C; i.e. the inert solid component does not chemically react with any of the lipid components described herein.
- the LNPsfurther contain an inert solid component
- the LNPshave a particle diameter in the range of between 50 nm to 1500 nm, advantageously in the range of between 50 nm to 300 nm, as measured by dynamic light scattering.
- the primary particle size of the inert solidis smaller than the particle diameter of the LNPs.
- the term “primary particle size”relates to the diameter of a single, non-aggregated particle.
- suitable organic polymersinclude polystyrene, polyethylene terephthalate (PET) and polyethene (PE).
- suitable metal oxidesinclude titania (TiO 2 ), alumina (Al 2 O 3 ), zirconia (ZrO 2 ) and ceria (CeO 2 ), advantageously titania (TiO2) and zirconia (ZrO 2 ).
- semiconductor oxiderelates to oxides of semiconductors, e.g. silica (SiO 2 ). The skilled person understands that silicon (Si) is a semiconductor whereas silica (SiO 2 ), i.e. a semiconductor oxide, is not a semiconductor.
- the term “semiconductor oxide”is not to be confused with the terms “semiconducting oxides” or “oxide semiconductors”, i.e. certain oxides that are semiconductors, e.g. zirconia (ZrO 2 ), zinc oxide (ZnO) and cadmium oxide (CdO).
- the inert solid componentis selected from the group consisting of titania (TiO2), zirconia (ZrO 2 ) and silica (SiO 2 ).
- the LNPscontain at least the following components: • an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol% and preferably selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more preferably selected from the group consisting of oleylamine and 306Oi10, most preferably 306Oi10; and • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO 2 ), alumina (Al 2 O 3 ), zirconia (ZrO 2 ) and ceria (CeO 2 )) and semiconductor oxides (preferably silica (SiO 2 )), preferably in an amount of between 5 wt% to 80 wt
- the analytical compositioncomprises additives.
- Ionic strength adjusting additivesIn certain advantageous embodiments, the analytical composition comprises ionic strength adjusting additives, including buffers and salts. Ionic strength adjusting additives may be added for example to adjust the zeta potential of a composition containing LNPs, thereby increasing the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs.
- ionic strength adjusting additivesA large variety of ionic strength adjusting additives is known in the field and suitable in the context of the present invention.
- suitable ionic strength adjusting additivesmay be selected from the group consisting of NaCl, such as an isotonic NaCl solution, PBS, citrate buffer, and isocitrate buffer.
- the analytical compositionhas a zeta potential in the range of between –30 mV to +30 mV as measured by electrophoretic light scattering, e.g. using a zetasizer.
- pH adjusting additivesThe skilled person understands that some additives, while having an effect on the ionic strength of the composition, may also be added to adjust the pH value. Adjusting the pH value may be performed e.g. to increase the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs.
- a large variety of pH adjusting additivesare known in the field and suitable in the context of the present invention, such as PBS, citrate buffer, and isocitrate buffer.
- the analytical compositionhas a pH value in the range of 6 to 8.
- Disaccharides and polysaccharidesIt has been surprisingly found that addition of disaccharides and polysaccharides increases the stability of the analytical composition upon drying.
- a large variety of disaccharides and polysaccharidesare known in the field and are suitable in the context of the present invention.
- the following disaccharides and polysaccharideshave been found particularly beneficial additives to increase the stability of the analytical composition upon drying:
- DisaccharidesTrehalose, sucrose, maltose, lactose, and mixtures thereof.
- PolysaccharidesAgarose, starch, dextran, and mixtures thereof.
- the wtthe wt.
- the analytical compositioncomprises PEG (also referred to as “free PEG”).
- PEGalso referred to as “free PEG”.
- free PEGis not a component of the LNPs but instead is added to the analytical composition and is present external to the LNPs.
- Addition of PEGhas been found to increase the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs.
- Addition of PEGhas also been found to increase the stability of the analytical composition upon drying.
- the analytical compositioncontains oleylamine, DSPC, cholesterol, and PEG1000, advantageously in a molar ratio of oleylamine:DSPC:cholesterol:PEG1000 of 55:28:12:5.
- the analytical compositioncontains oleylamine, DSPC, cholesterol and PEG2000, advantageously in a molar ratio of oleylamine:DSPC:cholesterol:PEG2000 of 57:29:12:2.
- the analytical compositioncontains hexadecylamine, DSPC, cholesterol and PEG600, advantageously in a molar ratio of hexadecylamine:DSPC:cholesterol:PEG600 of 56:26:11:7.
- the inventionrelates to a method for monitoring a disinfection procedure with an analytical composition as defined above for the first aspect.
- the methodcomprises the following steps: Step a: Applying the analytical composition onto a surface, advantageously air-drying the composition.
- at least 0.25 ng of the LNPs encapsulating the nucleic acidare applied on the surface, e.g.
- Step bAfter a pre-determined time period, taking a test sample from the surface or from a second surface to which at least part of the analytical composition has been transferred, thereby dissolving the test sample in an aqueous solution, e.g. water, to obtain an aqueous test sample, e.g. in one of the following manners: • adding water onto the (first) surface or the second surface, for example pipetting 200 ⁇ l water onto the surface, and recovering the water containing the aqueous test sample, e.g.
- the time period between step a and step bmay be varied depending on the cleaning strategy of the facility in which the analytical composition is used. For example, if the surface is scheduled to be disinfected every 24 h, the time period between step a and step b may also be 24 h.
- Step cSubjecting a portion A of the aqueous test sample to an enzymatic nuclease degradation step, in particular an enzymatic DNA degradation step, e.g. in the following manner: • Diluting a defined volume of the aqueous test sample, e.g. 10 ⁇ l, with a defined volume of Tris buffer (10 mM, pH 7) containing 20 mM MgCl2, e.g. 90 ⁇ l; • adding a suitable nuclease, e.g. 2.5 units of Benzonase; and • incubating the mixture, e.g. for 1 h at 37°C, • inactivating the nuclease, e.g.
- Step dExamining the portion A and a reference portion B of the aqueous test sample by a qPCR detection procedure using primers specific for the nucleic acid, in particular the DNA, encapsulated in the lipid nanoparticles, to differentiate with reference to portion B, whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A and was thus protected during the enzymatic DNA nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step.
- the following qPCR conditionsmay be used: • Preactivation for 240s at 95°C; • Amplification (40 cycles) with each cycle comprising 2 s at 95°C, followed by 12 s at 58°C, followed by 4 s at 72°C.
- the reference portion B of the aqueous test sampleis prepared by diluting a defined volume of the aqueous test sample, e.g. 10 ⁇ l, in a defined volume of water, e.g. 90 ⁇ l. It is understood that the reference portion B is not subjected to an enzymatic nuclease degradation step.
- steps b – dfurther comprise the following steps: Step b) additionally comprises taking a third test sample from a third additional surface, thereby dissolving the third test sample in an aqueous solution, e.g. water, to obtain a third aqueous test sample. Step c) additionally comprises subjecting a portion A’ of the third aqueous test sample to an enzymatic nuclease degradation step.
- Step b)additionally comprises taking a third test sample from a third additional surface, thereby dissolving the third test sample in an aqueous solution, e.g. water, to obtain a third aqueous test sample.
- Step c)additionally comprises subjecting a portion A’ of the third aqueous test sample to an enzymatic nuclease degradation step.
- Step d)additionally comprises examining the portion A’ and a reference portion B’ of the third aqueous test sample by a qPCR detection procedure using primers specific for the nucleic acid encapsulated in the lipid nanoparticles to differentiate with reference to portion B’, whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A’ and was thus protected during the enzymatic nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A’, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step, or • the analytical composition is not present on the third surface.
- the reference portion B’ of the third aqueous test sampleis prepared by diluting a defined volume of the third aqueous test sample, e.g. 10 ⁇ l, in a defined volume of water, e.g. 90 ⁇ l. It is understood that the reference portion B’ is not subjected to an enzymatic nuclease degradation step.
- the detected DNA from the test sample, or optionally the second test sampleoriginates from disintegrated LNPs if the difference between Ct values (A-B) – (A0-B0) > 2 with • A0 being a control portion of A, and • B0 being a control portion of B.
- differences in the nucleic acid concentrations between different samples or portions of a samplecan be determined by comparing Ct values measured by a qPCR detection procedure, e.g. qPCR or RT-qPCR. Ct values so measured can be converted to an absolute concentration of nucleic acid by means of a calibration curve. However, in the context of the instant invention, it is not required to convert the Ct value to an absolute nucleic acid concentration. Instead it is sufficient to compare Ct values between different samples or portions of a sample, i.e. a relative comparison of the nucleic acid concentrations is sufficient. Determining the Ct value from a sample or portion of a sample by a qPCR detection procedure, e.g.
- the detected DNA from the third test sampleoriginates from disintegrated LNPs if the difference between Ct values (A’-B’) – (A0-B0) > 2 with • A0 being a control portion of A, respectively A’, and • B0 being a control portion of B, respectively B’.
- the control portion A0is a control portion for both portion A and portion A’.
- the control portion B0is a control portion for both portion B and portion B’.
- a control portion B0is prepared from the analytical composition (i.e.
- a portion of the analytical composition that was stored before application) in the same manner as the reference portion Bis prepared from the aqueous test sample, e.g. by diluting a defined volume of the analytical composition, e.g. 10 ⁇ l, in a defined volume of water, e.g. 90 ⁇ l.
- a control portion A0is obtained by subjecting a portion of the analytical composition (i.e. a portion of the analytical composition that was stored before application) to the same enzymatic nuclease degradation step as portion A.
- a conclusion whether the (first) surface, and optionally the second surface and / or the third surface was disinfected between step a and step bmay be reached in the following manner: • Calculating the difference between the nucleic acid concentration in portion A and in the reference portion B; • Calculating the difference between the nucleic acid concentration in the control portion A0 and in the control portion B0; • Comparing the difference between the nucleic acid concentration in portion A and in the reference portion B, i.e. comparing the Ct values measured by a qPCR detection procedure, with the difference between the nucleic acid concentration in the control portion A0 and in the control portion B0, and • if the difference between Ct values A-B ⁇ A0-B0, i.e.
- the difference between Ct values (A’- B’) – (A0-B0) ⁇ 2the detected nucleic acid originates from intact lipid nanoparticles, i.e. the surface was not disinfected, and • optionally if the difference between Ct values A’-B ’ ⁇ A0-B0, i.e. the difference between Ct values (A’- B’) – (A0-B0) > 2 the detected nucleic acid originates from disintegrated lipid nanoparticles, i.e. the surface was disinfected or the analytical composition was not present on the third surface.
- steps a, c and dcomprise the following substeps:
- at least 0.25 ng of the LNPs encapsulating the nucleic acidare applied on the surface, e.g. 50 ⁇ l of the analytical composition containing the LNPs encapsulating the nucleic acid in a concentration of 0.005 ng/ ⁇ l.
- a-2Storing a defined volume of the analytical composition, e.g. 50 ⁇ l, thereby obtaining a stored analytical composition.
- c-2Subjecting a portion of the stored analytical composition to the same enzymatic nuclease degradation step as portion A, to thereby obtain a control portion A0.
- c-3Preparing a reference portion B of the aqueous test sample, e.g. by diluting a defined volume of the aqueous test sample, e.g. 10 ⁇ l, in a defined volume of water, e.g. 90 ⁇ l. It is understood that the reference portion B is not subjected to an enzymatic nuclease degradation step.
- c-4Preparing a control portion B0 of the stored analytical composition in the same manner as the reference potion B is prepared from the aqueous test sample, e.g. by diluting a defined volume of the stored analytical composition, e.g. 10 ⁇ l, in a defined volume of water, e.g. 90 ⁇ l. It is understood that the reference portion B0 is not subjected to an enzymatic nuclease degradation step.
- c-5Subjecting a portion A’ of the third aqueous test sample to the same enzymatic nuclease degradation step as portion A.
- c-6Preparing a reference portion B’ of the third aqueous test sample, e.g.
- qPCR conditionsmay be used: • Preactivation for 240 s at 95°C; • Amplification (40 cycles) with each cycle comprising 2 s at 95°C, followed by 12s at 58°C, followed by 4s at 72°C.
- d-2Quantifying the nucleic acid present in the control portion A0 and in the control portion B0 in the same manner as portion A and reference portion B.
- d-3Comparing the Ct values measured for the portion A, for the reference portion B, for the control portion A0 and for the control portion B0 to differentiate whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A and was thus protected during the enzymatic nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step.
- Optionally d-4Quantifying the nucleic acid, in particular the DNA, present in portion A’ and in the reference portion B’ of the third aqueous test sample by a qPCR detection procedure using primers that are specific for the encapsulated nucleic acid.
- Optionally d-5Comparing the Ct values measured for portion A’, for the reference portion B’, for the control portion A0 and for the control portion B0 to differentiate whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A’ and was thus protected during the enzymatic DNA nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A’, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic DNA nuclease degradation step.
- the inventionrelates to an analytical composition as described herein for the first aspect.
- the LNPs comprised within the analytical compositioncontain an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol%, and/or contain at least one of the following: • a phospholipid, preferably in an amount of between 10 mol% and 50 mol%, • cholesterol, preferably in an amount of between 10 mol% and 50 mol%, and • a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%; • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO 2 ), alumina (Al 2 O 3 ), zirconia (ZrO 2 ) and ceria (CeO 2 )) and semiconductor oxides (preferably silica (SiO 2 )), preferably in an amount of between 5 wt%
- the LNPscontain at least the following components: • an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol% and preferably selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more preferably selected from the group consisting of oleylamine and 306Oi10, most preferably 306Oi10; and • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO 2 ), alumina (Al 2 O 3 ), zirconia (ZrO 2 ) and ceria (CeO 2 )) and semiconductor oxides (preferably silica (SiO 2 )), preferably in an amount of 5 wt% to 80 wt%
- the inert solid componentis selected from the group consisting of titania (TiO 2 ), zirconia (ZrO 2 ) and silica (SiO 2 ).
- the inventionrelates to a kit of parts comprising an analytical composition as described herein for the first aspect, qPCR primers specific for the nucleic acid, and optionally one or more of the following: • a nuclease, • a DNA polymerase, • a qPCR buffer.
- ionisable lipid 306Oi10The ionisable lipid 306Oi10 was synthesised from 2-isodecyl acrylate (Sigma-Aldrich) and 3,3'-Diamino-N- methyldipropylamine (Aldrich-Fine Chemicals) in stochiometric amounts via stirring for 3 days at 90°C.
- Synthesis of TMAPS-functionalised silica particlesThe silica particles were functionalised by first sonicating a aqueous dispersion of silica nanoparticles (0.142 ⁇ m diameter, 5% w/v aq.
- LNP composition number 1LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10
- DOPEl,2-dioleoyl-sn-glycero-3- phosphoethanolamine, Avanti
- cholesterolSigma-Aldrich
- C14-PEG1000(1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000], ammonium salt, Avanti
- the previously synthesised ionisable lipid 306Oi10were dissolved in 90% ethanol and 10% 10 nM citrate buffer at molar ratios of 16 : 46.5 : 2.5 : 35.
- LNP composition number 2LNP comprising oleylamine, DSPC, cholesterol and PEG 600 First, oleylamine, DSPC, cholesterol and PEG 600 were dissolved in methanol at molar ratios of 54:27:11.5:7.5.
- an aqueous phasewas DNA prepared consisting of DNA (ds DNA, 62 nt; SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTTG AACACGAACAAACCTCTTT) in 0.1 M acetic acid with a final DNA concentration of 1 g/l. Both phases were preheated to 80°C and the methanol phase was added dropwise to the aqueous phase in 5:1 volumetric ratio (aqueous:methanol) and mixed by rapid pipette mixing. The mixture was allowed to cool down to room temperature.
- LNP composition number 3LNP comprising oleylamine First, oleylamine was dissolved in methanol.
- an aqueous phasewas DNA prepared consisting of DNA (ds DNA, 62 nt; SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTTGAACACGAACAAACC TCTTT) in 0.1 M acetic acid with a final DNA concentration of 1 g/l. Both phases were preheated to 80°C and the methanol phase was added dropwise to the aqueous phase in 2.5:1 volumetric ratio (aqueous:methanol) and mixed by rapid pipette mixing. The mixture was allowed to cool down to room temperature.
- LNP composition number 4LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10 with silica nanoparticles
- DOPEl,2-dioleoyl-sn-glycero-3- phosphoethanolamine, Avanti
- cholesterolSigma-Aldrich
- C14-PEG1000(1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000], ammonium salt, Avanti
- the previously synthesised ionisable lipid 306Oi10were dissolved in 90% ethanol and 10% 10 nM citrate buffer at molar ratios of 16 : 46.5 : 2.5 : 35.
- LNP composition number 5LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10 with silica nanoparticles via lipid rehydration
- DOPEl,2-dioleoyl-sn-glycero-3- phosphoethanolamine, Avanti
- cholesterolSigma-Aldrich
- C14-PEG10001,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000], ammonium salt, Avanti
- the previously synthesised ionisable lipid 306Oi10were dissolved in chloroform at molar ratios of 16 : 46.5 : 2.5 : 35.
- Example 2Preparation of analytical compositions To prepare an analytical composition, LNP dispersions were diluted in miliQ water to a final concentration of 0.005 ng/ ⁇ l. If additives were added, they were added in the respective amount to the LNP dispersion, which was subsequently diluted in miliQ water to a final concentration of 0.005 ng/ ⁇ l LNP.
- Example 3Disintegration of the LNPs in 70 vol.% ethanol The concentration of a LNP constituent lipid (i.e.
- any one of the lipid components being present in the LNP in an amount of at least 1 mol%)was measured in the continuous phase of an LNP dispersion comprising 150 ⁇ l LNP in miliQ water (0.2 ⁇ g/ ⁇ l constituent lipid; cf. example 2) in either 350 ⁇ l miliQ water (disintegration negative control D) or in 350 ⁇ l ethanol (resulting in 70% V/V ethanol; disintegration test sample C).
- the concentration of the lipid constituent in the continuous phasewas measured using a GC-MS-5977A series instrument (Agilent; column: DB-5MS 20 x 0.18 ⁇ m, oven temperature: 170°C ramped to 325°C at 8°C/min, with a final hold time of 20 min; detector temperature: 300°C). Results are shown in table 2 below.
- Step 1Application of the analytical composition on a surface. 50 ⁇ l of the analytical composition containing 0.005 ng/ ⁇ l LNPs encapsulating the DNA (SEQ ID NO: 1) were applied onto a clean glass surface (no residual DNA, LNPs on the surface). The analytical composition was air-dried on the surface. In addition, 200 ⁇ l of the same analytical composition containing 0.00125 ng/ ⁇ l LNPs encapsulating the DNA (sequence) were stored at 5°C for later use (referred to as the “stored analytical composition”). Step 2: Disinfection of the surface. The following disinfectants were tested, i.e.
- Step 3Taking a test sample from a surface.
- a test samplewas taken from the surface either • by pipetting 200 ⁇ l miliQ water onto the surface and recovering this liquid, or • by swabbing the surface with a premoistened swab and recovering the sample from the swab by dissolving it in 200 ⁇ l miliQ water.
- the results of the analysiswere comparable when either of the two methods of taking the test sample were used (step 3).
- the obtained test sampleis further referred to as the “aqueous test sample”.
- Step 4Enzymatic DNase digestion.
- a portion A of the aqueous test samplewas prepared by diluting 10 ⁇ l of the aqueous test sample with 90 ⁇ l of Tris buffer (10 mM, pH 7) containing 20 mM MgCl 2 .
- a reference portion B of the aqueous test samplewas prepared by diluting 10 ⁇ l of the aqueous test sample with 90 ⁇ l miliQ water. No nuclease digestion was performed on reference portion B (untreated sample).
- a control portion B0 of the stored analytical compositionwas prepared by diluting 10 ⁇ l of the stored analytical composition with 90 ⁇ l miliQ water. No nuclease digestion was performed on the control portion B0 (control for reference portion B).
- a control portion A0 of the stored analytical compositionwas prepared by diluting 10 ⁇ l of the stored analytical composition with 90 ⁇ l of Tris buffer (10mM, pH 7) containing 20 mM MgCl 2 . To this solution was added 2.5 units Benzonase and the mixture was incubated for 1 h at 37°C. After incubation, benzonase was inactivated by further incubation for 10 min at 75°C.
- Step 5qPCR analysis – assessment whether the surface was disinfected
- the following primerswere used: Forward primer (SEQ ID NO: 2): CTCTGCCCTTACGTTTATC Reverse primer (SEQ ID NO: 3): AGAGGTTTGTTCGTGTTC
- a qPCR master mixwas prepared for each sample by mixing 10 ⁇ l MasterMix (KAPA SYBR FAST for LightCycler 480, REF: KK4611 07959494001, kapa biosystems), 1 ⁇ l of each primer and 3 ⁇ l miliQ water.
- qPCRwas performed on a 96 well plate using a LightCycler 480 II, Roche Diagnostics by adding 15 ⁇ l of the respective qPCR master mix per well. Each sample (5 ⁇ l) was pipetted on the 96 well plate in duplicates and the plate was sealed. qPCR was performed according to the following protocol: Preactivation: 240 s at 95°C Amplification (40 cycles): 2 s at 95°C, 12 s at 58 °C, 4 s at 72 °C.
- a conclusion whether the surface was efficiently disinfected in step 2was reached in the following manner: • The difference between the Ct values measured for portion A and for the reference portion B was calculated; • The difference between the Ct values measured for the control portion A0 and for the control portion B0 was calculated; • The difference between the Ct values measured for portion A and for the reference portion B was compared to the difference between the Ct values measured for the control portion A0 and for the control portion B0; • if the difference between Ct values A-B ⁇ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) ⁇ 2, the detected nucleic acid originates from intact LNPs, i.e.
- LNP composition number 1LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10; cf. example 1
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description
Analytical composition for monitoring disinfection Technical Field The present invention relates to analytical compositions and kits, their use for monitoring disinfection procedures, and a method for monitoring a disinfection procedure. The analytical composition comprises lipid nanoparticles encapsulating a nucleic acid. Background Art In health care settings, it is crucial to lower the number of healthcare associated infections, which can lead to prolonged hospital stays, additive financial burden and excess deaths [World Health Organization. Report on the burden of endemic health care-associated infection world- wide. 2011]. Transmission events of pathogens via health care-worker's hands are well understood and increased hand hygiene is established as the most effective intervention strategy [Pittet et al., Lancet Infect. Dis. 6(10):641- 652, 2006]. Moreover, the importance of environmental surfaces in the endemic transmission of hospital pathogens is well-established [Otter et al., Am. J. Infect. Control, 41(5,Supplement):S6-Sll, 2013. Disinfection, Sterillzation and Antisepsis: Current Issues, New Research and New Technologies]. In order to combat healthcare associated infections, it is thus important to identify microbe transmission pathways via health care-worker's hands and environmental surfaces. Typically, microbial surrogate markers are used for monitoring the transmission of microorganisms. Examples of traditional surrogate markers are live non-pathogenic viruses, bacteriophages, such as bacteriophage MS2, or cauliflower mosaic virus DNA. However, such traditional surrogate markers often do not accurately mimic the dissemination of pathogens. In particular, traditional surrogate markers typically do not accurately reflect the susceptibility of pathogens to environmental disinfection [e.g. Alhmidi et al., Open Forum Infect. Dis., 4(3), 06, 2017]. In addition, use of live viruses is limited in sensitive areas, such as intensive care units. Furthermore, monitoring of surface disinfection procedures may be conducted e.g. via ATP testing or agar contact plates. However, use of these techniques is limited due to their relatively low sensitivity, a relatively long time until a result is obtained, and the dependence on the test result on several circumstantial factors, such as the temperature [R. E. Moon, ATP Testing: Use and Misuse in the Restoration Industry. The Cleaning Industry Research Institute 2021]. An innovative approach for monitoring of disinfection procedures and microbe transmission is the use of silica nanoparticles with encapsulated DNA (SPED) [e.g. Ullrich et al., Antimicrob. Resist. Infect. Control 11, Article number: 4, 2022]. However, despite representing a significant advancement in the field, SPED lack some physiochemical properties similar to microorganisms, in particular concerning the susceptibility to cleaning and disinfection procedures. Accordingly, whereas SPED is a sensitive tool to generally monitor pathogen transmission pathways, it may be improved to better reflect the influence of cleaning protocols on dissemination of pathogens. In view of the above, there is an unmet need to provide improved tools and methods for monitoring disinfection procedures and microbe transmission. Disclosure of the Invention The problem is solved by the subject matter of the independent claims. Further aspects of the invention are disclosed in the specification and independent claims, advantageous embodiments are disclosed in the specification and the dependent claims. The present invention will be described in more detail below. It is understood that the various embodiments, preferences and ranges as provided / disclosed in this specification may be combined at will. As used herein, the term "a", "an", "the" and similar terms used in the context of the present invention (especially in the context of the claims) are to be construed to cover both the singular and plural unless otherwise indicated herein or clearly contradicted by the context. As used herein, the terms "including", "containing" and "comprising" are used herein in their open, non-limiting sense. Advantageously, the term “monitoring a disinfection procedure” relates to an evaluation whether a successful disinfection of a surface was achieved. “Monitoring a disinfection procedure” thus includes validation of a disinfection procedure. Advantageously, the term further includes “monitoring transmission pathways of microorganisms between surfaces”, i.e. providing information whether microorganisms (the LNPs encapsulating a nucleic acid as described herein being a surrogate for microorganisms) have been transferred from a first surface to a second surface or a third surface, etc. Nucleic acid: The term "nucleic acid" (synonymously "polynucleotide") refers to a deoxyribonucleotide or ribonucleotide polymer, in linear or circular conformation, and in either single- or double-stranded form (ss or ds, respectively). The terms can encompass known analogs of natural nucleotides, as well as nucleotides that are modified in the base, sugar and/or phosphate moieties (e.g. phosphorothioate or phosphorodithioate backbones). Nucleotide: The term "nucleotide” refers to deoxyribonucleotides or ribonucleotides. The nucleotides may be standard nucleotides (i.e., adenosine, guanosine, cytidine, thymidine, and uridine) or nucleotide analogs. A nucleotide analog refers to a nucleotide having a modified purine or pyrimidine base or a modified ribose moiety. A nucleotide analog may be a naturally occurring nucleotide (e.g. inosine) or a non-naturally occurring nucleotide. Examples of non-naturally occurring nucleotides include nucleotides that are modified at the 2’-O position of the sugar such as 2’-OMe, 2’-MOE, 2’-F modified nucleotides, as well as locked nucleic acids (LNAs), peptide nucleic acids (PNA), and morpholinos. The skilled person understands that this list of modified nucleotides is not exhaustive and is able to select suitable modified nucleo- tides based on the present disclosure. Lipid: The term “lipid” is known in the field and advantageously relates to biomolecules that are soluble in non-polar solvents such as benzene, chloroform, acetone, etc. The term “lipid” includes for example fatty acids, waxes, sterols (e.g. cholesterol), fat-soluble vitamins (such as vitamins A, D, E, and K), monoglycerides, diglycerides, triglycerides, and phospholipids. In an aqueous environment, amphiphilic lipids (e.g. ionisable lipids and phospholipids) can form 3D structures such as lipid nanoparticles. Lipid nanoparticles: The term “lipid nanoparticle” (LNP) is known in the field [e.g. Hou et al., 2021, Nat Rev Mater 6, 1078–1094 and references therein]. LNPs are typically spherical with an average diameter between 20 nm and 1500 nm, e.g. between 50 nm to 1500 nm, such as between 50 nm to 300 nm. In the context of the present invention, components of the LNPs are typically selected from four types of lipids: an ionisable lipid (positive charge supports binding to negatively charged nucleic acid), a phospholipid (improves / stabilizes the structure of the LNPs), cholesterol (improves / stabilizes the structure of the LNPs), and a lipid anchored PEG (improves the stability of the LNP, e.g. by preventing aggregation). The skilled person understands that LNPs suitable for the present invention are non-toxic. Assessing the toxicity of LNPs is within the ordinary skill. Dispersion: The term “dispersion” is known in the field. In general, the term “dispersion” relates to a system in which distributed particles of one material are dispersed in a continuous phase of another material. In an aqueous dispersion, the continuous phase thus contains or consists of water. The term “dispersion” includes the terms “suspension” such as a “colloidal suspension”, “emulsion” and “solution”. Typically, a dispersion containing lipid nanoparticles relates to a suspension, usually a colloidal suspension. Moreover, lipid nanoparticle dispersions typically are aqueous suspensions, usually aqueous colloidal suspensions. Dispersed phase: The term “dispersed phase” is known in the field. In general, the term “dispersed phase” relates to colloids of a substance dispersed in another substance. For example, the dispersed phase of a lipid nanoparticle dispersion comprises of the lipid nanoparticles. Continuous phase: The term “continuous phase” is known in the field. In general, the term “continuous phase” relates to the fluid phase in a dispersion within which solid or fluid particles are distributed. For example, the continuous phase of a lipid nanoparticle dispersion is the aqueous liquid the lipid nanoparticles are dispersed in. N/P ratio: The term “N/P ratio” relates to the ratio of positively-chargeable polymer amine (N = nitrogen) groups to negatively-charged nucleic acid phosphate (P) groups. For example, such positively-chargeable polymer amine groups are typically present in ionisable lipids. Polyethylenglycol: The term “polyethylenglycol” (PEG; also referred to as polyethylene oxide) is known in the field and relates to a polymer, specifically a polyether, composed of ethylene oxide monomeric units. The structure of PEG is commonly expressed as H−(O−CH2−CH2)n−OH, wherein n is the number of ethylene oxide monomeric units. As is common in the field, a PEG polymer is defined by its molecular weight, e.g. PEG1000 refers to PEG having an average molecular weight of 1000 Da. The skilled person understands that the molecular weight of PEG, such as PEG1000, is an average molecular weight based on the polydispersity of PEG. Ionisable lipid: The term “ionisable lipid” is known in the field and relates to lipid molecules containing a chargeable group, typically an amine group, such as 306Oi10 [e.g. Hou et al., 2021, Nat Rev Mater 6, 1078–1094 and references therein]. Ionisable lipids remain neutral at physiological pH, but are protonated at low pH, making them positively charged, and thereby supporting condensation of nucleic acids, such as DNA or RNA, into LNPs. Phospholipid: The term “phospholipid” is known in the field and relates to amphiphilic lipid molecules containing a hydrophilic "head" containing a phosphate group and two hydrophobic "tails" derived from fatty acids, joined by an alcohol residue, typically a glycerol. The phosphate group may be further modified with organic molecules, typically serine (e.g. phosphatidylserine), choline (e.g. 1,2- distearoyl-sn-glycero-3-phosphocholin, DSPC), or ethanolamine (e.g. 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine; cf. e.g. Hou et al., 2021, Nat. Rev. Mater. 6, 1078–1094 and references therein). Phospholipids generally improve the structural stability of LNPs. Lipid anchored PEG: The term “lipid anchored PEG” is known in the field and relates to a PEG polymer covalently linked to a lipid (also referred to as PEG-derivatized lipids, PEG-lipids or PEGylated lipids). Lipid-anchored PEGs are generally described in, for example, U.S. 5,672,662 (also see e.g. Hou et al., 2021, Nat. Rev. Mater. 6, 1078–1094 and references therein). As is common in the field, a lipid anchored PEG is referred to in the following way: CxPEGy, wherein x is the number of carbon atoms in the fatty acid component of the lipid and y is the average molecular weight of the PEG component. The skilled person understands that the molecular weight of a lipid anchored PEG such as C14PEG1000 is an average molecular weight based on the polydispersity of PEG. Lipid anchored PEGs generally increase the stability of LNPs, e.g. by preventing aggregation. As an example, the chemical structure of C14PEG1000, also referred to as 1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)-1000] is shown below (ammonium salt):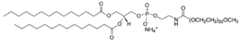 Disaccharide: The term “disaccharide” is known in the field and particularly describes carbohydrates composed of two monosaccharide units bound together by glycosidic linkages. It is understood that a disaccharide may comprise two identical monosaccharide units or may comprise two different monosaccharide units, which may also be chemically modified (e.g. amidated, sulphonated, acetylated, phosphorylated, etc). Typically found monosaccharides in said repeating units are cyclic or linear monosaccharides containing three to seven carbon atoms. Polysaccharide: The term “polysaccharide” is known in the field and particularly describes polymeric carbohydrates composed of monosaccharide units bound together by glycosidic linkages, either linear or branched. Such polysaccharides are characterized by their repeating units, each repeating unit described with their respective monosaccharide composition. Said repeating units include one or more monosaccharides which can also be chemically modified (e.g. amidated, sulphonated, acetylated, phos- phorylated, etc). Typically found monosaccharides in said repeating units are cyclic or linear monosaccharides containing three to seven carbon atoms. Throughout this specification a number of abbreviations are used, including: Delta Ct Difference between Ct values DLS Dynamic light scattering DNA Deoxyribonucleic acid DOPE 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine ds double-stranded DSPC 1,2-distearoyl-sn-glycero-3-phosphocholin GC-MS gas chromatography mass spectrometry HPLC high performance liquid chromatography LNP lipid nanoparticle MALDI-TOF Matrix-assisted laser desorption ionisation- time-of- flight mass spectrometry MMLV Moloney Murine Leukemia Virus PEG polyethylenglycol RNA ribonucleic acid SEM Scanning electron microscopy SPED silica nanoparticles with encapsulated DNA ss single-stranded qPCR quantitative polymerase chain reaction RT-qPCR reverse transcription quantitative polymerase chain reaction Wt.% weight % In a first aspect, the invention relates to the use of an analytical composition for monitoring disinfection procedures of surfaces. The analytical composition comprises lipid nanoparticles (LNPs) encapsulating a nucleic acid. The analytical composition further complies with the following characteristics: The LNPs disintegrate in 70% V/V ethanol. As used herein, the term “disintegration of lipid nanoparticles” relates to an increase in concentration of any one of the lipid components of the LNP in the continuous phase of at least 100% compared the continuous phase of a reference dispersion of the same lipid nanoparticles in water. Disintegration can be measured by determining in the continuous phase the concentration of any one of the lipids that are present in the respective LNP in signification amounts, i.e. any one of the lipid components being present in the LNP in an amount of at least 1 mol% (also referred to as “constituent lipid”), e.g. in an amount of between 1 mol% to 100 mol%, such as 1 mol% to 50 mol%. For example, the lipid component to be analysed may be cholesterol or an ionisable lipid, such as oleylamine, dodecylamine, hexadecylamine or 306Oi10. The concentration of any one of the lipids in the continuous phase can be measured by standard methods for lipid quantification, such as GC-MS, HPLC, MALDI-TOF. Advantageously, disintegration is measured by GC-MS, e.g. on a GC-MS-5977A series instrument (Agilent) fitted with a DB-5MS 20 x 0.18 µm column using the following conditions (cf. example 3): Time, oven temperature: 170°C ramped to 325°C at 8°C/min, with a final hold time of 20 min. Temperature of the detector: 300°C. The thereby determined concentration value is compared to the concentration of the same lipid in the continuous phase of a reference dispersion of the same LNPs in water. To measure disintegration of the LNPs, the dispersed phase is separated from the continuous phase by standard methods, usually employing centrifugation and filtration. Measuring the concentration of any one of the lipids in the continuous phase is within the ordinary skill. The specific quantification and separation methods used are not important as long as the same methods are used when measuring the concentration of the lipid in the continuous phase either of a dispersion of the LNP in water or of a dispersion of the same LNP in 70% V/V ethanol. Advantageously, the term “disintegration” relates to an increase in concentration by 100% of a lipid component of the LNPs (i.e. any lipid being present in the LNP in an amount of at least 1 mol%, e.g. in an amount of between 1 mol% to 100 mol%, such as 1 mol% to 50 mol%) in the continuous phase of a dispersion of the LNP in 70% V/V ethanol compared to the concentration of the same lipid in the continuous phase of a reference dispersion of the same lipid nanoparticles in water. It is understood that the reference dispersion does not contain ethanol. For example, disintegration may be measured according to the following protocol: • Diluting a sample of the LNP (containing min. 30 µg of the lipid component to be analysed) in either ethanol or miliQ water to yield two samples, one in water (disintegration control D) and one in 70% V/V ethanol solution (disintegration test sample C) • Separating the dispersed phase from the continuous phase of each sample via centrifugation (16000g for 30 min) followed by filtration (0.2 µm pores) • Measuring the concentration of the lipid component in the continuous phase via standard methods such as GC- MS, HPLC or MALDI-TOF, advantageously GC-MS, for each sample (disintegration test sample C: Value C; disintegration negative control D: Value D) • Calculating the increase in lipid concentration in the continuous phase: Increase = ((Value C - Value D) / Value D) * 100% • LNPs are regarded disintegrated in 70% V/V ethanol if the increase in the concentration lipid concentration in the continuous phase as calculated according to the above formula is at least 100%. In addition, the LNPs protect the nucleic acid encapsulated therein from nuclease degradation as measured by qPCR. The encapsulated nucleic acid has a length of between 60 – 200 nucleotides, advantageously of between 60 – 120 nucleotides. The wt. ratio between the nucleic acid and the LNPs in the analytical composition is in a range of between 0.01% to 15% nucleic acid, advantageously in a range of between 0.1% to 2% nucleic acid, such as 1% nucleic acid. The use of the analytical composition described above has the advantage that the encapsulated nucleic acid is protected from nuclease degradation as long as the LNPs are intact. However, upon contact with 70% V/V ethanol, i.e. the most common disinfection reagent, the LNPs disintegrate and the nucleic acid becomes susceptible to degradation, in particular nuclease degradation. It is considered beneficial that the nucleic acid can be quantified by well-established and sensitive detection methods, e.g. by quantitative polymerase chain reaction (qPCR). The LNPs encapsulating the nucleic acid as described above have the advantage that they more closely mimic common pathogens than the tools provided in the prior art. Based on the present disclosure, the skilled person understands that the chemical composition of the LNPs can be adapted to particularly mimic certain pathogens, e.g. multi-drug resistant bacteria such as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci. Such pathogens are of particular relevance in certain environments, e.g. in the intensive care unit of a hospital. The LNPs encapsulating the nucleic acid are thus improved surrogate markers for pathogens compared to the prior art. In particular, analytical compositions as described above have the advantage that the LNPs are susceptible to disinfection procedures. In consequence, the use of the analytical composition enables both monitoring the disinfection procedure of a single surface and monitoring the transmission of pathogens from a first surface to a further surface (e.g. a second surface, a third surface, etc.) while providing information whether the first surface or the one or more further surfaces have been disinfected. In addition, the use of the analytical composition as described above has the advantage that the LNPs are non- infectious and thus well suited for use in a hospital, e.g. in an intensive care unit. Further, the use of the analytical composition has the advantage that the nucleic acid can be detected with high sensitivity by established nucleic acid amplification techniques such as qPCR. In addition, the use of the analytical composition has the advantage that results can be obtained in a relatively short time period, typically results can be obtained within 2 hours. In an advantageous embodiment, the encapsulated nucleic acid is deoxyribonucleic acid (DNA). For example, the encapsulated DNA may have the nucleotide sequence according to SEQ ID NO: 1 shown below: SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTT GAACACGAACAAACCTCTTT In an alternative embodiment, the encapsulated nucleic acid is ribonucleic acid. In another embodiment, the nucleic acid comprises or consists of modified nucleotides. Based on the present disclosure, the skilled person understands that the nucleic acid may be either single- stranded (ss) or double-stranded (ds). Based on the present disclosure, the skilled person understands that the use of multiple analytical compositions, each containing LNPs encapsulating a nucleic acid, e.g. DNA, having a different sequence, enables monitoring of transmission pathways from multiple surfaces while providing information on the efficiency of the disinfection procedure. In an advantageous embodiment the lipid nanoparticles and the DNA encapsulated therein have a N/P ratio of between 1:1 and 10:1, e.g. between 5:1 and 10:1, such as 8:1 or 10:1. Advantageously, “protection from nuclease degradation”, such as protection from DNAse degradation, relates to a decrease of max. 75% of intact nucleic acid present in the analytical composition after treatment with a nuclease, e.g. benzonase, (protection test sample E) compared to a reference analytical composition that was not treated with the nuclease (protection negative control F), as measured by quantitative polymerase chain reaction (qPCR). The decrease in intact nucleic acid can be calculated from the difference between Ct values measured by qPCR according to the following formula: Decrease = 2^(Delta Ct). Thus, a difference between Ct-values measured for (protection test sample E) – (protection negative control F) of 2 corresponds to a 4-fold decrease in intact nucleic acid. Accordingly, a decrease of max. 75% intact nucleic acid corresponds to a delta Ct between protection test sample E and protection negative control F of max. 2. It is understood that the choice of the nuclease is adapted to the nucleic acid to be analysed. For example, DNase degradation may be measured by subjecting the analytical composition to digestion with benzonase. For example, protection from nuclease degradation may be measured according to the following protocol (cf. example 4): • Protection test sample E: 2.5 units Benzonase in 10 µl sample (1.25 ng/ml) diluted in 90 µl Tris buffer (10 mM, pH 7) with MgCl2 (20 mM) • Protection negative control F: 10 µl sample (1.25 ng/ml) diluted in 90 µl miliQ water • Incubating protection test sample E for 1 h at 37°C • Inactivating enzyme by incubation for 10 min at 75 °C • Measuring DNA concentration of protection test sample E and protection negative control F via qPCR (see Example 6, Step 5) • Calculating the concentration difference between protection test sample E and protection negative control F based on the difference between Ct values (delta Ct). However, the skilled person understands that if RNA is to be analysed, i.e. the encapsulated nucleic acid being RNA, a suitable RNase, e.g. RNase A, needs to be used to measure degradation of the nucleic acid. Moreover, the skilled person understands that qPCR measurement of RNA requires a reverse transcription step, e.g. using a reverse transcriptase such as Moloney Murine Leukemia Virus (MMLV) Reverse Transcriptase, prior to the PCR step (referred to as RT-qPCR). Performing qPCR or RT-qPCR is well known in the art and all required components are commercially available. The skilled person further understands that depending on the modified nucleotides, the qPCR step, and optionally the RT step, need to be adapted to these modified nucleotides, e.g. by varying the enzymes, buffer or temperature or by using an alternative qPCR / RT-qPCR technique such as e.g. chemical-ligation qPCR (e.g. Boos et al., 2013, Nucleic Acids Res 41(15):e145; Shivalingam et al., 2020, Angew. Chem. Int. Ed., 59:11416–11422). Advantageously, “stability upon drying on a surface” relates to a decrease of max. 75% of intact nucleic acid present in the analytical composition after drying on a glass surface and after treatment with a nuclease, e.g. benzonase, (stability test sample G) compared to a reference analytical composition that was also dried on a glass surface, but not treated with the nuclease (stability negative control H), as measured by qPCR. As for the “protection from nuclease degradation”, the decrease in intact nucleic acid can be calculated from the difference between Ct values measured by qPCR according to the following formula: Decrease = 2^(Delta Ct). Thus, a difference between Ct-values measured for (stability test sample G) – (stability negative control H) of 2 corresponds to a 4-fold decrease in intact nucleic acid. Accordingly, a decrease of max. 75% intact nucleic acid corresponds to a delta Ct between stability test sample G and stability negative control H of max. 2. For example, “stability upon drying on a surface” may be measured according to the following protocol (cf. example 5): • Air drying 50 µl of the analytical composition containing 0.005 ng/µl of the LNPs encapsulating the nucleic acid on a glass surface, • rehydrating the dried composition with 200 µl water, • Stability test sample G: 2.5 units Benzonase in 10 µl rehydrated sample (1.25 ng/ml) diluted in 90 µl Tris buffer (10 mM, pH 7) with MgCl2 (20 mM) • Stability negative control H: 10 µl rehydrated sample (1.25 ng/ml) diluted in 90 µl miliQ water • Incubating stability test sample G for 1 h at 37°C • Inactivating benzonase by incubation for 10 min at 75 °C • Measuring DNA concentration of stability test sample G and stability negative control H via qPCR (see Example 6, Step 5) • Calculating the concentration difference between stability test sample G and stability negative control H based on the difference between Ct values (delta Ct). In an advantageous embodiment, the lipid nanoparticles have a particle diameter in the range of between 50 nm to 1500 nm, advantageously in the range of between 50 nm to 300 nm, as measured by dynamic light scattering. In one embodiment, the LNPs encapsulating the nucleic acid are lyophilized prior to use. In this embodiment, the lyophilized LNPs are dissolved in a liquid prior to application on a surface. Typically the lyophilized LNPs are dissolved in water or buffer, optionally containing additives as described herein. The skilled person understands that additives as described herein may also be lyophilized together with the LNPs. Components of the lipid nanoparticles: Lipid components: As is known in the field, lipid nanoparticles contain at least one lipid component. In advantageous embodiments, the at least one lipid component is selected from the group consisting of ionisable lipids, phospholipids, cholesterol, lipid anchored PEGs, and mixtures thereof. In an advantageous embodiment, the lipid nanoparticles contain an ionisable lipid, advantageously in an amount of between 35 mol% and 100 mol%, e.g. 35 mol%, e.g. 50 mol% to 60 mol% such as 55 mol%, 56 mol%, 57 mol%, and 58 mol%, e.g. 100 mol%. Thus in an advantageous embodiment, the lipid nanoparticles consist of, or essentially consist of, ionisable lipids. In an advantageous embodiment, the lipid nanoparticles contain a phospholipid, advantageously in an amount of between 10 mol% and 50 mol%, e.g. 16 mol%, e.g. in a range of between 20 mol% to 35 mol% such as 26 mol%, 27 mol%, 28 mol%, 29 mol% and 30 mol%. In an advantageous embodiment, the lipid nanoparticles contain cholesterol, advantageously in an amount of between 10 mol% and 50 mol%, e.g. in a range of between 10 mol% to 15 mol% such as 10 mol%, 11 mol% and 12 mol%, e.g. in a range of between 45 mol% to 50 mol% such as 45 mol%, 46.5 mol% and 48 mol%. In an advantageous embodiment, the lipid nanoparticles contain a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%, advantageously in an amount of between 2 mol% and 8 mol% such as 2 mol%, 2.5 mol%, 5 mol% and 7 mol%. In an advantageous embodiment, the lipid nanoparticles contain an ionisable lipid, advantageously in an amount as described above and further contain at least one of the following: • a phospholipid, advantageously in an amount of between 10 mol% and 50 mol%, e.g. 16 mol%, e.g. in a range of between 20 mol% to 35 mol% such as 26 mol%, 28 mol% and 29 mol%; • cholesterol, advantageously in an amount of between 10 mol% and 50 mol%, e.g. in a range of between 10 mol% to 15 mol% such as 11 mol% and 12 mol%, e.g. in a range of between 45 mol% to 50 mol% such as 45 mol%, 46.5 mol% and 48 mol%; • a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%, advantageously in an amount of between 2 mol% and 8 mol% such as 2 mol%, 2.5 mol%, 5 mol% and 7 mol%. In an advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, and a phospholipid, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, and cholesterol, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, and a lipid anchored PEG, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, a phospholipid, advantageously in an amount as described above, and cholesterol, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, cholesterol, advantageously in an amount as described above and a lipid anchored PEG, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, a phospholipid, advantageously in an amount as described above, cholesterol, advantageously in an amount as described above, and a lipid anchored PEG, advantageously in an amount as described above. Advantageously, the ionisable lipid is selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more advantageously selected from the group consisting of oleylamine and 306Oi10, most advantageously 306Oi10. 306Oi10 is also referred to as tetrakis(8-methylnonyl) 3,3',3'',3'''-((methylazanediyl)bis(propane-3,1- diyl))bis(azanetriyl))-tetrapropionate (cf. e.g. Hou et al., 2021, Nat Rev Mater 6, 1078–1094). Advantageously, the phospholipid is selected from the group consisting of 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3- phosphocholin (DSPC), and mixtures thereof. Advantageously, the lipid anchored PEG is C14PEG1000, also referred to as 1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000]. In an embodiment, the lipid nanoparticles consist of, or essentially consist of, oleylamine. In another embodiment, the lipid nanoparticles consist of, or essentially consist of, oleylamine, advantageously in an amount of between 55 mol% to 60 mol% such as 55 mol% or 58 mol%, DSPC, advantageously in an amount of between 25 mol% to 35 mol% such as 28 mol%, 29 mol% or 30 mol%, and cholesterol, advantageously in an amount of between 10 mol% to 15 mol% such as 12 mol% or 13 mol%. Thus in an embodiment, the lipid nanoparticles consist of, or essentially consist of, 58 mol% oleylamine, 29 mol% DSPC, and 13 mol% cholesterol. In another embodiment, the lipid nanoparticles consist of, or essentially consist of, 58 mol% oleylamine, 30 mol% DSPC, and 12 mol% cholesterol. Non-lipid components of the lipid nanoparticles: In certain embodiments, in addition to the at least one lipid component, the lipid nanoparticles further contain an inert solid component, advantageously in an amount of 5 wt% to 80 wt%. As used herein, the term “inert solid component” relates to an organic polymer, a metal oxide or semiconductor oxide that complies with the following characteristics: • Being solid at 22 °C; • having a primary particle size in the range of between 50 nm and 1500 nm; • being insoluble in water, ethanol and mixtures thereof, such as 70% V/V ethanol; i.e. max. 10 mg of the inert solid component can be dissolved in either of 100 ml of water, ethanol or a mixture thereof; • being unreactive with any of the lipid components described herein when being present in water at 22°C; i.e. the inert solid component does not chemically react with any of the lipid components described herein. Accordingly, in case the LNPs further contain an inert solid component, the LNPs have a particle diameter in the range of between 50 nm to 1500 nm, advantageously in the range of between 50 nm to 300 nm, as measured by dynamic light scattering. It is understood that the primary particle size of the inert solid is smaller than the particle diameter of the LNPs. As used herein, the term “primary particle size” relates to the diameter of a single, non-aggregated particle. An advantage of including an inert solid component into the LNPs is that the LNPs have an increased stability upon drying compared to LNPs that do not contain an inert solid component as described herein. Examples of suitable organic polymers include polystyrene, polyethylene terephthalate (PET) and polyethene (PE). Examples of suitable metal oxides include titania (TiO2), alumina (Al2O3), zirconia (ZrO2) and ceria (CeO2), advantageously titania (TiO2) and zirconia (ZrO2). As used herein, the term “semiconductor oxide” relates to oxides of semiconductors, e.g. silica (SiO2). The skilled person understands that silicon (Si) is a semiconductor whereas silica (SiO2), i.e. a semiconductor oxide, is not a semiconductor. Thus, the term “semiconductor oxide” is not to be confused with the terms “semiconducting oxides” or “oxide semiconductors”, i.e. certain oxides that are semiconductors, e.g. zirconia (ZrO2), zinc oxide (ZnO) and cadmium oxide (CdO). In an advantageous embodiment, the inert solid component is selected from the group consisting of titania (TiO2), zirconia (ZrO2) and silica (SiO2). In certain embodiments, the LNPs contain at least the following components: • an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol% and preferably selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more preferably selected from the group consisting of oleylamine and 306Oi10, most preferably 306Oi10; and • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO2), alumina (Al2O3), zirconia (ZrO2) and ceria (CeO2)) and semiconductor oxides (preferably silica (SiO2)), preferably in an amount of between 5 wt% to 80 wt%. Additives In an advantageous embodiment, the analytical composition comprises additives. Ionic strength adjusting additives In certain advantageous embodiments, the analytical composition comprises ionic strength adjusting additives, including buffers and salts. Ionic strength adjusting additives may be added for example to adjust the zeta potential of a composition containing LNPs, thereby increasing the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs. A large variety of ionic strength adjusting additives is known in the field and suitable in the context of the present invention. For example, suitable ionic strength adjusting additives may be selected from the group consisting of NaCl, such as an isotonic NaCl solution, PBS, citrate buffer, and isocitrate buffer. Advantageously, the analytical composition has a zeta potential in the range of between –30 mV to +30 mV as measured by electrophoretic light scattering, e.g. using a zetasizer. pH adjusting additives The skilled person understands that some additives, while having an effect on the ionic strength of the composition, may also be added to adjust the pH value. Adjusting the pH value may be performed e.g. to increase the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs. A large variety of pH adjusting additives are known in the field and suitable in the context of the present invention, such as PBS, citrate buffer, and isocitrate buffer. Advantageously, the analytical composition has a pH value in the range of 6 to 8. Disaccharides and polysaccharides It has been surprisingly found that addition of disaccharides and polysaccharides increases the stability of the analytical composition upon drying. A large variety of disaccharides and polysaccharides are known in the field and are suitable in the context of the present invention. The following disaccharides and polysaccharides have been found particularly beneficial additives to increase the stability of the analytical composition upon drying: Disaccharides: Trehalose, sucrose, maltose, lactose, and mixtures thereof. Polysaccharides: Agarose, starch, dextran, and mixtures thereof. Advantageously, the wt. ratio between the lipid nanoparticles and the additives is in a range of between 1:0.1 to 1:20, more advantageously in a range of between 1:1 to 1:10 Polyethylenglycol In certain advantageous embodiments, the analytical composition comprises PEG (also referred to as “free PEG”). The skilled person understands that in contrast to lipid anchored PEG, free PEG is not a component of the LNPs but instead is added to the analytical composition and is present external to the LNPs. Addition of PEG has been found to increase the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs. Addition of PEG has also been found to increase the stability of the analytical composition upon drying. A large variety of PEGs are known in the field and are suitable in the context of the present invention. The following PEGs have been found particularly beneficial additives to increase the stability of the analytical composition upon drying: PEG600, PEG1000, PEG2000, PEG6000. In an advantageous embodiment, the analytical composition contains oleylamine, DSPC, cholesterol, and PEG1000, advantageously in a molar ratio of oleylamine:DSPC:cholesterol:PEG1000 of 55:28:12:5. In another advantageous embodiment, the analytical composition contains oleylamine, DSPC, cholesterol and PEG2000, advantageously in a molar ratio of oleylamine:DSPC:cholesterol:PEG2000 of 57:29:12:2. In another advantageous embodiment, the analytical composition contains hexadecylamine, DSPC, cholesterol and PEG600, advantageously in a molar ratio of hexadecylamine:DSPC:cholesterol:PEG600 of 56:26:11:7. In a second aspect, the invention relates to a method for monitoring a disinfection procedure with an analytical composition as defined above for the first aspect. The method comprises the following steps: Step a: Applying the analytical composition onto a surface, advantageously air-drying the composition. Advantageously, at least 0.25 ng of the LNPs encapsulating the nucleic acid are applied on the surface, e.g. 50 µl of the analytical composition containing 0.005 ng/µl of the LNPs encapsulating the nucleic acid. Step b: After a pre-determined time period, taking a test sample from the surface or from a second surface to which at least part of the analytical composition has been transferred, thereby dissolving the test sample in an aqueous solution, e.g. water, to obtain an aqueous test sample, e.g. in one of the following manners: • adding water onto the (first) surface or the second surface, for example pipetting 200 µl water onto the surface, and recovering the water containing the aqueous test sample, e.g. by pipetting; or • swabbing the (first) surface or the second surface with a premoistened absorbent means, e.g. a swab, and recovering the test sample from the absorbent means by dissolving it in water, e.g. in 200 µl water. Advantageously, the skilled person understands that “dissolving the test sample” also includes “obtaining a suspension of the test sample”. Advantageously, the time period between step a and step b may be varied depending on the cleaning strategy of the facility in which the analytical composition is used. For example, if the surface is scheduled to be disinfected every 24 h, the time period between step a and step b may also be 24 h. Step c: Subjecting a portion A of the aqueous test sample to an enzymatic nuclease degradation step, in particular an enzymatic DNA degradation step, e.g. in the following manner: • Diluting a defined volume of the aqueous test sample, e.g. 10 µl, with a defined volume of Tris buffer (10 mM, pH 7) containing 20 mM MgCl2, e.g. 90 µl; • adding a suitable nuclease, e.g. 2.5 units of Benzonase; and • incubating the mixture, e.g. for 1 h at 37°C, • inactivating the nuclease, e.g. by incubating for 10 min at 75°C. Step d: Examining the portion A and a reference portion B of the aqueous test sample by a qPCR detection procedure using primers specific for the nucleic acid, in particular the DNA, encapsulated in the lipid nanoparticles, to differentiate with reference to portion B, whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A and was thus protected during the enzymatic DNA nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step. For example, the following qPCR conditions may be used: • Preactivation for 240s at 95°C; • Amplification (40 cycles) with each cycle comprising 2 s at 95°C, followed by 12 s at 58°C, followed by 4 s at 72°C. Advantageously, the reference portion B of the aqueous test sample is prepared by diluting a defined volume of the aqueous test sample, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B is not subjected to an enzymatic nuclease degradation step. In an advantageous embodiment, steps b – d further comprise the following steps: Step b) additionally comprises taking a third test sample from a third additional surface, thereby dissolving the third test sample in an aqueous solution, e.g. water, to obtain a third aqueous test sample. Step c) additionally comprises subjecting a portion A’ of the third aqueous test sample to an enzymatic nuclease degradation step. Step d) additionally comprises examining the portion A’ and a reference portion B’ of the third aqueous test sample by a qPCR detection procedure using primers specific for the nucleic acid encapsulated in the lipid nanoparticles to differentiate with reference to portion B’, whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A’ and was thus protected during the enzymatic nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A’, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step, or • the analytical composition is not present on the third surface. Advantageously, the reference portion B’ of the third aqueous test sample is prepared by diluting a defined volume of the third aqueous test sample, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B’ is not subjected to an enzymatic nuclease degradation step. In an advantageous embodiment, it is concluded that the detected DNA from the test sample, or optionally the second test sample, originates from disintegrated LNPs if the difference between Ct values (A-B) – (A0-B0) > 2 with • A0 being a control portion of A, and • B0 being a control portion of B. As is common in the field, differences in the nucleic acid concentrations between different samples or portions of a sample can be determined by comparing Ct values measured by a qPCR detection procedure, e.g. qPCR or RT-qPCR. Ct values so measured can be converted to an absolute concentration of nucleic acid by means of a calibration curve. However, in the context of the instant invention, it is not required to convert the Ct value to an absolute nucleic acid concentration. Instead it is sufficient to compare Ct values between different samples or portions of a sample, i.e. a relative comparison of the nucleic acid concentrations is sufficient. Determining the Ct value from a sample or portion of a sample by a qPCR detection procedure, e.g. qPCR or RT-qPCR, is within the ordinary skill. Furthermore, establishing a calibration curve is within the ordinary skill. In a further advantageous embodiment, it is concluded that the detected DNA from the third test sample originates from disintegrated LNPs if the difference between Ct values (A’-B’) – (A0-B0) > 2 with • A0 being a control portion of A, respectively A’, and • B0 being a control portion of B, respectively B’. The skilled person understands that the control portion A0 is a control portion for both portion A and portion A’. The skilled person further understands that the control portion B0 is a control portion for both portion B and portion B’. Advantageously, a control portion B0 is prepared from the analytical composition (i.e. a portion of the analytical composition that was stored before application) in the same manner as the reference portion B is prepared from the aqueous test sample, e.g. by diluting a defined volume of the analytical composition, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B0 is not subjected to an enzymatic nuclease degradation step. Advantageously, a control portion A0 is obtained by subjecting a portion of the analytical composition (i.e. a portion of the analytical composition that was stored before application) to the same enzymatic nuclease degradation step as portion A. For example, a conclusion whether the (first) surface, and optionally the second surface and / or the third surface was disinfected between step a and step b may be reached in the following manner: • Calculating the difference between the nucleic acid concentration in portion A and in the reference portion B; • Calculating the difference between the nucleic acid concentration in the control portion A0 and in the control portion B0; • Comparing the difference between the nucleic acid concentration in portion A and in the reference portion B, i.e. comparing the Ct values measured by a qPCR detection procedure, with the difference between the nucleic acid concentration in the control portion A0 and in the control portion B0, and • if the difference between Ct values A-B ≈ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) < 2 the detected nucleic acid originates from intact lipid nanoparticles, i.e. the surface was not disinfected; • if the difference between Ct values A-B ≫ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) > 2 the detected nucleic acid originates from disintegrated lipid nanoparticles, i.e. the surface was disinfected; and • optionally if the difference between Ct values A’-B’ ≈ A0-B0, i.e. the difference between Ct values (A’- B’) – (A0-B0) < 2 the detected nucleic acid originates from intact lipid nanoparticles, i.e. the surface was not disinfected, and • optionally if the difference between Ct values A’-B ’ ≫ A0-B0, i.e. the difference between Ct values (A’- B’) – (A0-B0) > 2 the detected nucleic acid originates from disintegrated lipid nanoparticles, i.e. the surface was disinfected or the analytical composition was not present on the third surface. In a further advantageous embodiment, steps a, c and d comprise the following substeps: Step a: a-1: Applying the analytical composition onto a surface, advantageously air-drying the composition. Advantageously, at least 0.25 ng of the LNPs encapsulating the nucleic acid are applied on the surface, e.g. 50 µl of the analytical composition containing the LNPs encapsulating the nucleic acid in a concentration of 0.005 ng/µl. a-2: Storing a defined volume of the analytical composition, e.g. 50 µl, thereby obtaining a stored analytical composition. Step c: c-1: Subjecting a portion A of the aqueous test sample to an enzymatic nuclease degradation step, in particular an enzymatic DNA degradation step, e.g. in the following manner: • Diluting a defined volume, e.g. 10 µl, of the aqueous test sample with a defined volume, e.g. 90 µl, of Tris buffer (10 mM, pH 7) containing 20 mM MgCl2; • adding a suitable nuclease, e.g. 2.5 units Benzonase; and • incubating the mixture, e.g. for 1 h at 37°C, • inactivating the enzyme, e.g. by incubating for 10 min at 75°C. c-2: Subjecting a portion of the stored analytical composition to the same enzymatic nuclease degradation step as portion A, to thereby obtain a control portion A0. c-3: Preparing a reference portion B of the aqueous test sample, e.g. by diluting a defined volume of the aqueous test sample, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B is not subjected to an enzymatic nuclease degradation step. c-4: Preparing a control portion B0 of the stored analytical composition in the same manner as the reference potion B is prepared from the aqueous test sample, e.g. by diluting a defined volume of the stored analytical composition, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B0 is not subjected to an enzymatic nuclease degradation step. Optionally c-5: Subjecting a portion A’ of the third aqueous test sample to the same enzymatic nuclease degradation step as portion A. Optionally c-6: Preparing a reference portion B’ of the third aqueous test sample, e.g. by diluting a defined volume of the third aqueous test sample, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B’ is not subjected to an enzymatic nuclease degradation step. Step d: d-1: Quantifying the nucleic acid, in particular the DNA, present in portion A and in the reference portion B by a qPCR detection procedure using primers that are specific for the encapsulated nucleic acid. For example, the following qPCR conditions may be used: • Preactivation for 240 s at 95°C; • Amplification (40 cycles) with each cycle comprising 2 s at 95°C, followed by 12s at 58°C, followed by 4s at 72°C. d-2: Quantifying the nucleic acid present in the control portion A0 and in the control portion B0 in the same manner as portion A and reference portion B. d-3: Comparing the Ct values measured for the portion A, for the reference portion B, for the control portion A0 and for the control portion B0 to differentiate whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A and was thus protected during the enzymatic nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step. Optionally d-4: Quantifying the nucleic acid, in particular the DNA, present in portion A’ and in the reference portion B’ of the third aqueous test sample by a qPCR detection procedure using primers that are specific for the encapsulated nucleic acid. Optionally d-5: Comparing the Ct values measured for portion A’, for the reference portion B’, for the control portion A0 and for the control portion B0 to differentiate whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A’ and was thus protected during the enzymatic DNA nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A’, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic DNA nuclease degradation step. In a third aspect, the invention relates to an analytical composition as described herein for the first aspect. Advantageously, the LNPs comprised within the analytical composition contain an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol%, and/or contain at least one of the following: • a phospholipid, preferably in an amount of between 10 mol% and 50 mol%, • cholesterol, preferably in an amount of between 10 mol% and 50 mol%, and • a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%; • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO2), alumina (Al2O3), zirconia (ZrO2) and ceria (CeO2)) and semiconductor oxides (preferably silica (SiO2)), preferably in an amount of between 5 wt% to 80 wt%. In an advantageous embodiment, the LNPs contain at least the following components: • an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol% and preferably selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more preferably selected from the group consisting of oleylamine and 306Oi10, most preferably 306Oi10; and • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO2), alumina (Al2O3), zirconia (ZrO2) and ceria (CeO2)) and semiconductor oxides (preferably silica (SiO2)), preferably in an amount of 5 wt% to 80 wt%. In an advantageous embodiment, the inert solid component is selected from the group consisting of titania (TiO2), zirconia (ZrO2) and silica (SiO2). In a fourth aspect, the invention relates to a kit of parts comprising an analytical composition as described herein for the first aspect, qPCR primers specific for the nucleic acid, and optionally one or more of the following: • a nuclease, • a DNA polymerase, • a qPCR buffer. Brief Description of the Drawings The invention will be better understood and objects other than those set forth above will become apparent from the annexed drawings and the detailed description thereof. Fig. 1: Scheme of LNP and exemplary components Legend: 1 – Schematic representation of LNP 2 – Schematic representation of encapsulated DNA within the LNP 3 – Schematic representation of lipid anchored PEG 4 - Schematic representation of ionizable lipid 5 - Schematic representation of cholesterol 6 - Schematic representation of DNA Fig. 2: Scheme of workflow to determine the amount/ratio of intact LNPs in a sample. Legend: 1 – sample of LNPs 2 – DNAse / Benzonase treatment 3 – qPCR quantification 4 – Fluorescence 5 – PCR cycle number 6 – difference in cycle threshold, Delta Ct A – sample portion A B – sample portion B Fig. 3: Scheme of workflow to determine if a successful disinfection procedure was carried out. Legend: 1 – Surface with dried LNPs 2 – treatment 3 – disinfection with ethanol 4 – no disinfection, wiping with water 5 – rehydration to recover sample from surface 6 – analysis to determine the amount of intact LNPs in the sample (see fig.1) 7 – fluorescence 8 – PCR cycle number 9 - difference in cycle threshold, Delta Ct 10 – cycle threshold value from qPCR measurement A – sample portion A B – sample portion B
Disaccharide: The term “disaccharide” is known in the field and particularly describes carbohydrates composed of two monosaccharide units bound together by glycosidic linkages. It is understood that a disaccharide may comprise two identical monosaccharide units or may comprise two different monosaccharide units, which may also be chemically modified (e.g. amidated, sulphonated, acetylated, phosphorylated, etc). Typically found monosaccharides in said repeating units are cyclic or linear monosaccharides containing three to seven carbon atoms. Polysaccharide: The term “polysaccharide” is known in the field and particularly describes polymeric carbohydrates composed of monosaccharide units bound together by glycosidic linkages, either linear or branched. Such polysaccharides are characterized by their repeating units, each repeating unit described with their respective monosaccharide composition. Said repeating units include one or more monosaccharides which can also be chemically modified (e.g. amidated, sulphonated, acetylated, phos- phorylated, etc). Typically found monosaccharides in said repeating units are cyclic or linear monosaccharides containing three to seven carbon atoms. Throughout this specification a number of abbreviations are used, including: Delta Ct Difference between Ct values DLS Dynamic light scattering DNA Deoxyribonucleic acid DOPE 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine ds double-stranded DSPC 1,2-distearoyl-sn-glycero-3-phosphocholin GC-MS gas chromatography mass spectrometry HPLC high performance liquid chromatography LNP lipid nanoparticle MALDI-TOF Matrix-assisted laser desorption ionisation- time-of- flight mass spectrometry MMLV Moloney Murine Leukemia Virus PEG polyethylenglycol RNA ribonucleic acid SEM Scanning electron microscopy SPED silica nanoparticles with encapsulated DNA ss single-stranded qPCR quantitative polymerase chain reaction RT-qPCR reverse transcription quantitative polymerase chain reaction Wt.% weight % In a first aspect, the invention relates to the use of an analytical composition for monitoring disinfection procedures of surfaces. The analytical composition comprises lipid nanoparticles (LNPs) encapsulating a nucleic acid. The analytical composition further complies with the following characteristics: The LNPs disintegrate in 70% V/V ethanol. As used herein, the term “disintegration of lipid nanoparticles” relates to an increase in concentration of any one of the lipid components of the LNP in the continuous phase of at least 100% compared the continuous phase of a reference dispersion of the same lipid nanoparticles in water. Disintegration can be measured by determining in the continuous phase the concentration of any one of the lipids that are present in the respective LNP in signification amounts, i.e. any one of the lipid components being present in the LNP in an amount of at least 1 mol% (also referred to as “constituent lipid”), e.g. in an amount of between 1 mol% to 100 mol%, such as 1 mol% to 50 mol%. For example, the lipid component to be analysed may be cholesterol or an ionisable lipid, such as oleylamine, dodecylamine, hexadecylamine or 306Oi10. The concentration of any one of the lipids in the continuous phase can be measured by standard methods for lipid quantification, such as GC-MS, HPLC, MALDI-TOF. Advantageously, disintegration is measured by GC-MS, e.g. on a GC-MS-5977A series instrument (Agilent) fitted with a DB-5MS 20 x 0.18 µm column using the following conditions (cf. example 3): Time, oven temperature: 170°C ramped to 325°C at 8°C/min, with a final hold time of 20 min. Temperature of the detector: 300°C. The thereby determined concentration value is compared to the concentration of the same lipid in the continuous phase of a reference dispersion of the same LNPs in water. To measure disintegration of the LNPs, the dispersed phase is separated from the continuous phase by standard methods, usually employing centrifugation and filtration. Measuring the concentration of any one of the lipids in the continuous phase is within the ordinary skill. The specific quantification and separation methods used are not important as long as the same methods are used when measuring the concentration of the lipid in the continuous phase either of a dispersion of the LNP in water or of a dispersion of the same LNP in 70% V/V ethanol. Advantageously, the term “disintegration” relates to an increase in concentration by 100% of a lipid component of the LNPs (i.e. any lipid being present in the LNP in an amount of at least 1 mol%, e.g. in an amount of between 1 mol% to 100 mol%, such as 1 mol% to 50 mol%) in the continuous phase of a dispersion of the LNP in 70% V/V ethanol compared to the concentration of the same lipid in the continuous phase of a reference dispersion of the same lipid nanoparticles in water. It is understood that the reference dispersion does not contain ethanol. For example, disintegration may be measured according to the following protocol: • Diluting a sample of the LNP (containing min. 30 µg of the lipid component to be analysed) in either ethanol or miliQ water to yield two samples, one in water (disintegration control D) and one in 70% V/V ethanol solution (disintegration test sample C) • Separating the dispersed phase from the continuous phase of each sample via centrifugation (16000g for 30 min) followed by filtration (0.2 µm pores) • Measuring the concentration of the lipid component in the continuous phase via standard methods such as GC- MS, HPLC or MALDI-TOF, advantageously GC-MS, for each sample (disintegration test sample C: Value C; disintegration negative control D: Value D) • Calculating the increase in lipid concentration in the continuous phase: Increase = ((Value C - Value D) / Value D) * 100% • LNPs are regarded disintegrated in 70% V/V ethanol if the increase in the concentration lipid concentration in the continuous phase as calculated according to the above formula is at least 100%. In addition, the LNPs protect the nucleic acid encapsulated therein from nuclease degradation as measured by qPCR. The encapsulated nucleic acid has a length of between 60 – 200 nucleotides, advantageously of between 60 – 120 nucleotides. The wt. ratio between the nucleic acid and the LNPs in the analytical composition is in a range of between 0.01% to 15% nucleic acid, advantageously in a range of between 0.1% to 2% nucleic acid, such as 1% nucleic acid. The use of the analytical composition described above has the advantage that the encapsulated nucleic acid is protected from nuclease degradation as long as the LNPs are intact. However, upon contact with 70% V/V ethanol, i.e. the most common disinfection reagent, the LNPs disintegrate and the nucleic acid becomes susceptible to degradation, in particular nuclease degradation. It is considered beneficial that the nucleic acid can be quantified by well-established and sensitive detection methods, e.g. by quantitative polymerase chain reaction (qPCR). The LNPs encapsulating the nucleic acid as described above have the advantage that they more closely mimic common pathogens than the tools provided in the prior art. Based on the present disclosure, the skilled person understands that the chemical composition of the LNPs can be adapted to particularly mimic certain pathogens, e.g. multi-drug resistant bacteria such as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci. Such pathogens are of particular relevance in certain environments, e.g. in the intensive care unit of a hospital. The LNPs encapsulating the nucleic acid are thus improved surrogate markers for pathogens compared to the prior art. In particular, analytical compositions as described above have the advantage that the LNPs are susceptible to disinfection procedures. In consequence, the use of the analytical composition enables both monitoring the disinfection procedure of a single surface and monitoring the transmission of pathogens from a first surface to a further surface (e.g. a second surface, a third surface, etc.) while providing information whether the first surface or the one or more further surfaces have been disinfected. In addition, the use of the analytical composition as described above has the advantage that the LNPs are non- infectious and thus well suited for use in a hospital, e.g. in an intensive care unit. Further, the use of the analytical composition has the advantage that the nucleic acid can be detected with high sensitivity by established nucleic acid amplification techniques such as qPCR. In addition, the use of the analytical composition has the advantage that results can be obtained in a relatively short time period, typically results can be obtained within 2 hours. In an advantageous embodiment, the encapsulated nucleic acid is deoxyribonucleic acid (DNA). For example, the encapsulated DNA may have the nucleotide sequence according to SEQ ID NO: 1 shown below: SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTT GAACACGAACAAACCTCTTT In an alternative embodiment, the encapsulated nucleic acid is ribonucleic acid. In another embodiment, the nucleic acid comprises or consists of modified nucleotides. Based on the present disclosure, the skilled person understands that the nucleic acid may be either single- stranded (ss) or double-stranded (ds). Based on the present disclosure, the skilled person understands that the use of multiple analytical compositions, each containing LNPs encapsulating a nucleic acid, e.g. DNA, having a different sequence, enables monitoring of transmission pathways from multiple surfaces while providing information on the efficiency of the disinfection procedure. In an advantageous embodiment the lipid nanoparticles and the DNA encapsulated therein have a N/P ratio of between 1:1 and 10:1, e.g. between 5:1 and 10:1, such as 8:1 or 10:1. Advantageously, “protection from nuclease degradation”, such as protection from DNAse degradation, relates to a decrease of max. 75% of intact nucleic acid present in the analytical composition after treatment with a nuclease, e.g. benzonase, (protection test sample E) compared to a reference analytical composition that was not treated with the nuclease (protection negative control F), as measured by quantitative polymerase chain reaction (qPCR). The decrease in intact nucleic acid can be calculated from the difference between Ct values measured by qPCR according to the following formula: Decrease = 2^(Delta Ct). Thus, a difference between Ct-values measured for (protection test sample E) – (protection negative control F) of 2 corresponds to a 4-fold decrease in intact nucleic acid. Accordingly, a decrease of max. 75% intact nucleic acid corresponds to a delta Ct between protection test sample E and protection negative control F of max. 2. It is understood that the choice of the nuclease is adapted to the nucleic acid to be analysed. For example, DNase degradation may be measured by subjecting the analytical composition to digestion with benzonase. For example, protection from nuclease degradation may be measured according to the following protocol (cf. example 4): • Protection test sample E: 2.5 units Benzonase in 10 µl sample (1.25 ng/ml) diluted in 90 µl Tris buffer (10 mM, pH 7) with MgCl2 (20 mM) • Protection negative control F: 10 µl sample (1.25 ng/ml) diluted in 90 µl miliQ water • Incubating protection test sample E for 1 h at 37°C • Inactivating enzyme by incubation for 10 min at 75 °C • Measuring DNA concentration of protection test sample E and protection negative control F via qPCR (see Example 6, Step 5) • Calculating the concentration difference between protection test sample E and protection negative control F based on the difference between Ct values (delta Ct). However, the skilled person understands that if RNA is to be analysed, i.e. the encapsulated nucleic acid being RNA, a suitable RNase, e.g. RNase A, needs to be used to measure degradation of the nucleic acid. Moreover, the skilled person understands that qPCR measurement of RNA requires a reverse transcription step, e.g. using a reverse transcriptase such as Moloney Murine Leukemia Virus (MMLV) Reverse Transcriptase, prior to the PCR step (referred to as RT-qPCR). Performing qPCR or RT-qPCR is well known in the art and all required components are commercially available. The skilled person further understands that depending on the modified nucleotides, the qPCR step, and optionally the RT step, need to be adapted to these modified nucleotides, e.g. by varying the enzymes, buffer or temperature or by using an alternative qPCR / RT-qPCR technique such as e.g. chemical-ligation qPCR (e.g. Boos et al., 2013, Nucleic Acids Res 41(15):e145; Shivalingam et al., 2020, Angew. Chem. Int. Ed., 59:11416–11422). Advantageously, “stability upon drying on a surface” relates to a decrease of max. 75% of intact nucleic acid present in the analytical composition after drying on a glass surface and after treatment with a nuclease, e.g. benzonase, (stability test sample G) compared to a reference analytical composition that was also dried on a glass surface, but not treated with the nuclease (stability negative control H), as measured by qPCR. As for the “protection from nuclease degradation”, the decrease in intact nucleic acid can be calculated from the difference between Ct values measured by qPCR according to the following formula: Decrease = 2^(Delta Ct). Thus, a difference between Ct-values measured for (stability test sample G) – (stability negative control H) of 2 corresponds to a 4-fold decrease in intact nucleic acid. Accordingly, a decrease of max. 75% intact nucleic acid corresponds to a delta Ct between stability test sample G and stability negative control H of max. 2. For example, “stability upon drying on a surface” may be measured according to the following protocol (cf. example 5): • Air drying 50 µl of the analytical composition containing 0.005 ng/µl of the LNPs encapsulating the nucleic acid on a glass surface, • rehydrating the dried composition with 200 µl water, • Stability test sample G: 2.5 units Benzonase in 10 µl rehydrated sample (1.25 ng/ml) diluted in 90 µl Tris buffer (10 mM, pH 7) with MgCl2 (20 mM) • Stability negative control H: 10 µl rehydrated sample (1.25 ng/ml) diluted in 90 µl miliQ water • Incubating stability test sample G for 1 h at 37°C • Inactivating benzonase by incubation for 10 min at 75 °C • Measuring DNA concentration of stability test sample G and stability negative control H via qPCR (see Example 6, Step 5) • Calculating the concentration difference between stability test sample G and stability negative control H based on the difference between Ct values (delta Ct). In an advantageous embodiment, the lipid nanoparticles have a particle diameter in the range of between 50 nm to 1500 nm, advantageously in the range of between 50 nm to 300 nm, as measured by dynamic light scattering. In one embodiment, the LNPs encapsulating the nucleic acid are lyophilized prior to use. In this embodiment, the lyophilized LNPs are dissolved in a liquid prior to application on a surface. Typically the lyophilized LNPs are dissolved in water or buffer, optionally containing additives as described herein. The skilled person understands that additives as described herein may also be lyophilized together with the LNPs. Components of the lipid nanoparticles: Lipid components: As is known in the field, lipid nanoparticles contain at least one lipid component. In advantageous embodiments, the at least one lipid component is selected from the group consisting of ionisable lipids, phospholipids, cholesterol, lipid anchored PEGs, and mixtures thereof. In an advantageous embodiment, the lipid nanoparticles contain an ionisable lipid, advantageously in an amount of between 35 mol% and 100 mol%, e.g. 35 mol%, e.g. 50 mol% to 60 mol% such as 55 mol%, 56 mol%, 57 mol%, and 58 mol%, e.g. 100 mol%. Thus in an advantageous embodiment, the lipid nanoparticles consist of, or essentially consist of, ionisable lipids. In an advantageous embodiment, the lipid nanoparticles contain a phospholipid, advantageously in an amount of between 10 mol% and 50 mol%, e.g. 16 mol%, e.g. in a range of between 20 mol% to 35 mol% such as 26 mol%, 27 mol%, 28 mol%, 29 mol% and 30 mol%. In an advantageous embodiment, the lipid nanoparticles contain cholesterol, advantageously in an amount of between 10 mol% and 50 mol%, e.g. in a range of between 10 mol% to 15 mol% such as 10 mol%, 11 mol% and 12 mol%, e.g. in a range of between 45 mol% to 50 mol% such as 45 mol%, 46.5 mol% and 48 mol%. In an advantageous embodiment, the lipid nanoparticles contain a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%, advantageously in an amount of between 2 mol% and 8 mol% such as 2 mol%, 2.5 mol%, 5 mol% and 7 mol%. In an advantageous embodiment, the lipid nanoparticles contain an ionisable lipid, advantageously in an amount as described above and further contain at least one of the following: • a phospholipid, advantageously in an amount of between 10 mol% and 50 mol%, e.g. 16 mol%, e.g. in a range of between 20 mol% to 35 mol% such as 26 mol%, 28 mol% and 29 mol%; • cholesterol, advantageously in an amount of between 10 mol% and 50 mol%, e.g. in a range of between 10 mol% to 15 mol% such as 11 mol% and 12 mol%, e.g. in a range of between 45 mol% to 50 mol% such as 45 mol%, 46.5 mol% and 48 mol%; • a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%, advantageously in an amount of between 2 mol% and 8 mol% such as 2 mol%, 2.5 mol%, 5 mol% and 7 mol%. In an advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, and a phospholipid, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, and cholesterol, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, and a lipid anchored PEG, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, a phospholipid, advantageously in an amount as described above, and cholesterol, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, cholesterol, advantageously in an amount as described above and a lipid anchored PEG, advantageously in an amount as described above. In another advantageous embodiment, the lipid nanoparticles contain or consist of an ionisable lipid, advantageously in an amount as described above, a phospholipid, advantageously in an amount as described above, cholesterol, advantageously in an amount as described above, and a lipid anchored PEG, advantageously in an amount as described above. Advantageously, the ionisable lipid is selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more advantageously selected from the group consisting of oleylamine and 306Oi10, most advantageously 306Oi10. 306Oi10 is also referred to as tetrakis(8-methylnonyl) 3,3',3'',3'''-((methylazanediyl)bis(propane-3,1- diyl))bis(azanetriyl))-tetrapropionate (cf. e.g. Hou et al., 2021, Nat Rev Mater 6, 1078–1094). Advantageously, the phospholipid is selected from the group consisting of 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3- phosphocholin (DSPC), and mixtures thereof. Advantageously, the lipid anchored PEG is C14PEG1000, also referred to as 1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000]. In an embodiment, the lipid nanoparticles consist of, or essentially consist of, oleylamine. In another embodiment, the lipid nanoparticles consist of, or essentially consist of, oleylamine, advantageously in an amount of between 55 mol% to 60 mol% such as 55 mol% or 58 mol%, DSPC, advantageously in an amount of between 25 mol% to 35 mol% such as 28 mol%, 29 mol% or 30 mol%, and cholesterol, advantageously in an amount of between 10 mol% to 15 mol% such as 12 mol% or 13 mol%. Thus in an embodiment, the lipid nanoparticles consist of, or essentially consist of, 58 mol% oleylamine, 29 mol% DSPC, and 13 mol% cholesterol. In another embodiment, the lipid nanoparticles consist of, or essentially consist of, 58 mol% oleylamine, 30 mol% DSPC, and 12 mol% cholesterol. Non-lipid components of the lipid nanoparticles: In certain embodiments, in addition to the at least one lipid component, the lipid nanoparticles further contain an inert solid component, advantageously in an amount of 5 wt% to 80 wt%. As used herein, the term “inert solid component” relates to an organic polymer, a metal oxide or semiconductor oxide that complies with the following characteristics: • Being solid at 22 °C; • having a primary particle size in the range of between 50 nm and 1500 nm; • being insoluble in water, ethanol and mixtures thereof, such as 70% V/V ethanol; i.e. max. 10 mg of the inert solid component can be dissolved in either of 100 ml of water, ethanol or a mixture thereof; • being unreactive with any of the lipid components described herein when being present in water at 22°C; i.e. the inert solid component does not chemically react with any of the lipid components described herein. Accordingly, in case the LNPs further contain an inert solid component, the LNPs have a particle diameter in the range of between 50 nm to 1500 nm, advantageously in the range of between 50 nm to 300 nm, as measured by dynamic light scattering. It is understood that the primary particle size of the inert solid is smaller than the particle diameter of the LNPs. As used herein, the term “primary particle size” relates to the diameter of a single, non-aggregated particle. An advantage of including an inert solid component into the LNPs is that the LNPs have an increased stability upon drying compared to LNPs that do not contain an inert solid component as described herein. Examples of suitable organic polymers include polystyrene, polyethylene terephthalate (PET) and polyethene (PE). Examples of suitable metal oxides include titania (TiO2), alumina (Al2O3), zirconia (ZrO2) and ceria (CeO2), advantageously titania (TiO2) and zirconia (ZrO2). As used herein, the term “semiconductor oxide” relates to oxides of semiconductors, e.g. silica (SiO2). The skilled person understands that silicon (Si) is a semiconductor whereas silica (SiO2), i.e. a semiconductor oxide, is not a semiconductor. Thus, the term “semiconductor oxide” is not to be confused with the terms “semiconducting oxides” or “oxide semiconductors”, i.e. certain oxides that are semiconductors, e.g. zirconia (ZrO2), zinc oxide (ZnO) and cadmium oxide (CdO). In an advantageous embodiment, the inert solid component is selected from the group consisting of titania (TiO2), zirconia (ZrO2) and silica (SiO2). In certain embodiments, the LNPs contain at least the following components: • an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol% and preferably selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more preferably selected from the group consisting of oleylamine and 306Oi10, most preferably 306Oi10; and • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO2), alumina (Al2O3), zirconia (ZrO2) and ceria (CeO2)) and semiconductor oxides (preferably silica (SiO2)), preferably in an amount of between 5 wt% to 80 wt%. Additives In an advantageous embodiment, the analytical composition comprises additives. Ionic strength adjusting additives In certain advantageous embodiments, the analytical composition comprises ionic strength adjusting additives, including buffers and salts. Ionic strength adjusting additives may be added for example to adjust the zeta potential of a composition containing LNPs, thereby increasing the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs. A large variety of ionic strength adjusting additives is known in the field and suitable in the context of the present invention. For example, suitable ionic strength adjusting additives may be selected from the group consisting of NaCl, such as an isotonic NaCl solution, PBS, citrate buffer, and isocitrate buffer. Advantageously, the analytical composition has a zeta potential in the range of between –30 mV to +30 mV as measured by electrophoretic light scattering, e.g. using a zetasizer. pH adjusting additives The skilled person understands that some additives, while having an effect on the ionic strength of the composition, may also be added to adjust the pH value. Adjusting the pH value may be performed e.g. to increase the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs. A large variety of pH adjusting additives are known in the field and suitable in the context of the present invention, such as PBS, citrate buffer, and isocitrate buffer. Advantageously, the analytical composition has a pH value in the range of 6 to 8. Disaccharides and polysaccharides It has been surprisingly found that addition of disaccharides and polysaccharides increases the stability of the analytical composition upon drying. A large variety of disaccharides and polysaccharides are known in the field and are suitable in the context of the present invention. The following disaccharides and polysaccharides have been found particularly beneficial additives to increase the stability of the analytical composition upon drying: Disaccharides: Trehalose, sucrose, maltose, lactose, and mixtures thereof. Polysaccharides: Agarose, starch, dextran, and mixtures thereof. Advantageously, the wt. ratio between the lipid nanoparticles and the additives is in a range of between 1:0.1 to 1:20, more advantageously in a range of between 1:1 to 1:10 Polyethylenglycol In certain advantageous embodiments, the analytical composition comprises PEG (also referred to as “free PEG”). The skilled person understands that in contrast to lipid anchored PEG, free PEG is not a component of the LNPs but instead is added to the analytical composition and is present external to the LNPs. Addition of PEG has been found to increase the stability of the LNPs, e.g. by preventing or mitigating aggregation of the LNPs. Addition of PEG has also been found to increase the stability of the analytical composition upon drying. A large variety of PEGs are known in the field and are suitable in the context of the present invention. The following PEGs have been found particularly beneficial additives to increase the stability of the analytical composition upon drying: PEG600, PEG1000, PEG2000, PEG6000. In an advantageous embodiment, the analytical composition contains oleylamine, DSPC, cholesterol, and PEG1000, advantageously in a molar ratio of oleylamine:DSPC:cholesterol:PEG1000 of 55:28:12:5. In another advantageous embodiment, the analytical composition contains oleylamine, DSPC, cholesterol and PEG2000, advantageously in a molar ratio of oleylamine:DSPC:cholesterol:PEG2000 of 57:29:12:2. In another advantageous embodiment, the analytical composition contains hexadecylamine, DSPC, cholesterol and PEG600, advantageously in a molar ratio of hexadecylamine:DSPC:cholesterol:PEG600 of 56:26:11:7. In a second aspect, the invention relates to a method for monitoring a disinfection procedure with an analytical composition as defined above for the first aspect. The method comprises the following steps: Step a: Applying the analytical composition onto a surface, advantageously air-drying the composition. Advantageously, at least 0.25 ng of the LNPs encapsulating the nucleic acid are applied on the surface, e.g. 50 µl of the analytical composition containing 0.005 ng/µl of the LNPs encapsulating the nucleic acid. Step b: After a pre-determined time period, taking a test sample from the surface or from a second surface to which at least part of the analytical composition has been transferred, thereby dissolving the test sample in an aqueous solution, e.g. water, to obtain an aqueous test sample, e.g. in one of the following manners: • adding water onto the (first) surface or the second surface, for example pipetting 200 µl water onto the surface, and recovering the water containing the aqueous test sample, e.g. by pipetting; or • swabbing the (first) surface or the second surface with a premoistened absorbent means, e.g. a swab, and recovering the test sample from the absorbent means by dissolving it in water, e.g. in 200 µl water. Advantageously, the skilled person understands that “dissolving the test sample” also includes “obtaining a suspension of the test sample”. Advantageously, the time period between step a and step b may be varied depending on the cleaning strategy of the facility in which the analytical composition is used. For example, if the surface is scheduled to be disinfected every 24 h, the time period between step a and step b may also be 24 h. Step c: Subjecting a portion A of the aqueous test sample to an enzymatic nuclease degradation step, in particular an enzymatic DNA degradation step, e.g. in the following manner: • Diluting a defined volume of the aqueous test sample, e.g. 10 µl, with a defined volume of Tris buffer (10 mM, pH 7) containing 20 mM MgCl2, e.g. 90 µl; • adding a suitable nuclease, e.g. 2.5 units of Benzonase; and • incubating the mixture, e.g. for 1 h at 37°C, • inactivating the nuclease, e.g. by incubating for 10 min at 75°C. Step d: Examining the portion A and a reference portion B of the aqueous test sample by a qPCR detection procedure using primers specific for the nucleic acid, in particular the DNA, encapsulated in the lipid nanoparticles, to differentiate with reference to portion B, whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A and was thus protected during the enzymatic DNA nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step. For example, the following qPCR conditions may be used: • Preactivation for 240s at 95°C; • Amplification (40 cycles) with each cycle comprising 2 s at 95°C, followed by 12 s at 58°C, followed by 4 s at 72°C. Advantageously, the reference portion B of the aqueous test sample is prepared by diluting a defined volume of the aqueous test sample, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B is not subjected to an enzymatic nuclease degradation step. In an advantageous embodiment, steps b – d further comprise the following steps: Step b) additionally comprises taking a third test sample from a third additional surface, thereby dissolving the third test sample in an aqueous solution, e.g. water, to obtain a third aqueous test sample. Step c) additionally comprises subjecting a portion A’ of the third aqueous test sample to an enzymatic nuclease degradation step. Step d) additionally comprises examining the portion A’ and a reference portion B’ of the third aqueous test sample by a qPCR detection procedure using primers specific for the nucleic acid encapsulated in the lipid nanoparticles to differentiate with reference to portion B’, whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A’ and was thus protected during the enzymatic nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A’, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step, or • the analytical composition is not present on the third surface. Advantageously, the reference portion B’ of the third aqueous test sample is prepared by diluting a defined volume of the third aqueous test sample, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B’ is not subjected to an enzymatic nuclease degradation step. In an advantageous embodiment, it is concluded that the detected DNA from the test sample, or optionally the second test sample, originates from disintegrated LNPs if the difference between Ct values (A-B) – (A0-B0) > 2 with • A0 being a control portion of A, and • B0 being a control portion of B. As is common in the field, differences in the nucleic acid concentrations between different samples or portions of a sample can be determined by comparing Ct values measured by a qPCR detection procedure, e.g. qPCR or RT-qPCR. Ct values so measured can be converted to an absolute concentration of nucleic acid by means of a calibration curve. However, in the context of the instant invention, it is not required to convert the Ct value to an absolute nucleic acid concentration. Instead it is sufficient to compare Ct values between different samples or portions of a sample, i.e. a relative comparison of the nucleic acid concentrations is sufficient. Determining the Ct value from a sample or portion of a sample by a qPCR detection procedure, e.g. qPCR or RT-qPCR, is within the ordinary skill. Furthermore, establishing a calibration curve is within the ordinary skill. In a further advantageous embodiment, it is concluded that the detected DNA from the third test sample originates from disintegrated LNPs if the difference between Ct values (A’-B’) – (A0-B0) > 2 with • A0 being a control portion of A, respectively A’, and • B0 being a control portion of B, respectively B’. The skilled person understands that the control portion A0 is a control portion for both portion A and portion A’. The skilled person further understands that the control portion B0 is a control portion for both portion B and portion B’. Advantageously, a control portion B0 is prepared from the analytical composition (i.e. a portion of the analytical composition that was stored before application) in the same manner as the reference portion B is prepared from the aqueous test sample, e.g. by diluting a defined volume of the analytical composition, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B0 is not subjected to an enzymatic nuclease degradation step. Advantageously, a control portion A0 is obtained by subjecting a portion of the analytical composition (i.e. a portion of the analytical composition that was stored before application) to the same enzymatic nuclease degradation step as portion A. For example, a conclusion whether the (first) surface, and optionally the second surface and / or the third surface was disinfected between step a and step b may be reached in the following manner: • Calculating the difference between the nucleic acid concentration in portion A and in the reference portion B; • Calculating the difference between the nucleic acid concentration in the control portion A0 and in the control portion B0; • Comparing the difference between the nucleic acid concentration in portion A and in the reference portion B, i.e. comparing the Ct values measured by a qPCR detection procedure, with the difference between the nucleic acid concentration in the control portion A0 and in the control portion B0, and • if the difference between Ct values A-B ≈ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) < 2 the detected nucleic acid originates from intact lipid nanoparticles, i.e. the surface was not disinfected; • if the difference between Ct values A-B ≫ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) > 2 the detected nucleic acid originates from disintegrated lipid nanoparticles, i.e. the surface was disinfected; and • optionally if the difference between Ct values A’-B’ ≈ A0-B0, i.e. the difference between Ct values (A’- B’) – (A0-B0) < 2 the detected nucleic acid originates from intact lipid nanoparticles, i.e. the surface was not disinfected, and • optionally if the difference between Ct values A’-B ’ ≫ A0-B0, i.e. the difference between Ct values (A’- B’) – (A0-B0) > 2 the detected nucleic acid originates from disintegrated lipid nanoparticles, i.e. the surface was disinfected or the analytical composition was not present on the third surface. In a further advantageous embodiment, steps a, c and d comprise the following substeps: Step a: a-1: Applying the analytical composition onto a surface, advantageously air-drying the composition. Advantageously, at least 0.25 ng of the LNPs encapsulating the nucleic acid are applied on the surface, e.g. 50 µl of the analytical composition containing the LNPs encapsulating the nucleic acid in a concentration of 0.005 ng/µl. a-2: Storing a defined volume of the analytical composition, e.g. 50 µl, thereby obtaining a stored analytical composition. Step c: c-1: Subjecting a portion A of the aqueous test sample to an enzymatic nuclease degradation step, in particular an enzymatic DNA degradation step, e.g. in the following manner: • Diluting a defined volume, e.g. 10 µl, of the aqueous test sample with a defined volume, e.g. 90 µl, of Tris buffer (10 mM, pH 7) containing 20 mM MgCl2; • adding a suitable nuclease, e.g. 2.5 units Benzonase; and • incubating the mixture, e.g. for 1 h at 37°C, • inactivating the enzyme, e.g. by incubating for 10 min at 75°C. c-2: Subjecting a portion of the stored analytical composition to the same enzymatic nuclease degradation step as portion A, to thereby obtain a control portion A0. c-3: Preparing a reference portion B of the aqueous test sample, e.g. by diluting a defined volume of the aqueous test sample, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B is not subjected to an enzymatic nuclease degradation step. c-4: Preparing a control portion B0 of the stored analytical composition in the same manner as the reference potion B is prepared from the aqueous test sample, e.g. by diluting a defined volume of the stored analytical composition, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B0 is not subjected to an enzymatic nuclease degradation step. Optionally c-5: Subjecting a portion A’ of the third aqueous test sample to the same enzymatic nuclease degradation step as portion A. Optionally c-6: Preparing a reference portion B’ of the third aqueous test sample, e.g. by diluting a defined volume of the third aqueous test sample, e.g. 10 µl, in a defined volume of water, e.g. 90 µl. It is understood that the reference portion B’ is not subjected to an enzymatic nuclease degradation step. Step d: d-1: Quantifying the nucleic acid, in particular the DNA, present in portion A and in the reference portion B by a qPCR detection procedure using primers that are specific for the encapsulated nucleic acid. For example, the following qPCR conditions may be used: • Preactivation for 240 s at 95°C; • Amplification (40 cycles) with each cycle comprising 2 s at 95°C, followed by 12s at 58°C, followed by 4s at 72°C. d-2: Quantifying the nucleic acid present in the control portion A0 and in the control portion B0 in the same manner as portion A and reference portion B. d-3: Comparing the Ct values measured for the portion A, for the reference portion B, for the control portion A0 and for the control portion B0 to differentiate whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A and was thus protected during the enzymatic nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step. Optionally d-4: Quantifying the nucleic acid, in particular the DNA, present in portion A’ and in the reference portion B’ of the third aqueous test sample by a qPCR detection procedure using primers that are specific for the encapsulated nucleic acid. Optionally d-5: Comparing the Ct values measured for portion A’, for the reference portion B’, for the control portion A0 and for the control portion B0 to differentiate whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A’ and was thus protected during the enzymatic DNA nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A’, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic DNA nuclease degradation step. In a third aspect, the invention relates to an analytical composition as described herein for the first aspect. Advantageously, the LNPs comprised within the analytical composition contain an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol%, and/or contain at least one of the following: • a phospholipid, preferably in an amount of between 10 mol% and 50 mol%, • cholesterol, preferably in an amount of between 10 mol% and 50 mol%, and • a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%; • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO2), alumina (Al2O3), zirconia (ZrO2) and ceria (CeO2)) and semiconductor oxides (preferably silica (SiO2)), preferably in an amount of between 5 wt% to 80 wt%. In an advantageous embodiment, the LNPs contain at least the following components: • an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol% and preferably selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, more preferably selected from the group consisting of oleylamine and 306Oi10, most preferably 306Oi10; and • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania (TiO2), alumina (Al2O3), zirconia (ZrO2) and ceria (CeO2)) and semiconductor oxides (preferably silica (SiO2)), preferably in an amount of 5 wt% to 80 wt%. In an advantageous embodiment, the inert solid component is selected from the group consisting of titania (TiO2), zirconia (ZrO2) and silica (SiO2). In a fourth aspect, the invention relates to a kit of parts comprising an analytical composition as described herein for the first aspect, qPCR primers specific for the nucleic acid, and optionally one or more of the following: • a nuclease, • a DNA polymerase, • a qPCR buffer. Brief Description of the Drawings The invention will be better understood and objects other than those set forth above will become apparent from the annexed drawings and the detailed description thereof. Fig. 1: Scheme of LNP and exemplary components Legend: 1 – Schematic representation of LNP 2 – Schematic representation of encapsulated DNA within the LNP 3 – Schematic representation of lipid anchored PEG 4 - Schematic representation of ionizable lipid 5 - Schematic representation of cholesterol 6 - Schematic representation of DNA Fig. 2: Scheme of workflow to determine the amount/ratio of intact LNPs in a sample. Legend: 1 – sample of LNPs 2 – DNAse / Benzonase treatment 3 – qPCR quantification 4 – Fluorescence 5 – PCR cycle number 6 – difference in cycle threshold, Delta Ct A – sample portion A B – sample portion B Fig. 3: Scheme of workflow to determine if a successful disinfection procedure was carried out. Legend: 1 – Surface with dried LNPs 2 – treatment 3 – disinfection with ethanol 4 – no disinfection, wiping with water 5 – rehydration to recover sample from surface 6 – analysis to determine the amount of intact LNPs in the sample (see fig.1) 7 – fluorescence 8 – PCR cycle number 9 - difference in cycle threshold, Delta Ct 10 – cycle threshold value from qPCR measurement A – sample portion A B – sample portion B

Examples Example 1 – Preparation of LNPs encapsulating DNA Synthesis of ionisable lipid 306Oi10: The ionisable lipid 306Oi10 was synthesised from 2-isodecyl acrylate (Sigma-Aldrich) and 3,3'-Diamino-N- methyldipropylamine (Aldrich-Fine Chemicals) in stochiometric amounts via stirring for 3 days at 90°C. Synthesis of TMAPS-functionalised silica particles: The silica particles were functionalised by first sonicating a aqueous dispersion of silica nanoparticles (0.142 µm diameter, 5% w/v aq. sol., Lot: SiO2-R-L3205- 23/1, microparticles GmbH) for 10 min, then adding 1 V/V % TMAPS (N-Trimethoxysilylpropyl-N,N,N-trimethylammonium chloride, 50% solution in methanol, abcr) and stirring for 12 h at 900 rpm. LNP composition number 1: LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10 First the lipids DOPE (l,2-dioleoyl-sn-glycero-3- phosphoethanolamine, Avanti), cholesterol (Sigma-Aldrich), C14-PEG1000 (1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000], ammonium salt, Avanti) and the previously synthesised ionisable lipid 306Oi10 were dissolved in 90% ethanol and 10% 10 nM citrate buffer at molar ratios of 16 : 46.5 : 2.5 : 35. Then, an aqueous phase (pH = 5) was prepared consisting of DNA (ds DNA, 62 nt; SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTTG AACACGAACAAACCTCTTT), 10 mM Tris base buffer and 10 nM citrate buffer, with a final DNA concentration of 1 g/l. Both phases were preheated to 37°C and the ethanol phase was added dropwise to the aqueous phase in a 3:1 volumetric ratio (aqueous:ethanol) and mixed by rapid pipette mixing. The formed LNPs were stabilised with 300 mM NaCl citrate buffer (three times the volume of LNPs) and dialysed against PBS for 1 h at room temperature using a MWCO 20000 cassette (gibco). A DNA blank sample was prepared with the aqueous phase and pure ethanol as the ethanol phase. LNP composition number 2: LNP comprising oleylamine, DSPC, cholesterol and PEG 600 First, oleylamine, DSPC, cholesterol and PEG 600 were dissolved in methanol at molar ratios of 54:27:11.5:7.5. Then an aqueous phase was DNA prepared consisting of DNA (ds DNA, 62 nt; SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTTG AACACGAACAAACCTCTTT) in 0.1 M acetic acid with a final DNA concentration of 1 g/l. Both phases were preheated to 80°C and the methanol phase was added dropwise to the aqueous phase in 5:1 volumetric ratio (aqueous:methanol) and mixed by rapid pipette mixing. The mixture was allowed to cool down to room temperature. LNP composition number 3: LNP comprising oleylamine First, oleylamine was dissolved in methanol. Then an aqueous phase was DNA prepared consisting of DNA (ds DNA, 62 nt; SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTTGAACACGAACAAACC TCTTT) in 0.1 M acetic acid with a final DNA concentration of 1 g/l. Both phases were preheated to 80°C and the methanol phase was added dropwise to the aqueous phase in 2.5:1 volumetric ratio (aqueous:methanol) and mixed by rapid pipette mixing. The mixture was allowed to cool down to room temperature. LNP composition number 4: LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10 with silica nanoparticles First the lipids DOPE (l,2-dioleoyl-sn-glycero-3- phosphoethanolamine, Avanti), cholesterol (Sigma-Aldrich), C14-PEG1000 (1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000], ammonium salt, Avanti) and the previously synthesised ionisable lipid 306Oi10 were dissolved in 90% ethanol and 10% 10 nM citrate buffer at molar ratios of 16 : 46.5 : 2.5 : 35. Then, an aqueous phase (pH = 5) was prepared consisting of DNA (ds DNA, 62 nt; SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTTGAACACGAACAAACC TCTTT), TMAPS-functionalised silica particles, 10 mM Tris base buffer and 10 nM citrate buffer, with a final DNA concentration of 0.1 g/l and a final silica particle concentration of 4 g/l, where the DNA is bound on the surface of the silica particles. Both phases were preheated to 37°C and the ethanol phase was added dropwise to the aqueous phase in a 3:1 volumetric ratio (aqueous:ethanol) and mixed by rapid pipette mixing. The formed LNPs were stabilised with 300 mM NaCl citrate buffer (three times the volume of LNPs). A DNA blank sample was prepared with the aqueous phase and pure ethanol as the ethanol phase. LNP composition number 5: LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10 with silica nanoparticles via lipid rehydration
First the lipids DOPE (l,2-dioleoyl-sn-glycero-3- phosphoethanolamine, Avanti), cholesterol (Sigma-Aldrich), C14-PEG1000 (1,2-dimyristoyl-sn-glycero-3- phosphoethanolamine-N-[methoxy(polyethylene glycol)- 1000], ammonium salt, Avanti) and the previously synthesised ionisable lipid 306Oi10 were dissolved in chloroform at molar ratios of 16 : 46.5 : 2.5 : 35. The dissolved lipids were dried in vacuo and rehydrated with 0.25 M Phosphate buffered saline in a final lipid concentration of 10 g/l, followed by sonication for 10min.Then, an aqueous phase (pH = 7) was prepared consisting of DNA (ds DNA, 62 nt; SEQ ID NO: 1: TTCTCTGCCCTTACGTTTATCTTAAGGGCCGGTCCACCAGTTGAACACGAACAAACC TCTTT), TMAPS-functionalised silica particles in milli Q water, with a final DNA concentration of 1 mg/l and a final silica particle concentration of 2 g/l, where the DNA is bound on the surface of the silica particles. Both phases were combined in volumetric ratio 1:1 by rapid pipette mixing, sonicated for 15 min and shaken at 450 rpm at 37°C for 1h. Results are shown in table 1 below. Table 1: ) ) ) M) Example 2 – Preparation of analytical compositions To prepare an analytical composition, LNP dispersions were diluted in miliQ water to a final concentration of 0.005 ng/µl. If additives were added, they were added in the respective amount to the LNP dispersion, which was subsequently diluted in miliQ water to a final concentration of 0.005 ng/µl LNP. Example 3 – Disintegration of the LNPs in 70 vol.% ethanol The concentration of a LNP constituent lipid (i.e. any one of the lipid components being present in the LNP in an amount of at least 1 mol%) was measured in the continuous phase of an LNP dispersion comprising 150 µl LNP in miliQ water (0.2 µg/µl constituent lipid; cf. example 2) in either 350 µl miliQ water (disintegration negative control D) or in 350 µl ethanol (resulting in 70% V/V ethanol; disintegration test sample C). After separating the dispersed phase from the continuous phase by centrifugation (16000 g for 30 min) and filtration (0.2 µm pore size), the concentration of the lipid constituent in the continuous phase was measured using a GC-MS-5977A series instrument (Agilent; column: DB-5MS 20 x 0.18 µm, oven temperature: 170°C ramped to 325°C at 8°C/min, with a final hold time of 20 min; detector temperature: 300°C). Results are shown in table 2 below. Table 2: re nt n l] ol % e
Example 2 – Preparation of analytical compositions To prepare an analytical composition, LNP dispersions were diluted in miliQ water to a final concentration of 0.005 ng/µl. If additives were added, they were added in the respective amount to the LNP dispersion, which was subsequently diluted in miliQ water to a final concentration of 0.005 ng/µl LNP. Example 3 – Disintegration of the LNPs in 70 vol.% ethanol The concentration of a LNP constituent lipid (i.e. any one of the lipid components being present in the LNP in an amount of at least 1 mol%) was measured in the continuous phase of an LNP dispersion comprising 150 µl LNP in miliQ water (0.2 µg/µl constituent lipid; cf. example 2) in either 350 µl miliQ water (disintegration negative control D) or in 350 µl ethanol (resulting in 70% V/V ethanol; disintegration test sample C). After separating the dispersed phase from the continuous phase by centrifugation (16000 g for 30 min) and filtration (0.2 µm pore size), the concentration of the lipid constituent in the continuous phase was measured using a GC-MS-5977A series instrument (Agilent; column: DB-5MS 20 x 0.18 µm, oven temperature: 170°C ramped to 325°C at 8°C/min, with a final hold time of 20 min; detector temperature: 300°C). Results are shown in table 2 below. Table 2: re nt n l] ol % e
 *Below detection limit Example 4 – Protection of encapsulated DNA from nuclease degradation Protection from nuclease degradation was measured according to the following protocol: • Protection test sample E: 2.5 units Benzonase in 10 µl sample diluted (1.25 ng/ml) in 90 µl Tris buffer (10 mM, pH 7) with MgCl2 (20 mM) • Protection negative control F: 10 µl sample (1.25 ng/ml) diluted in 90 µl miliQ water • Incubation of protection test sample E for 1 h at 37°C • Inactivation of benzonase by incubation for 10 min at 75°C • DNA concentration in protection test sample E (Ct value E) and protection negative control F (Ct value F) was measured by qPCR (see Example 6, Step 5) • Difference of cycle thresholds between protection test sample E and protection negative control F was calculated according to the following formula: Delta Ct = Ct value E – Ct value F Results are shown in table 3 below. Table 3: F
*Below detection limit Example 4 – Protection of encapsulated DNA from nuclease degradation Protection from nuclease degradation was measured according to the following protocol: • Protection test sample E: 2.5 units Benzonase in 10 µl sample diluted (1.25 ng/ml) in 90 µl Tris buffer (10 mM, pH 7) with MgCl2 (20 mM) • Protection negative control F: 10 µl sample (1.25 ng/ml) diluted in 90 µl miliQ water • Incubation of protection test sample E for 1 h at 37°C • Inactivation of benzonase by incubation for 10 min at 75°C • DNA concentration in protection test sample E (Ct value E) and protection negative control F (Ct value F) was measured by qPCR (see Example 6, Step 5) • Difference of cycle thresholds between protection test sample E and protection negative control F was calculated according to the following formula: Delta Ct = Ct value E – Ct value F Results are shown in table 3 below. Table 3: F Example 5 - Stability of LNPs upon air drying Stability upon drying on a surface was measured according to the following protocol: • air drying 50 µl of the analytical composition containing 0.005 ng/µl of the LNPs encapsulating the nucleic acid on a glass surface, • rehydrating the dried composition with 200 µl miliQ water, • Stability test sample G: 2.5 units Benzonase in 10 µl sample (1.25 ng/ml) diluted in 90 µl Tris buffer (10 mM, pH 7) with MgCl2 (20 mM) • Stability negative control H: 10 µl sample (1.25 ng/ml) diluted in 90 miliQ water • Incubation of stability test sample G for 1 h at 37°C • Inactivation of Benzonase by incubation for 10 min at 75 °C • Measure DNA concentration of stability test sample G (Ct value G) and stability negative control H (Ct value H) via qPCR (see Example 6, Step 5) • Calculate difference of cycle thresholds between stability test sample G and stability negative control H: Delta Ct = Ct value G – Ct value H Results are shown in table 4 below. Table 4: H
Example 5 - Stability of LNPs upon air drying Stability upon drying on a surface was measured according to the following protocol: • air drying 50 µl of the analytical composition containing 0.005 ng/µl of the LNPs encapsulating the nucleic acid on a glass surface, • rehydrating the dried composition with 200 µl miliQ water, • Stability test sample G: 2.5 units Benzonase in 10 µl sample (1.25 ng/ml) diluted in 90 µl Tris buffer (10 mM, pH 7) with MgCl2 (20 mM) • Stability negative control H: 10 µl sample (1.25 ng/ml) diluted in 90 miliQ water • Incubation of stability test sample G for 1 h at 37°C • Inactivation of Benzonase by incubation for 10 min at 75 °C • Measure DNA concentration of stability test sample G (Ct value G) and stability negative control H (Ct value H) via qPCR (see Example 6, Step 5) • Calculate difference of cycle thresholds between stability test sample G and stability negative control H: Delta Ct = Ct value G – Ct value H Results are shown in table 4 below. Table 4: H Example 6 – Disinfection tests Disinfection tests were performed according to the following general protocol: Step 1: Application of the analytical composition on a surface. 50 µl of the analytical composition containing 0.005 ng/µl LNPs encapsulating the DNA (SEQ ID NO: 1) were applied onto a clean glass surface (no residual DNA, LNPs on the surface). The analytical composition was air-dried on the surface. In addition, 200 µl of the same analytical composition containing 0.00125 ng/µl LNPs encapsulating the DNA (sequence) were stored at 5°C for later use (referred to as the “stored analytical composition”). Step 2: Disinfection of the surface. The following disinfectants were tested, i.e. the surface was wiped with a KimTECH wipe premoistened with 100 µl of either • miliQ water (control) • ethanol (20% V/V, 70% V/V, 85% V/V, 100% V/V) • SDS (5 g/l) • Triton X-100 (5 g/l). The disinfectant or water (control) was allowed to air-dry on the surface before proceeding to step 3. Step 3: Taking a test sample from a surface. A test sample was taken from the surface either • by pipetting 200 µl miliQ water onto the surface and recovering this liquid, or • by swabbing the surface with a premoistened swab and recovering the sample from the swab by dissolving it in 200 µl miliQ water. The results of the analysis were comparable when either of the two methods of taking the test sample were used (step 3). The obtained test sample is further referred to as the “aqueous test sample”. Step 4: Enzymatic DNase digestion. A portion A of the aqueous test sample was prepared by diluting 10 µl of the aqueous test sample with 90 µl of Tris buffer (10 mM, pH 7) containing 20 mM MgCl2. To this solution was added 2.5 units Benzonase and the mixture was incubated for 1 h at 37°C. After incubation, benzonase was inactivated by further incubation for 10 min at 75°C. A reference portion B of the aqueous test sample was prepared by diluting 10 µl of the aqueous test sample with 90 µl miliQ water. No nuclease digestion was performed on reference portion B (untreated sample). A control portion B0 of the stored analytical composition was prepared by diluting 10 µl of the stored analytical composition with 90 µl miliQ water. No nuclease digestion was performed on the control portion B0 (control for reference portion B). A control portion A0 of the stored analytical composition was prepared by diluting 10 µl of the stored analytical composition with 90 µl of Tris buffer (10mM, pH 7) containing 20 mM MgCl2. To this solution was added 2.5 units Benzonase and the mixture was incubated for 1 h at 37°C. After incubation, benzonase was inactivated by further incubation for 10 min at 75°C. Step 5: qPCR analysis – assessment whether the surface was disinfected The following primers were used: Forward primer (SEQ ID NO: 2): CTCTGCCCTTACGTTTATC Reverse primer (SEQ ID NO: 3): AGAGGTTTGTTCGTGTTC A qPCR master mix was prepared for each sample by mixing 10 µl MasterMix (KAPA SYBR FAST for LightCycler 480, REF: KK4611 07959494001, kapa biosystems), 1 µl of each primer and 3 µl miliQ water. From this qPCR master mix, a qPCR was performed on a 96 well plate using a LightCycler 480 II, Roche Diagnostics by adding 15 µl of the respective qPCR master mix per well. Each sample (5 µl) was pipetted on the 96 well plate in duplicates and the plate was sealed. qPCR was performed according to the following protocol: Preactivation: 240 s at 95°C Amplification (40 cycles): 2 s at 95°C, 12 s at 58 °C, 4 s at 72 °C. Interpretation of results: A conclusion whether the surface was efficiently disinfected in step 2 was reached in the following manner: • The difference between the Ct values measured for portion A and for the reference portion B was calculated; • The difference between the Ct values measured for the control portion A0 and for the control portion B0 was calculated; • The difference between the Ct values measured for portion A and for the reference portion B was compared to the difference between the Ct values measured for the control portion A0 and for the control portion B0; • if the difference between Ct values A-B ≈ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) < 2, the detected nucleic acid originates from intact LNPs, i.e. the surface was not disinfected; • if the difference between Ct values A-B ≫ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) > 2, the detected nucleic acid originates from disintegrated LNPs, i.e. the surface was disinfected. Results for LNP composition number 1 (LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10; cf. example 1) are shown in table 5 below. Table 5: io l?
Example 6 – Disinfection tests Disinfection tests were performed according to the following general protocol: Step 1: Application of the analytical composition on a surface. 50 µl of the analytical composition containing 0.005 ng/µl LNPs encapsulating the DNA (SEQ ID NO: 1) were applied onto a clean glass surface (no residual DNA, LNPs on the surface). The analytical composition was air-dried on the surface. In addition, 200 µl of the same analytical composition containing 0.00125 ng/µl LNPs encapsulating the DNA (sequence) were stored at 5°C for later use (referred to as the “stored analytical composition”). Step 2: Disinfection of the surface. The following disinfectants were tested, i.e. the surface was wiped with a KimTECH wipe premoistened with 100 µl of either • miliQ water (control) • ethanol (20% V/V, 70% V/V, 85% V/V, 100% V/V) • SDS (5 g/l) • Triton X-100 (5 g/l). The disinfectant or water (control) was allowed to air-dry on the surface before proceeding to step 3. Step 3: Taking a test sample from a surface. A test sample was taken from the surface either • by pipetting 200 µl miliQ water onto the surface and recovering this liquid, or • by swabbing the surface with a premoistened swab and recovering the sample from the swab by dissolving it in 200 µl miliQ water. The results of the analysis were comparable when either of the two methods of taking the test sample were used (step 3). The obtained test sample is further referred to as the “aqueous test sample”. Step 4: Enzymatic DNase digestion. A portion A of the aqueous test sample was prepared by diluting 10 µl of the aqueous test sample with 90 µl of Tris buffer (10 mM, pH 7) containing 20 mM MgCl2. To this solution was added 2.5 units Benzonase and the mixture was incubated for 1 h at 37°C. After incubation, benzonase was inactivated by further incubation for 10 min at 75°C. A reference portion B of the aqueous test sample was prepared by diluting 10 µl of the aqueous test sample with 90 µl miliQ water. No nuclease digestion was performed on reference portion B (untreated sample). A control portion B0 of the stored analytical composition was prepared by diluting 10 µl of the stored analytical composition with 90 µl miliQ water. No nuclease digestion was performed on the control portion B0 (control for reference portion B). A control portion A0 of the stored analytical composition was prepared by diluting 10 µl of the stored analytical composition with 90 µl of Tris buffer (10mM, pH 7) containing 20 mM MgCl2. To this solution was added 2.5 units Benzonase and the mixture was incubated for 1 h at 37°C. After incubation, benzonase was inactivated by further incubation for 10 min at 75°C. Step 5: qPCR analysis – assessment whether the surface was disinfected The following primers were used: Forward primer (SEQ ID NO: 2): CTCTGCCCTTACGTTTATC Reverse primer (SEQ ID NO: 3): AGAGGTTTGTTCGTGTTC A qPCR master mix was prepared for each sample by mixing 10 µl MasterMix (KAPA SYBR FAST for LightCycler 480, REF: KK4611 07959494001, kapa biosystems), 1 µl of each primer and 3 µl miliQ water. From this qPCR master mix, a qPCR was performed on a 96 well plate using a LightCycler 480 II, Roche Diagnostics by adding 15 µl of the respective qPCR master mix per well. Each sample (5 µl) was pipetted on the 96 well plate in duplicates and the plate was sealed. qPCR was performed according to the following protocol: Preactivation: 240 s at 95°C Amplification (40 cycles): 2 s at 95°C, 12 s at 58 °C, 4 s at 72 °C. Interpretation of results: A conclusion whether the surface was efficiently disinfected in step 2 was reached in the following manner: • The difference between the Ct values measured for portion A and for the reference portion B was calculated; • The difference between the Ct values measured for the control portion A0 and for the control portion B0 was calculated; • The difference between the Ct values measured for portion A and for the reference portion B was compared to the difference between the Ct values measured for the control portion A0 and for the control portion B0; • if the difference between Ct values A-B ≈ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) < 2, the detected nucleic acid originates from intact LNPs, i.e. the surface was not disinfected; • if the difference between Ct values A-B ≫ A0-B0, i.e. the difference between Ct values (A-B) – (A0-B0) > 2, the detected nucleic acid originates from disintegrated LNPs, i.e. the surface was disinfected. Results for LNP composition number 1 (LNP comprising DOPE, cholesterol, C14-PEG1000 and 306Oi10; cf. example 1) are shown in table 5 below. Table 5: io l? – >
– > For all measurements, (A0 – B0) = 1.7.
For all measurements, (A0 – B0) = 1.7.







Claims
Claims 1. Use of an analytical composition for monitoring disinfection procedures of surfaces, the composition comprising lipid nanoparticles encapsulating a nucleic acid, wherein the analytical composition complies with the following characteristics: • the lipid nanoparticles disintegrate in 70% V/V ethanol as measured by GC-MS, • the lipid nanoparticles protect the nucleic acid encapsulated therein from nuclease degradation as measured by qPCR, and • the lipid nanoparticles are stable upon drying on a surface as measured by qPCR, • the encapsulated nucleic acid has a length of between 60 – 200 nucleotides; and • the wt. ratio between the nucleic acid and the lipid nanoparticles in the analytical composition is in a range of between 0.01% to 15% nucleic acid. 2. The use according to claim 1, wherein the nucleic acid is DNA. 3. The use according to claim 1 or 2, wherein • the lipid nanoparticles comprise at least one lipid component and disintegration is defined as an increase in concentration of any one of the lipid components of the lipid nanoparticles in a continuous phase of a dispersion of the lipid nanoparticles in 70% V/V ethanol compared to the concentration of the same lipid component in a continuous phase of a reference dispersion of the same lipid nanoparticles in water as measured by GC-MS, wherein the increase in concentration is at least 100%, and/or • protection from nuclease degradation, preferably DNAse degradation, is defined as a decrease of max. 75% of intact nucleic acid present in the analytical composition after treatment with a nuclease compared to a reference analytical composition that was not treated with the nuclease as measured by qPCR, and/or • stability upon drying on a surface is defined as a decrease of max. 75% of intact nucleic acid present in the analytical composition after drying on a glass surface and after treatment with a nuclease compared to a reference analytical composition that was subjected to drying on the glass surface but not treated with the nuclease as measured by qPCR. 4. The use according to any one of the preceding claims, wherein the lipid nanoparticles contain an ionisable lipid, preferably in an amount of between 35 mol% and 100 mol%, and/or contain at least one of the following components: • a phospholipid, preferably in an amount of between 10 mol% and 50 mol%, • cholesterol, preferably in an amount of between 10 mol% and 50 mol%, and • a lipid anchored PEG, preferably in an amount of between 1 mol% and 10 mol%; • an inert solid component selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania, alumina, zirconia and ceria) and semiconductor oxides (preferably silica), preferably in an amount of between 5 wt% to 80 wt%. 5. The use according to claim 4, wherein the ionisable lipid is selected from the group consisting of oleylamine, dodecylamine, hexadecylamine, 306Oi10, and mixtures thereof, preferably selected from the group consisting of oleylamine and 306Oi10, more preferably 306Oi10. 6. The use according to any one of the preceding claims, wherein the lipid nanoparticles contain a phospholipid selected from the group consisting of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, 1,2- distearoyl-sn-glycero-3-phosphocholin, and mixtures thereof. 7. The use according to any one of the preceding claims, wherein the lipid nanoparticles contain cholesterol. 8. The use according to any one of the preceding claims, wherein the lipid nanoparticles contain lipid- anchored PEG, preferably C14PEG1000. 9. The use according to any one of the preceding claims, wherein the lipid nanoparticles contain, preferably consist of, one of the following: • oleylamine; • oleylamine, 1,2-distearoyl-sn-glycero-3- phosphocholin and cholesterol; • hexadecylamine, 1,2-distearoyl-sn-glycero-3- phosphocholin, and cholesterol; or • 306Oi10, 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine, cholesterol, and C14PEG1000. 10. The use according to any one of the preceding claims, wherein the analytical composition additionally contains additives selected from the group consisting of disaccharides, polysaccharides, PEG, and mixtures thereof, preferably wherein the wt. ratio between the lipid nanoparticles and the additives is in a range of between 1:0.1 to 1:20. 11. A method for monitoring a disinfection procedure with an analytical composition as defined in any one of the preceding claims, wherein the method comprises the steps: a) applying the analytical composition onto a surface, preferably followed by air-drying the composition on the surface, b) after a pre-determined time period, taking a test sample from the surface or taking a test sample from a second surface to which at least part of the analytical composition has been transferred, thereby dissolving the test sample in an aqueous solution to obtain an aqueous test sample, c) subjecting a portion A of the aqueous test sample to an enzymatic nuclease degradation step, in particular an enzymatic DNA degradation step, d) examining the portion A and a reference portion B of the aqueous test sample by a qPCR detection procedure using primers specific for the nucleic acid, in particular the DNA, encapsulated in the lipid nanoparticles to differentiate with reference to portion B, whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A and was thus protected during the enzymatic nuclease degradation step or, • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step. 12. The method according to claim 11, wherein • step b) additionally comprises taking a third test sample from a third surface, thereby dissolving the third test sample in a third aqueous solution to obtain a third aqueous test sample, • step c) additionally comprises subjecting a portion A’ of the third aqueous test sample to an enzymatic nuclease degradation step • step d) additionally comprises examining the portion A’ and a reference portion B’ of the third aqueous test sample by a qPCR detection procedure using primers specific for the nucleic acid encapsulated in the lipid nanoparticles to differentiate with reference to portion B’, whether • the detected nucleic acid originates from an intact lipid nanoparticle of portion A’ and was thus protected during the enzymatic nuclease degradation step, or • the detected nucleic acid originates from a disintegrated lipid nanoparticle of portion A’, wherein the lipid nanoparticle encapsulation was destroyed by the disinfection procedure and thus the nucleic acid was not protected during the enzymatic nuclease degradation step, or • the analytical composition is not present on the third surface. 13. The method according to claim 11 or 12, wherein if the difference between Ct values (A-B) – (A0-B0) > 2 or if the difference between Ct values (A’-B’) – (A0-B0) > 2, with • A0 being a control portion of A, respectively A’, and • B0 being a control portion of B, respectively B’, the detected nucleic acid originates from disintegrated lipid nanoparticles. 14. An analytical composition as defined in any one of claims 1 – 10, wherein the lipid nanoparticles additionally contain an inert solid component, said inert solid component: • Being selected from the group consisting of organic polymers (preferably selected from the group consisting of polystyrene, polyethylene terephthalate and polyethene), metal oxides (preferably selected from the group consisting of titania, alumina, zirconia and ceria) and semiconductor oxides (preferably silica), • being present in an amount of between 5 wt% to 80 wt%, • and having a primary particle size in the range of between 50 nm to 1500 nm. 15. A kit of parts comprising an analytical composition as defined in any one of claims 1 – 10, qPCR primers specific for the nucleic acid and optionally one or more of the following: • a nuclease, • a DNA polymerase, • a qPCR buffer.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23810403.8AEP4627114A1 (en) | 2022-11-28 | 2023-11-27 | Analytical composition for monitoring disinfection |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22209965 | 2022-11-28 | ||
| EP22209965.7 | 2022-11-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024115353A1true WO2024115353A1 (en) | 2024-06-06 |
Family
ID=84363935
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2023/083109CeasedWO2024115353A1 (en) | 2022-11-28 | 2023-11-27 | Analytical composition for monitoring disinfection |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP4627114A1 (en) |
| WO (1) | WO2024115353A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5672662A (en) | 1995-07-07 | 1997-09-30 | Shearwater Polymers, Inc. | Poly(ethylene glycol) and related polymers monosubstituted with propionic or butanoic acids and functional derivatives thereof for biotechnical applications |
| WO2017023431A1 (en)* | 2015-08-03 | 2017-02-09 | Safetraces, Inc. | Pathogen surrogates based on encapsulated tagged dna for verification of sanitation and wash water systems for fresh produce |
| US20200345641A1 (en)* | 2018-01-18 | 2020-11-05 | Etherna Immunotherapies Nv | Lipid nanoparticles |
- 2023
- 2023-11-27WOPCT/EP2023/083109patent/WO2024115353A1/ennot_activeCeased
- 2023-11-27EPEP23810403.8Apatent/EP4627114A1/enactivePending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5672662A (en) | 1995-07-07 | 1997-09-30 | Shearwater Polymers, Inc. | Poly(ethylene glycol) and related polymers monosubstituted with propionic or butanoic acids and functional derivatives thereof for biotechnical applications |
| WO2017023431A1 (en)* | 2015-08-03 | 2017-02-09 | Safetraces, Inc. | Pathogen surrogates based on encapsulated tagged dna for verification of sanitation and wash water systems for fresh produce |
| US20200345641A1 (en)* | 2018-01-18 | 2020-11-05 | Etherna Immunotherapies Nv | Lipid nanoparticles |
Non-Patent Citations (16)
| Title |
|---|
| ALHMIDI ET AL., OPEN FORUM INFECT. DIS., vol. 4, no. 3, 2017, pages 06 |
| BALL REBECCA L. ET AL: "Lipid Nanoparticle Formulations for Enhanced Co-delivery of siRNA and mRNA", vol. 18, no. 6, 25 April 2018 (2018-04-25), US, pages 3814 - 3822, XP055873781, ISSN: 1530-6984, Retrieved from the Internet <URL:https://pubs.acs.org/doi/pdf/10.1021/acs.nanolett.8b01101> DOI: 10.1021/acs.nanolett.8b01101* |
| BOOS ET AL., NUCLEIC ACIDS RES, vol. 41, no. 15, 2013, pages e145 |
| CASINI BEATRICE ET AL: "Evaluation of the Cleaning Procedure Efficacy in Prevention of Nosocomial Infections in Healthcare Facilities Using Cultural Method Associated with High Sensitivity Luminometer for ATP Detection", vol. 7, no. 3, 31 August 2018 (2018-08-31), pages 71, XP093046202, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6161163/pdf/pathogens-07-00071.pdf> DOI: 10.3390/pathogens7030071* |
| FERREIRA ADRIANO MENIS ET AL: "Assessment of disinfection of hospital surfaces using different monitoring methods", vol. 23, no. 3, 1 May 2015 (2015-05-01), pages 466 - 474, XP093046209, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4547070/pdf/0104-1169-rlae-23-03-00466.pdf> DOI: 10.1590/0104-1169.0094.2577* |
| GRIFFITH C.: "Surface Sampling and the Detection of Contamination", HANDBOOK OF HYGIENE CONTROL IN THE FOOD INDUSTRY., 15 July 2016 (2016-07-15), pages 673 - 696, XP093046218, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7152397/pdf/main.pdf> [retrieved on 20230511], DOI: https://doi.org/10.1016/B978-0-08-100155-4.00044-3* |
| HOU ET AL., NAT REV MATER, vol. 6, 2021, pages 1078 - 1094 |
| HOU ET AL., NAT. REV. MATER., vol. 6, 2021, pages 1078 - 1094 |
| NOCKER ET AL: "Molecular monitoring of disinfection efficacy using propidium monoazide in combination with quantitative PCR", JOURNAL OF MICROBIOLOGICAL METHODS, ELSEVIER, AMSTERDAM, NL, vol. 70, no. 2, 10 July 2007 (2007-07-10), pages 252 - 260, XP022146173, ISSN: 0167-7012, DOI: 10.1016/J.MIMET.2007.04.014* |
| OTTER ET AL., AM. J. INFECT. CONTROL, vol. 41, 2013 |
| PFUDERER LARA ET AL: "Synthetic Microbial Surrogates Consisting of Lipid Nanoparticles Encapsulating DNA for the Validation of Surface Disinfection Procedures", ACS APPLIED BIO MATERIALS, 20 March 2023 (2023-03-20), United States, pages 1252 - 1259, XP093045940, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10031560/pdf/mt3c00004.pdf> [retrieved on 20230510], DOI: 10.1021/acsabm.3c00004* |
| PITTET ET AL., LANCET INFECT. DIS., vol. 6, no. 10, 2006, pages 641 - 652 |
| SANTOS-JUNIOR AIRES G. ET AL: "Effectiveness of Surface Cleaning and Disinfection in a Brazilian Healthcare Facility", vol. 12, no. 1, 26 March 2018 (2018-03-26), pages 36 - 44, XP093046204, ISSN: 1874-4346, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5876921/pdf/TONURSJ-12-36.pdf> DOI: 10.2174/1874434601812010036* |
| SHIVALINGAM ET AL., ANGEW. CHEM. INT. ED., vol. 59, 2020, pages 11416 - 11422 |
| TIZIANA SANNA ET AL: "ATP bioluminescence assay for evaluating cleaning practices in operating theatres: applicability and limitations", BMC INFECTIOUS DISEASES, BIOMED CENTRAL LTD, LONDON, UK, vol. 18, no. 1, 19 November 2018 (2018-11-19), pages 1 - 7, XP021262746, DOI: 10.1186/S12879-018-3505-Y* |
| ULLRICH ET AL., ANTIMICROB. RESIST. INFECT. CONTROL, vol. 11, no. 4, 2022 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4627114A1 (en) | 2025-10-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Li et al. | Comparison of three common DNA concentration measurement methods | |
| AU2016329919B2 (en) | Quencher containing water soluble polymer-conjugated nanomaterial and use thereof | |
| Yan et al. | Influence of molecular structure on the antimicrobial function of phenylenevinylene conjugated oligoelectrolytes | |
| Wu et al. | Nano-graphene oxide with antisense vicR RNA reduced exopolysaccharide synthesis and biofilm aggregation for Streptococcus mutans | |
| Lu et al. | Selection of an aptamer against Muscovy duck parvovirus for highly sensitive rapid visual detection by label-free aptasensor | |
| WO2015120406A1 (en) | Oligonucleotide-based probes and methods for detection of microbes | |
| EP3431598B1 (en) | Method for collecting nucleic acid | |
| AU2001284203A1 (en) | Use | |
| WO2002018635A2 (en) | Use of nonviable particles comprising an internal control (ic) nucleic acid | |
| Yu et al. | Short-term oral administration of mesoporous silica nanoparticles potentially induced colon inflammation in rats through alteration of gut microbiota | |
| US10837051B2 (en) | Enzyme reaction reagent having dried form and method for preparing the same | |
| WO2019226648A1 (en) | Methods and control compositions for sequencing and chemical analyses | |
| JP2014027934A (en) | Method for enhancing reaction efficiency and sensitivity of nucleic acid amplification from biological material using ionic liquid | |
| CN108070583B (en) | Application of hydroxypropyl-beta-cyclodextrin in preparation of nucleic acid freeze-drying protective agent | |
| EP4060047A1 (en) | Method for detecting or quantifying oligonucleotide | |
| EP4627114A1 (en) | Analytical composition for monitoring disinfection | |
| Ramos et al. | Remodeling of the enterococcal cell envelope during surface penetration promotes intrinsic resistance to stress | |
| Lee et al. | Semi-automatic instrumentation for nucleic acid extraction and purification to quantify pathogens on surfaces | |
| AU2018277094A1 (en) | DNA stabilization of RNA | |
| CN103160512B (en) | A nucleic acid, a drug carrier, a drug composition and a preparation method thereof | |
| CA3006158A1 (en) | Sol-gel derived biohybrid materials incorporating long-chain dna aptamers | |
| ES2932660T3 (en) | Reagent to extract and amplify nucleic acid | |
| JP4546115B2 (en) | Quantitative determination of ATP in cells | |
| JP2020058270A (en) | Collection method of nucleic acid | |
| CN120099151B (en) | Buffer solution supporting multiplex fluorescence quantitative PCR (polymerase chain reaction) and preparation method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:23810403 Country of ref document:EP Kind code of ref document:A1 | |
| WWE | Wipo information: entry into national phase | Ref document number:2023810403 Country of ref document:EP | |
| NENP | Non-entry into the national phase | Ref country code:DE | |
| ENP | Entry into the national phase | Ref document number:2023810403 Country of ref document:EP Effective date:20250630 |