WO2023284872A1 - Polymorph of compound having dpp4-inhibitory activity and preparation method therefor - Google Patents
Polymorph of compound having dpp4-inhibitory activity and preparation method thereforDownload PDFInfo
- Publication number
- WO2023284872A1 WO2023284872A1PCT/CN2022/106082CN2022106082WWO2023284872A1WO 2023284872 A1WO2023284872 A1WO 2023284872A1CN 2022106082 WCN2022106082 WCN 2022106082WWO 2023284872 A1WO2023284872 A1WO 2023284872A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal form
- formula
- preparation
- compound shown
- following
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


























Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/78—Ring systems having three or more relevant rings
- C07D311/92—Naphthopyrans; Hydrogenated naphthopyrans
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- the inventionbelongs to the field of medicinal chemistry.
- the present inventionrelates to (2S,3R)-2-amino-9-methoxy-3-(2,4,5-trifluorophenyl)-2,3-dihydro-1H-benzo[ f] Polymorphic forms of the maleate salt of chroman-8-carbonitrile (HL012) and a process for their preparation.
- Polymorphismrefers to the phenomenon that solid substances form solid states with different physical and chemical properties in two or more different spatial arrangements.
- polymorphismincludes multi-component crystal forms such as organic solvates and hydrates.
- Drug polymorphismwidely exists in the drug development process and is an inherent characteristic of small organic molecules. Theoretically, small-molecule drugs can have an infinite number of crystal packing methods - polymorphic forms. Studies have shown that the number of drug polymorphic forms discovered is proportional to the time and resources invested in research. For example, Lipitor, the drug with the highest sales in the world so far, has as many as 35 crystal forms for patent protection.
- Polymorphismis not only controlled by internal factors such as the spatial structure and functional group properties of the molecule itself, intramolecular and intermolecular interactions, but also by drug synthesis process design, crystallization and purification conditions, preparation excipients selection, preparation process, etc. Routes and granulation methods, as well as storage conditions, packaging materials and other factors. Different crystal forms have different colors, melting points, solubility, dissolution properties, chemical stability, reactivity, mechanical stability, etc. These physical and chemical properties or processability sometimes directly affect the safety and effectiveness of drugs. Therefore, crystal form research and control has become an important research content in the process of drug development.
- (2S,3R)-2-Amino-9-methoxy-3-(2,4,5-trifluorophenyl)-2,3-dihydro-1H-benzo[f]chroman-8- Nitrileis an excellent inhibitor of dipeptidyl peptidase-4 (DPP-4). It can increase the activity of GLP-1 and GIP, promote insulin secretion, thereby lowering blood sugar levels and increasing glucose tolerance without side effects such as weight gain and hypoglycemia. The compound can replace the existing hypoglycemic drugs, has great industrialization and commercialization prospects and market value, and has remarkable economic benefits.
- the object of the present inventionis to provide (2S,3R)-2-amino-9-methoxy-3-(2,4,5-trifluorophenyl)-2,3-dihydro-1H-benzo[f ]
- the polymorphic forms of the maleate salt of chroman-8-nitrile, these crystal formshave higher crystallinity, lower hygroscopicity and excellent stability, and the preparation process of these crystal forms is simple and convenient simultaneously, thereby is conducive to The improvement of process treatment and physical and chemical properties of drugs, and the improvement of drug performance.
- the present inventionprovides the polymorphic form of the maleate salt of the compound shown in formula I
- the present inventionprovides the monohydrate B crystal form of the maleate salt of the compound represented by formula I.
- the X-ray powder diffraction pattern of the B crystal formhas characteristic peaks at the following 2 ⁇ angles: 8.5 ⁇ 0.2, 13.8 ⁇ 0.2, 18.3 ⁇ 0.2, and 25.9 ⁇ 0.2°.
- the X-ray powder diffraction pattern of the B crystal formhas characteristic peaks at the following 2 ⁇ angles: 8.5 ⁇ 0.2, 10.3 ⁇ 0.2, 13.8 ⁇ 0.2, 14.5 ⁇ 0.2, 18.3 ⁇ 0.2 and 25.9 ⁇ 0.2 °.
- the X-ray powder diffraction pattern of the B crystal formhas characteristic peaks at the following 2 ⁇ angles: 8.5 ⁇ 0.2, 10.0 ⁇ 0.2, 10.3 ⁇ 0.2, 13.8 ⁇ 0.2, 14.5 ⁇ 0.2, 17.2 ⁇ 0.2 , 17.7 ⁇ 0.2, 18.3 ⁇ 0.2, 22.2 ⁇ 0.2, and 25.9 ⁇ 0.2°.
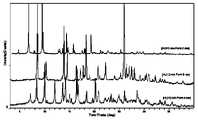
- the X-ray powder diffraction pattern of the B crystal formis shown in FIG. 9 .
- the peak positions and intensities of the characteristic peaksare shown in the following table:
- the X-ray powder diffraction pattern of the B crystal formis obtained by Cu K ⁇ ray diffraction.
- said Form Bshows decomposition prior to melting (158° C.) in differential scanning calorimetry.
- the differential scanning calorimetry curve of the crystal form Bis shown in FIG. 11 .
- thermogravimetric analysis (TG) diagram of the crystal form Bis shown in FIG. 10 .
- the infrared spectrogram of the B crystal formis shown in Figure 13 .
- the Raman spectrum of the crystal form Bis shown in FIG. 14 .
- the polarized photo of the crystal form Bis shown in FIG. 8 .
- the hygroscopicity analysis (DVS) diagram of the B crystal formis shown in FIG. 12 .
- the hygroscopicity of the crystal form Bis 0.5% under the condition of 80% humidity.
- the structure of the crystal form Bis shown in FIG. 28 .
- the present inventionprovides the preparation method of the B crystal form, the method comprising the following steps:
- step 1)is carried out at 25°C-50°C.
- step 1)the suspension is stirred; more preferably the suspension is stirred for at least 24 hours.
- the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solventis 25:1 to 15:1 g/l; more preferably 20:1 g/l.
- the organic solventis one or more selected from the following group: esters (including but not limited to: ethyl acetate, isopropyl ethyl ester), C1-6 Alcohols (including but not limited to: methanol, ethanol, propanol, isopropanol, butanol, isobutanol), C3-6 ketones (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone ), C6-C8 alkanes (including but not limited to hexane, heptane), substituted C1-6 alkanes (including but not limited to nitromethane, halogenated C1-6 alkanes, such as dichloromethane, chloroform), C2- 6 Nitriles (including but not limited to: acetonitrile).
- estersincluding but not limited to: ethyl acetate, isopropyl ethyl ester
- C1-6 Alcoholsincluding but not
- the methodincludes the following steps:
- step 3Collect and optionally dry the solid obtained in step 2).
- step 2)is carried out at 25°C-50°C.
- the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solventis 20:1 to 5:1 g/l; more preferably 10:1 g/l.
- the organic solventis one or more selected from the group consisting of C2-6 nitriles (including but not limited to: acetonitrile), C3-6 ketones (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone), tetrahydrofuran.
- the methodincludes the following steps:
- step 3Collect and optionally dry the solid obtained in step 2).
- the organic solventis one or more selected from the following group: C1-6 alcohol (including but not limited to: methanol, ethanol, propanol, isopropanol, butanol, isobutanol ); preferably methanol.
- C1-6 alcoholincluding but not limited to: methanol, ethanol, propanol, isopropanol, butanol, isobutanol ; preferably methanol.
- the present inventionprovides the monohydrate C crystal form of the maleate salt of the compound represented by formula I.
- the X-ray powder diffraction pattern of the C crystal formhas characteristic peaks at the following 2 ⁇ angles: 6.7 ⁇ 0.2, 9.5 ⁇ 0.2, 14.3 ⁇ 0.2 and 19.3 ⁇ 0.2°.
- the X-ray powder diffraction pattern of the C crystal formhas characteristic peaks at the following 2 ⁇ angles: 4.7 ⁇ 0.2, 6.7 ⁇ 0.2, 9.5 ⁇ 0.2, 14.3 ⁇ 0.2, 15.8 ⁇ 0.2 and 19.3 ⁇ 0.2 °.
- the X-ray powder diffraction pattern of the C crystal formhas characteristic peaks at the following 2 ⁇ angles: 4.7 ⁇ 0.2, 6.7 ⁇ 0.2, 9.5 ⁇ 0.2, 13.0 ⁇ 0.2, 13.6 ⁇ 0.2, 14.3 ⁇ 0.2 , 14.7 ⁇ 0.2, 15.8 ⁇ 0.2, 18.0 ⁇ 0.2, and 19.3 ⁇ 0.2°.
- the X-ray powder diffraction pattern of the C crystal formis shown in FIG. 16 .
- the peak positions and intensities of the characteristic peaksare shown in the following table:
- the X-ray powder diffraction pattern of the C crystal formis obtained by Cu K ⁇ ray diffraction.
- said Form Cshows decomposition prior to melting (155° C.) in differential scanning calorimetry.
- the differential scanning calorimetry curve of the C crystal formis shown in FIG. 18 .
- thermogravimetric analysis (TG) diagram of the crystal form Cis shown in FIG. 17 .
- the infrared spectrum of the crystal form Cis shown in Figure 20.
- the Raman spectrum of the crystal form Cis shown in FIG. 21 .
- the polarized photo of the crystal form Cis shown in FIG. 15 .
- the hygroscopicity analysis (DVS) diagram of the C crystal formis shown in FIG. 19 .
- the hygroscopicity of the crystal form Cis 0.7% under the condition of 80% humidity.
- the crystal form Cis a columnar crystal.
- the present inventionprovides a method for preparing the crystal form C, the method comprising the following steps:
- step 1)is performed at room temperature, such as 25°C.
- step 1)the obtained suspension is stirred for 1-3 days; more preferably, the obtained suspension is stirred for 3 days.
- the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solventis 40:1 to 5:1 g/l; more preferably 15:1 g/l.
- the organic solventis one or more selected from the group consisting of: esters (including but not limited to: ethyl acetate, isopropyl ethyl ester), C1-6 alcohols (including but not limited to Limited to: methanol, ethanol, propanol, isopropanol, butanol, isobutanol), aromatics (including but not limited to: benzene, toluene).
- estersincluding but not limited to: ethyl acetate, isopropyl ethyl ester
- C1-6 alcoholsincluding but not limited to Limited to: methanol, ethanol, propanol, isopropanol, butanol, isobutanol
- aromaticsincluding but not limited to: benzene, toluene.
- the methodincludes the following steps:
- step 3Collect and optionally dry the solid obtained in step 2).
- the organic solventis one or more selected from the group consisting of C3-6 ketones (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone).
- the methodincludes the following steps:
- step 3Collect and optionally dry the solid obtained in step 2).
- step 2)is carried out at 25°C-50°C.
- the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solventis 20:1 to 5:1 g/l; more preferably 10:1 g/l.
- the organic solventis one or more mixed solvents selected from the following group, preferably a plurality of mixed solvents: C1-6 alcohol (including but not limited to: methanol, ethanol, propanol, Isopropanol, butanol, isobutanol), C2-6 nitriles (including but not limited to: acetonitrile), C6-C8 alkanes (including but not limited to hexane, heptane), C2-6 ethers (including but not limited to : methyl ether, diethyl ether, methyl tert-butyl ether).
- C1-6 alcoholincluding but not limited to: methanol, ethanol, propanol, Isopropanol, butanol, isobutanol
- C2-6 nitrilesincluding but not limited to: acetonitrile
- C6-C8 alkanesincluding but not limited to hexane, heptane
- the present inventionprovides the anhydrous A crystal form of the maleate salt of the compound represented by formula I.
- the X-ray powder diffraction pattern of the A crystal formhas characteristic peaks at the following 2 ⁇ angles: 8.3 ⁇ 0.2, 10.0 ⁇ 0.2, 14.2 ⁇ 0.2 and 16.4 ⁇ 0.2°.
- the X-ray powder diffraction pattern of the crystal form Ahas characteristic peaks at the following 2 ⁇ angles: 8.3 ⁇ 0.2, 10.0 ⁇ 0.2, 12.0 ⁇ 0.2, 13.6 ⁇ 0.2, 14.2 ⁇ 0.2 and 16.4 ⁇ 0.2 °.
- the X-ray powder diffraction pattern of the crystal form Ahas characteristic peaks at the following 2 ⁇ angles: 8.3 ⁇ 0.2, 10.0 ⁇ 0.2, 12.0 ⁇ 0.2, 13.6 ⁇ 0.2, 14.2 ⁇ 0.2, 16.4 ⁇ 0.2 , 16.7 ⁇ 0.2, 18.3 ⁇ 0.2, 20.1 ⁇ 0.2, and 26.0 ⁇ 0.2°.
- the X-ray powder diffraction pattern of the crystal form Ais shown in FIG. 2 .
- the peak positions and intensities of the characteristic peaksare shown in the following table:
- the X-ray powder diffraction pattern of the A crystal formis obtained by Au K ⁇ ray diffraction.
- the crystalline Form Ashows onset of melting at about 188.2°C in differential scanning calorimetry analysis.
- said crystalline form Ashows decomposition starting at 190° C. in differential scanning calorimetry analysis.
- the differential scanning calorimetry curve of the crystal form Ais shown in FIG. 4 .
- thermogravimetric analysis (TG) diagram of the crystal form Ais shown in FIG. 3 .
- the infrared spectrum of the crystal form Ais shown in FIG. 6 .
- the Raman spectrum of the crystal form Ais shown in FIG. 7 .
- the polarized photo of the crystal form Ais shown in FIG. 1 .
- the hygroscopicity analysis (DVS) diagram of the crystal form Ais shown in FIG. 5 .
- the hygroscopicity of the crystal form Ais 0.5% under the condition of 80% humidity.
- the crystal form Ais a columnar crystal.
- the present inventionprovides the preparation method of the A crystal form, the method comprising the following steps:
- step 2Collect and optionally dry the solid precipitated in step 1).
- the methodincludes the following steps:
- step 1)suspending the maleate salt of the compound represented by formula I in an organic solvent at 25-50°C, preferably 50°C, and then stirring to obtain a suspension for 1-3 days; more The resulting suspension is preferably stirred for 3 days.
- the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solventis 40:1 to 5:1 g/l; more preferably 15:1 g/l.
- the organic solventis one or more selected from the following group: esters (including but not limited to: ethyl acetate, isopropyl ethyl ester).
- the methodincludes the following steps:
- step 3Collect and optionally dry the solid obtained in step 2).
- the maleate of the compound shown in formula Iis combined with C3-6 ketone (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone), methyl ethyl ketone or
- C3-6 ketoneincluding but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone, methyl ethyl ketone or
- the mass volume ratio of acetonitrileis 100:1 to 20:1 g/l; more preferably 50:1 g/l.
- the present inventionprovides a pharmaceutical composition comprising the polymorphic form of any one of the first, second, fourth and sixth aspects and a pharmaceutically acceptable carrier or excipient.
- the present inventionprovides the use of the polymorphic form described in any one of the first, second, fourth and sixth aspects in the preparation of a DPP4 inhibitor.
- the DPP4 inhibitoris a hypoglycemic drug or a diabetes treatment drug.
- the present inventionprovides the polymorphic form of any one of the first, second, fourth and sixth aspects, which is used for preparing a DPP4 inhibitor.
- the DPP4 inhibitoris a hypoglycemic drug or a diabetes treatment drug.
- the present inventionprovides a treatment method for lowering blood sugar or treating diabetes, the method comprising treating an effective amount of the polymorphic form described in any one of the first, second, fourth and sixth aspects or the eighth
- the pharmaceutical composition of the aspectis administered to a subject in need thereof.
- the subjectis a human.
- Fig. 1has shown the polarized photo of A crystal form of the present invention
- Figure 2shows the X-ray powder diffraction (XRPD) pattern of the A crystal form of the present invention
- Fig. 3shows the thermogravimetric analysis (TG) figure of A crystal form of the present invention
- Fig. 4has shown the differential scanning calorimetry (DSC) figure of A crystal form of the present invention
- Figure 5shows the hygroscopicity analysis (DVS) figure of the A crystal form of the present invention
- Figure 6shows the infrared spectrum (IR) figure of the A crystal form of the present invention
- Figure 7shows the Raman spectrum (Raman) figure of the A crystal form of the present invention
- Fig. 8shows the polarized photo of B crystal form of the present invention
- Figure 9shows an X-ray powder diffraction (XRPD) pattern of Form B of the present invention
- FIG. 10shows the thermogravimetric analysis (TG) figure of B crystal form of the present invention
- FIG. 11shows the differential scanning calorimetry (DSC) diagram of the B crystal form of the present invention
- Figure 12shows the hygroscopicity analysis (DVS) figure of B crystal form of the present invention
- Figure 13shows the infrared spectrum (IR) figure of B crystal form of the present invention
- Figure 14shows the Raman spectrum (Raman) figure of B crystal form of the present invention
- Fig. 15has shown the polarized photo of C crystal form of the present invention.
- Figure 16shows the X-ray powder diffraction (XRPD) pattern of the C crystal form of the present invention
- FIG. 17shows the thermogravimetric analysis (TG) diagram of the C crystal form of the present invention.
- FIG. 18shows the differential scanning calorimetry (DSC) diagram of the C crystal form of the present invention
- Figure 19shows the hygroscopicity analysis (DVS) diagram of the C crystal form of the present invention
- Figure 20shows the infrared spectrum (IR) diagram of the C crystal form of the present invention
- Figure 21shows the Raman spectrum (Raman) figure of C crystal form of the present invention
- Figure 22shows the X-ray powder diffraction (XRPD) overlay comparison of the A crystal form, the B crystal form and the C crystal form of the present invention
- FIG. 23shows the thermogravimetric analysis (TG) spectrum overlay comparison of A crystal form, B crystal form and C crystal form of the present invention
- Figure 24shows the differential scanning calorimetry (DSC) overlay comparison of Form A, Form B and Form C of the present invention
- Figure 25shows the overlay comparison of hygroscopicity analysis (DVS) of A crystal form, B crystal form and C crystal form of the present invention
- Figure 26shows the overlay comparison of the Ramam spectrum of the A crystal form, the B crystal form and the C crystal form of the present invention
- Figure 27shows the overlay comparison of the IR spectra of Form A, Form B and Form C of the present invention.
- Figure 28shows the structure of Form B.
- the inventorsAfter extensive and in-depth research, the inventors have systematically screened possible crystal forms by using different crystallization conditions and experimental means in view of the polymorphic form of the maleate salt of the compound represented by formula I. Finally, it was found that the maleate salt of the compound represented by formula I exists in three different solid forms, which are anhydrous Form A, monohydrate Form B, and monohydrate Form C. These new crystal forms have high crystallinity, low hygroscopicity and excellent stability, and at the same time, the preparation process of these crystal forms is simple, which is beneficial to the process treatment of drugs and the improvement of physical and chemical properties, and improves the drug-making performance. The present invention has been accomplished on this basis.
- Crystal form researchincludes two stages of crystal discovery and crystal form optimization.
- various crystallization methodsare mainly used, such as melt crystallization, solution volatilization, rapid cooling and suspension crystallization methods.
- solventsBy changing crystallization conditions, solvents , external factors such as temperature, speed and suspension solvent ratio affect drug crystallization.
- a high-throughput sample preparation platformis used to prepare hundreds of crystallization tests at the same time, using micro-sample preparation technology and analysis and testing methods. Preparation and discovery of new crystal forms.
- crystal form optimization stageit is necessary to explore new crystal form process amplification and preparation conditions, and use various solid characterization methods, such as x-ray diffraction, solid state nuclear magnetic resonance, Raman spectroscopy, infrared spectroscopy and other means of crystal form crystal characterization
- various solid characterization methodssuch as x-ray diffraction, solid state nuclear magnetic resonance, Raman spectroscopy, infrared spectroscopy and other means of crystal form crystal characterization
- DSC, TGA, DVS, HPLC, etc.should be used to study the physical and chemical properties of the crystal form, and to compare the hygroscopicity, chemical stability, physical state stability, and processability of different crystal forms.
- the most preferred solid formis selected for development.
- the present inventionprovides three stable new crystal forms.
- the present inventionprovides the monohydrate B crystal form of the maleate salt of the compound shown in formula I, and the X-ray powder diffraction pattern of the B crystal form has a characteristic peak at the following 2 ⁇ angle: 8.5 ⁇ 0.2, 10.0 ⁇ 0.2, 10.3 ⁇ 0.2, 13.8 ⁇ 0.2, 14.5 ⁇ 0.2, 17.2 ⁇ 0.2, 17.7 ⁇ 0.2, 18.3 ⁇ 0.2, 22.2 ⁇ 0.2, and 25.9 ⁇ 0.2°.
- the B crystal formwas shown to decompose before melting (158°C) in differential scanning calorimetry analysis, and the hygroscopicity was 0.5% under the condition of 80% humidity.
- the present inventionprovides the monohydrate C crystal form of the maleate salt of the compound shown in formula I, and the X-ray powder diffraction pattern of the C crystal form has a characteristic peak at the following 2 ⁇ angle: 4.7 ⁇ 0.2, 6.7 ⁇ 0.2, 9.5 ⁇ 0.2, 13.0 ⁇ 0.2, 13.6 ⁇ 0.2, 14.3 ⁇ 0.2, 14.7 ⁇ 0.2, 15.8 ⁇ 0.2, 18.0 ⁇ 0.2, and 19.3 ⁇ 0.2°.
- the crystal formis a columnar crystal, which is decomposed before melting (155° C.) in differential scanning calorimetry analysis; and has a hygroscopicity of 0.7% under 80% humidity conditions .
- the present inventionprovides the anhydrous A crystal form of the maleate salt of the compound represented by formula I, and the X-ray powder diffraction pattern of the A crystal form has a characteristic peak at the following 2 ⁇ angle: 8.3 ⁇ 0.2 , 10.0 ⁇ 0.2, 12.0 ⁇ 0.2, 13.6 ⁇ 0.2, 14.2 ⁇ 0.2, 16.4 ⁇ 0.2, 16.7 ⁇ 0.2, 18.3 ⁇ 0.2, 20.1 ⁇ 0.2, and 26.0 ⁇ 0.2°.
- the crystal formis a columnar crystal, which starts to melt at about 188.2°C and decomposes at 190°C in differential scanning calorimetry analysis; Moisture is 0.5%.
- the present inventionfurther provides a pharmaceutical composition comprising the polymorphic form. Since the polymorphic form of the present invention has the advantages of high crystallinity, low hygroscopicity, and regular crystal form, the pharmaceutical composition of the present invention has excellent drug-making properties.
- the pharmaceutical composition of the present inventioncan be used as a DPP4 inhibitor because the compound represented by formula I has the ability to inhibit DPP4.
- the pharmaceutical composition of the present inventioncan be used as a hypoglycemic drug or a diabetes treatment drug
- compositionalso includes an optional pharmaceutically acceptable carrier.
- compositionis intended to cover a product comprising the specified ingredients in the specified amounts, and any product resulting directly or indirectly from the combination of the specified ingredients in the specified amounts; A carrier, diluent, or excipient that does not cause significant irritation and does not interfere with the biological activity and properties of the administered compound; that is, the carrier, diluent, or excipient must be compatible with the other ingredients of the formulation and The recipient is harmless.
- the pharmaceutical composition of the present inventioncan be prepared by methods known to those skilled in the art.
- the compound of the present inventioncan be mixed with a pharmaceutically acceptable carrier, diluent or excipient to prepare the corresponding pharmaceutical composition.
- those skilled in the artcan prepare the compound or pharmaceutical composition of the present invention into various suitable dosage forms. According to the desired dosage form, those skilled in the art can also select corresponding pharmaceutically acceptable carriers, diluents or excipients.
- a safe and effective amount of the polymorphic formmay be included in the pharmaceutical composition of the present invention.
- the "safe and effective amount”refers to: the amount of the compound (or crystal form) is sufficient to significantly improve the condition without causing serious side effects.
- “Pharmaceutically acceptable carrier”refers to: one or more compatible solid or liquid fillers or gel substances, which are suitable for human use, and must have sufficient purity and low enough toxicity. "Compatibility” here means that each component in the composition can be blended with the active ingredient of the present invention and with each other without significantly reducing the efficacy of the active ingredient.
- Examples of pharmaceutically acceptable carrier partsinclude cellulose and derivatives thereof (such as sodium carboxymethylcellulose, sodium ethylcellulose, cellulose acetate, etc.), gelatin, talc, solid lubricants (such as stearic acid , magnesium stearate), calcium sulfate, vegetable oil (such as soybean oil, sesame oil, peanut oil, olive oil, etc.), polyols (such as propylene glycol, glycerin, mannitol, sorbitol, etc.), emulsifiers (such as Tween ), wetting agent (such as sodium lauryl sulfate), coloring agent, flavoring agent, stabilizer, antioxidant, preservative, pyrogen-free water, etc.
- cellulose and derivatives thereofsuch as sodium carboxymethylcellulose, sodium ethylcellulose, cellulose acetate, etc.
- gelatinsuch as talc
- solid lubricantssuch as stearic acid , magnesium stearate
- the mode of administration of the polymorph or pharmaceutical composition of the present inventionis not particularly limited, and representative modes of administration include (but are not limited to): oral, rectal, parenteral (intravenous, intramuscular or subcutaneous), and topical administration medicine.
- Solid dosage forms for oral administrationinclude capsules, tablets, pills, powders and granules.
- the active ingredientis admixed with at least one conventional inert excipient (or carrier), such as sodium citrate or dicalcium phosphate, or with: (a) fillers or extenders, for example, Starch, lactose, sucrose, glucose, mannitol and silicic acid; (b) binders such as hydroxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose and acacia; (c) humectants, For example, glycerol; (d) disintegrants, such as agar, calcium carbonate, potato starch or tapioca starch, alginic acid, certain complex silicates, and sodium carbonate; (e) slow agents, such as paraffin; (f) Absorption accelerators such as quaternary ammonium compounds; (g) wetting agents such as cetyl alcohol and glyceryl mono
- Solid dosage formssuch as tablets, dragees, capsules, pills, and granules can be prepared with coatings and shell materials, such as enteric coatings and others well known in the art. They may contain opacifying agents and the release of the active ingredient from such compositions may be in a certain part of the alimentary canal in a delayed manner.
- coatings and shell materialssuch as enteric coatings and others well known in the art. They may contain opacifying agents and the release of the active ingredient from such compositions may be in a certain part of the alimentary canal in a delayed manner.
- Examples of usable embedding componentsare polymeric substances and waxy substances.
- the active ingredientcan also be in the form of microcapsules with one or more of the above-mentioned excipients, if necessary.
- Liquid dosage forms for oral administrationinclude pharmaceutically acceptable emulsions, solutions, suspensions, syrups or tinctures.
- liquid dosage formsmay contain inert diluents conventionally used in the art, such as water or other solvents, solubilizers and emulsifiers, for example, ethanol, isopropanol, ethyl carbonate, ethyl acetate, propylene glycol, 1 , 3-butanediol, dimethylformamide and oils, especially cottonseed oil, peanut oil, corn germ oil, olive oil, castor oil and sesame oil or mixtures of these substances, etc.
- inert diluentsconventionally used in the art, such as water or other solvents, solubilizers and emulsifiers, for example, ethanol, isopropanol, ethyl carbonate, ethyl acetate, propylene glycol, 1 , 3-butanediol, dimethylformamide and
- compositionscan also contain adjuvants, such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- adjuvantssuch as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- Suspensionsin addition to the active ingredient, may contain suspending agents, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum methoxide and agar, mixtures of these substances, and the like.
- suspending agentsfor example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum methoxide and agar, mixtures of these substances, and the like.
- compositions for parenteral injectionmay comprise physiologically acceptable sterile aqueous or anhydrous solutions, dispersions, suspensions or emulsions, and sterile powders for reconstitution into sterile injectable solutions or dispersions.
- Suitable aqueous and non-aqueous carriers, diluents, solvents or vehiclesinclude water, ethanol, polyols, and suitable mixtures thereof.
- Dosage forms of the polymorphs of the invention for topical administrationinclude ointments, powders, patches, sprays and inhalants.
- the active ingredientis mixed under sterile conditions with a physiologically acceptable carrier and any preservatives, buffers, or propellants which may be required, if necessary.
- polymorphic form of the present inventioncan be used as a DPP4 inhibitor
- those skilled in the artcan understand that the polymorphic form or pharmaceutical composition of the present invention can be used to lower blood sugar or treat diabetes in a subject.
- the method of the present invention for lowering blood sugar or treating diabetes in a subjectcomprises administering to a subject in need thereof a therapeutically effective amount of said polymorphic form or composition of matter.
- Such subjectsinclude, but are not limited to, humans.
- the polymorphic form of the present inventionhas higher crystallinity, lower hygroscopicity and excellent stability;
- the preparation process of the polymorphic form of the present inventionis simple and convenient, thereby being conducive to the improvement of the process treatment and physical and chemical properties of the medicine;
- the detection method of instrument involved in the present invention and gained crystal formis as follows:
- HL012-MA50mg was dissolved in 1mL of methyl ethyl ketone at 50 degrees, slowly cooled to room temperature, and kept at room temperature for 3 days. After filtration, the resulting solid was dried under reduced pressure at room temperature. 30 mg of a crystalline powder (type A) was obtained, and the yield was 60%.
- Preparation methodSlowly volatilize in acetonitrile solution at room temperature to obtain Form B single crystal.
- the single crystal data information of Form Bis shown in the following table:
- the crystal structureis shown in Figure 28.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Engineering & Computer Science (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
Translated fromChinese本发明属于药物化学领域。具体地说,本发明涉及(2S,3R)-2-氨基-9-甲氧基-3-(2,4,5-三氟苯基)-2,3-二氢-1H-苯并[f]色满-8-腈(HL012)的马来酸盐的多晶型及其制备方法。The invention belongs to the field of medicinal chemistry. In particular, the present invention relates to (2S,3R)-2-amino-9-methoxy-3-(2,4,5-trifluorophenyl)-2,3-dihydro-1H-benzo[ f] Polymorphic forms of the maleate salt of chroman-8-carbonitrile (HL012) and a process for their preparation.
多晶型现象是指固体物质以两种或两种以上的不同空间排列方式,形成的具有不同物理化学性质的固体状态的现象。在药物研究领域,多晶型包括了有机溶剂化物、水合物等多组分晶体形式。药物多晶现象在药物开发过程中广泛存在,是有机小分子化合物固有的特性。理论上小分子药物可以有无限多的晶体堆积方式-多晶型,研究表明,药物多晶型的发现数量与其投入的研究的时间和资源成正比例。如世界上迄今为止销售额最高的药物-Lipitor,申请专利保护的晶型就多达35种。多晶型现象不光受到分子本身的空间结构和官能基团性能,分子内和分子间的相互作用等内在因素的控制,它还受药物合成工艺设计、结晶和纯化条件、制剂辅料选择、制剂工艺路线和制粒方法、以及储存条件、包装材料等诸方面因素的影响。不同晶型具有不同的颜色、熔点、溶解、溶出性能、化学稳定性、反应性、机械稳定性等,这些物理化学性能或可加工性能有时直接影响到药物的安全、有效性能。因此晶型研究和控制成为药物研发过程中的重要研究内容。Polymorphism refers to the phenomenon that solid substances form solid states with different physical and chemical properties in two or more different spatial arrangements. In the field of pharmaceutical research, polymorphism includes multi-component crystal forms such as organic solvates and hydrates. Drug polymorphism widely exists in the drug development process and is an inherent characteristic of small organic molecules. Theoretically, small-molecule drugs can have an infinite number of crystal packing methods - polymorphic forms. Studies have shown that the number of drug polymorphic forms discovered is proportional to the time and resources invested in research. For example, Lipitor, the drug with the highest sales in the world so far, has as many as 35 crystal forms for patent protection. Polymorphism is not only controlled by internal factors such as the spatial structure and functional group properties of the molecule itself, intramolecular and intermolecular interactions, but also by drug synthesis process design, crystallization and purification conditions, preparation excipients selection, preparation process, etc. Routes and granulation methods, as well as storage conditions, packaging materials and other factors. Different crystal forms have different colors, melting points, solubility, dissolution properties, chemical stability, reactivity, mechanical stability, etc. These physical and chemical properties or processability sometimes directly affect the safety and effectiveness of drugs. Therefore, crystal form research and control has become an important research content in the process of drug development.
(2S,3R)-2-氨基-9-甲氧基-3-(2,4,5-三氟苯基)-2,3-二氢-1H-苯并[f]色满-8-腈是一种优异的二肽基肽酶-4(Dipeptidyl peptidase-4,DPP-4)抑制剂。它可以提高GLP-1和GIP的活性,促进胰岛素分泌,从而降低血糖水平,增加葡萄糖耐量,且没有体重增加和低血糖等副作用。该化合物够替代现有的降血糖药物,具备极大的产业化和商品化前景以及市场价值,经济效益显著。(2S,3R)-2-Amino-9-methoxy-3-(2,4,5-trifluorophenyl)-2,3-dihydro-1H-benzo[f]chroman-8- Nitrile is an excellent inhibitor of dipeptidyl peptidase-4 (DPP-4). It can increase the activity of GLP-1 and GIP, promote insulin secretion, thereby lowering blood sugar levels and increasing glucose tolerance without side effects such as weight gain and hypoglycemia. The compound can replace the existing hypoglycemic drugs, has great industrialization and commercialization prospects and market value, and has remarkable economic benefits.
发明内容Contents of the invention
本发明的目的在于提供(2S,3R)-2-氨基-9-甲氧基-3-(2,4,5-三氟苯基)-2,3-二氢-1H-苯并[f]色满-8-腈的马来酸盐的多晶型,这些晶型具有较高的结晶度、较低的吸湿性以及优异的稳定性,同时这些晶型的制备工艺简便,从而有利于药物的工艺处理和物化性能的改善,并提高成药性能。The object of the present invention is to provide (2S,3R)-2-amino-9-methoxy-3-(2,4,5-trifluorophenyl)-2,3-dihydro-1H-benzo[f ] The polymorphic forms of the maleate salt of chroman-8-nitrile, these crystal forms have higher crystallinity, lower hygroscopicity and excellent stability, and the preparation process of these crystal forms is simple and convenient simultaneously, thereby is conducive to The improvement of process treatment and physical and chemical properties of drugs, and the improvement of drug performance.
在第一方面,本发明提供式I所示化合物的马来酸盐的多晶型In the first aspect, the present invention provides the polymorphic form of the maleate salt of the compound shown in formula I

在第二方面,本发明提供式I所示化合物的马来酸盐的一水合物B晶型,所述B晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:8.5±0.2、13.8±0.2、18.3±0.2和25.9±0.2°。In a second aspect, the present invention provides the monohydrate B crystal form of the maleate salt of the compound represented by formula I. The X-ray powder diffraction pattern of the B crystal form has characteristic peaks at the following 2θ angles: 8.5±0.2, 13.8±0.2, 18.3±0.2, and 25.9±0.2°.
在优选的实施方式中,所述B晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:8.5±0.2、10.3±0.2、13.8±0.2、14.5±0.2、18.3±0.2和25.9±0.2°。In a preferred embodiment, the X-ray powder diffraction pattern of the B crystal form has characteristic peaks at the following 2θ angles: 8.5±0.2, 10.3±0.2, 13.8±0.2, 14.5±0.2, 18.3±0.2 and 25.9±0.2 °.
在优选的实施方式中,所述B晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:8.5±0.2、10.0±0.2、10.3±0.2、13.8±0.2、14.5±0.2、17.2±0.2、17.7±0.2、18.3±0.2、22.2±0.2和25.9±0.2°。In a preferred embodiment, the X-ray powder diffraction pattern of the B crystal form has characteristic peaks at the following 2θ angles: 8.5±0.2, 10.0±0.2, 10.3±0.2, 13.8±0.2, 14.5±0.2, 17.2±0.2 , 17.7±0.2, 18.3±0.2, 22.2±0.2, and 25.9±0.2°.
在优选的实施方式中,所述B晶型的X-射线粉末衍射图谱如图9所示。In a preferred embodiment, the X-ray powder diffraction pattern of the B crystal form is shown in FIG. 9 .
在优选的实施方式中,所述B晶型的X射线粉末衍射图谱中,特征峰的峰位置及强度如下表所示:In a preferred embodiment, in the X-ray powder diffraction pattern of the B crystal form, the peak positions and intensities of the characteristic peaks are shown in the following table:


在优选的实施方式中,所述B晶型的X-射线粉末衍射图谱采用Cu Kα射线衍射得到。In a preferred embodiment, the X-ray powder diffraction pattern of the B crystal form is obtained by Cu Kα ray diffraction.
在优选的实施方式中,所述B晶型在差示扫描量热分析中显示在熔融前(158℃)分解。In a preferred embodiment, said Form B shows decomposition prior to melting (158° C.) in differential scanning calorimetry.
在优选的实施方式中,所述B晶型的差示扫描量热曲线如图11所示。In a preferred embodiment, the differential scanning calorimetry curve of the crystal form B is shown in FIG. 11 .
在优选的实施方式中,所述B晶型的热失重分析(TG)图如图10所示。In a preferred embodiment, the thermogravimetric analysis (TG) diagram of the crystal form B is shown in FIG. 10 .
在优选的实施方式中,所述B晶型的红外光谱图如图13所示。In a preferred embodiment, the infrared spectrogram of the B crystal form is shown in Figure 13 .
在优选的实施方式中,所述B晶型的拉曼光谱图如图14所示。In a preferred embodiment, the Raman spectrum of the crystal form B is shown in FIG. 14 .
在优选的实施方式中,所述B晶型的偏光照片如图8所示。In a preferred embodiment, the polarized photo of the crystal form B is shown in FIG. 8 .
在优选的实施方式中,所述B晶型的吸湿性分析(DVS)图如图12所示。In a preferred embodiment, the hygroscopicity analysis (DVS) diagram of the B crystal form is shown in FIG. 12 .
在优选的实施方式中,所述B晶型是一水合物;无色柱状晶体;空间群为P 212121;晶胞参数(a,b,c,α,β,γ)为

在优选的实施方式中,所述B晶型在80%湿度条件下引湿性为0.5%。In a preferred embodiment, the hygroscopicity of the crystal form B is 0.5% under the condition of 80% humidity.
在优选的实施方式中,所述B晶型的结构如图28所示。In a preferred embodiment, the structure of the crystal form B is shown in FIG. 28 .
在第三方面,本发明提供所述的B晶型的制备方法,所述方法包括以下步骤:In a third aspect, the present invention provides the preparation method of the B crystal form, the method comprising the following steps:
1)将式I所示化合物的马来酸盐混悬于有机溶剂中,从而得到一混悬液;1) suspending the maleate salt of the compound shown in formula I in an organic solvent to obtain a suspension;
2)过滤步骤1)得到的混悬液,从而得到固体部分;2) filtering the suspension obtained in step 1) to obtain a solid portion;
3)收集并任选干燥得到的固体部分。3) Collect and optionally dry the resulting solid portion.
在优选的实施方式中,步骤1)在25℃-50℃下进行。In a preferred embodiment, step 1) is carried out at 25°C-50°C.
在优选的实施方式中,步骤1)中,搅拌得到混悬液;更优选搅拌得到的混悬液至少24小时。In a preferred embodiment, in step 1), the suspension is stirred; more preferably the suspension is stirred for at least 24 hours.
在优选的实施方式中,步骤1)中,式I所示化合物的马来酸盐与有机溶剂的质量体积比为25:1到15:1g/l;更优选20:1g/l。In a preferred embodiment, in step 1), the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solvent is 25:1 to 15:1 g/l; more preferably 20:1 g/l.
在优选的实施方式中,步骤1)中,所述有机溶剂是选自下组的一种或多种:酯类(包括但不限于:乙酸乙酯、异丙基乙酯)、C1-6醇(包括但不限于:甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇)、C3-6酮(包括但不限于:丙酮、甲基乙基酮、甲基异丁基酮)、C6-C8烷烃(包 括但不限于己烷、庚烷)、取代的C1-6烷烃(包括但不限于硝基甲烷,卤代C1-6烷烃,例如二氯甲烷、氯仿)、C2-6腈(包括但不限于:乙腈)。In a preferred embodiment, in step 1), the organic solvent is one or more selected from the following group: esters (including but not limited to: ethyl acetate, isopropyl ethyl ester), C1-6 Alcohols (including but not limited to: methanol, ethanol, propanol, isopropanol, butanol, isobutanol), C3-6 ketones (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone ), C6-C8 alkanes (including but not limited to hexane, heptane), substituted C1-6 alkanes (including but not limited to nitromethane, halogenated C1-6 alkanes, such as dichloromethane, chloroform), C2- 6 Nitriles (including but not limited to: acetonitrile).
在具体的实施方式中,所述方法包括以下步骤:In a specific embodiment, the method includes the following steps:
1)将式I所示化合物的马来酸盐溶解于有机溶剂中,从而得到一溶液;1) dissolving the maleate of the compound shown in formula I in an organic solvent to obtain a solution;
2)将步骤1)所得的溶液挥发溶剂至干;2) volatilize the solvent from the solution obtained in step 1) to dryness;
3)收集并任选干燥步骤2)得到的固体。3) Collect and optionally dry the solid obtained in step 2).
在优选的实施方式中,步骤2)在25℃-50℃下进行。In a preferred embodiment, step 2) is carried out at 25°C-50°C.
在优选的实施方式中,式I所示化合物的马来酸盐与有机溶剂的质量体积比为20:1到5:1g/l;更优选10:1g/l。In a preferred embodiment, the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solvent is 20:1 to 5:1 g/l; more preferably 10:1 g/l.
在优选的实施方式中,所述有机溶剂是选自下组的一种或多种:C2-6腈(包括但不限于:乙腈)、C3-6酮(包括但不限于:丙酮、甲基乙基酮、甲基异丁基酮)、四氢呋喃。In a preferred embodiment, the organic solvent is one or more selected from the group consisting of C2-6 nitriles (including but not limited to: acetonitrile), C3-6 ketones (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone), tetrahydrofuran.
在具体的实施方式中,所述方法包括以下步骤:In a specific embodiment, the method includes the following steps:
1)在升高的温度下,将式I所示化合物的马来酸盐溶解于有机溶剂中,从而得到一热溶液;1) at elevated temperature, the maleate salt of the compound shown in formula I is dissolved in an organic solvent, thereby obtaining a hot solution;
2)将步骤1)所得的热溶液降温,从而析出固体;2) cooling the hot solution obtained in step 1) to precipitate solids;
3)收集并任选干燥步骤2)得到的固体。3) Collect and optionally dry the solid obtained in step 2).
在优选的实施方式中,所述有机溶剂是选自下组的一种或多种:C1-6醇(包括但不限于:甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇);优选甲醇。In a preferred embodiment, the organic solvent is one or more selected from the following group: C1-6 alcohol (including but not limited to: methanol, ethanol, propanol, isopropanol, butanol, isobutanol ); preferably methanol.
在第四方面,本发明提供式I所示化合物的马来酸盐的一水合物C晶型,所述C晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:6.7±0.2、9.5±0.2、14.3±0.2和19.3±0.2°。In the fourth aspect, the present invention provides the monohydrate C crystal form of the maleate salt of the compound represented by formula I. The X-ray powder diffraction pattern of the C crystal form has characteristic peaks at the following 2θ angles: 6.7±0.2, 9.5±0.2, 14.3±0.2 and 19.3±0.2°.
在优选的实施方式中,所述C晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:4.7±0.2、6.7±0.2、9.5±0.2、14.3±0.2、15.8±0.2和19.3±0.2°。In a preferred embodiment, the X-ray powder diffraction pattern of the C crystal form has characteristic peaks at the following 2θ angles: 4.7±0.2, 6.7±0.2, 9.5±0.2, 14.3±0.2, 15.8±0.2 and 19.3±0.2 °.
在优选的实施方式中,所述C晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:4.7±0.2、6.7±0.2、9.5±0.2、13.0±0.2、13.6±0.2、14.3±0.2、14.7±0.2、15.8±0.2、18.0±0.2和19.3±0.2°。In a preferred embodiment, the X-ray powder diffraction pattern of the C crystal form has characteristic peaks at the following 2θ angles: 4.7±0.2, 6.7±0.2, 9.5±0.2, 13.0±0.2, 13.6±0.2, 14.3±0.2 , 14.7±0.2, 15.8±0.2, 18.0±0.2, and 19.3±0.2°.
在优选的实施方式中,所述C晶型的X-射线粉末衍射图谱如图16所示。In a preferred embodiment, the X-ray powder diffraction pattern of the C crystal form is shown in FIG. 16 .
在优选的实施方式中,所述C晶型的X射线粉末衍射图谱中,特征峰的峰位置及强度如下表所示:In a preferred embodiment, in the X-ray powder diffraction pattern of the C crystal form, the peak positions and intensities of the characteristic peaks are shown in the following table:


在优选的实施方式中,所述C晶型的X-射线粉末衍射图谱采用Cu Kα射线衍射得到。In a preferred embodiment, the X-ray powder diffraction pattern of the C crystal form is obtained by Cu Kα ray diffraction.
在优选的实施方式中,所述C晶型在差示扫描量热分析中显示在熔融前(155℃)分解。In a preferred embodiment, said Form C shows decomposition prior to melting (155° C.) in differential scanning calorimetry.
在优选的实施方式中,所述C晶型的差示扫描量热曲线如图18所示。In a preferred embodiment, the differential scanning calorimetry curve of the C crystal form is shown in FIG. 18 .
在优选的实施方式中,所述C晶型的热失重分析(TG)图如图17所示。In a preferred embodiment, the thermogravimetric analysis (TG) diagram of the crystal form C is shown in FIG. 17 .
在优选的实施方式中,所述C晶型的红外光谱图如图20所示。In a preferred embodiment, the infrared spectrum of the crystal form C is shown in Figure 20.
在优选的实施方式中,所述C晶型的拉曼光谱图如图21所示。In a preferred embodiment, the Raman spectrum of the crystal form C is shown in FIG. 21 .
在优选的实施方式中,所述C晶型的偏光照片如图15所示。In a preferred embodiment, the polarized photo of the crystal form C is shown in FIG. 15 .
在优选的实施方式中,所述C晶型的吸湿性分析(DVS)图如图19所示。In a preferred embodiment, the hygroscopicity analysis (DVS) diagram of the C crystal form is shown in FIG. 19 .
在优选的实施方式中,所述C晶型在80%湿度条件下引湿性为0.7%。In a preferred embodiment, the hygroscopicity of the crystal form C is 0.7% under the condition of 80% humidity.
在优选的实施方式中,所述C晶型是柱状晶体。In a preferred embodiment, the crystal form C is a columnar crystal.
在第五方面,本发明提供所述的C晶型的制备方法,所述方法包括以下步骤:In a fifth aspect, the present invention provides a method for preparing the crystal form C, the method comprising the following steps:
1)将式I所示化合物的马来酸盐混悬于有机溶剂中,从而得到一混悬液;1) suspending the maleate salt of the compound shown in formula I in an organic solvent to obtain a suspension;
2)过滤步骤1)得到的混悬液,从而得到固体部分;2) filtering the suspension obtained in step 1) to obtain a solid portion;
3)收集并任选干燥得到的固体部分。3) Collect and optionally dry the resulting solid portion.
在优选的实施方式中,步骤1)在室温,例如25℃下进行。In a preferred embodiment, step 1) is performed at room temperature, such as 25°C.
在优选的实施方式中,步骤1)中,搅拌得到混悬液1-3天;更优选搅拌得到的混悬液3天。In a preferred embodiment, in step 1), the obtained suspension is stirred for 1-3 days; more preferably, the obtained suspension is stirred for 3 days.
在优选的实施方式中,式I所示化合物的马来酸盐与有机溶剂的质量体积比为40:1到5:1g/l;更优选15:1g/l。In a preferred embodiment, the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solvent is 40:1 to 5:1 g/l; more preferably 15:1 g/l.
在优选的实施方式中,所述有机溶剂是选自下组的一种或多种:酯类(包括但不限于:乙酸乙酯、异丙基乙酯)、C1-6醇(包括但不限于:甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇)、芳烃(包括但不限于:苯、甲苯)。In a preferred embodiment, the organic solvent is one or more selected from the group consisting of: esters (including but not limited to: ethyl acetate, isopropyl ethyl ester), C1-6 alcohols (including but not limited to Limited to: methanol, ethanol, propanol, isopropanol, butanol, isobutanol), aromatics (including but not limited to: benzene, toluene).
在具体的实施方式中,所述方法包括以下步骤:In a specific embodiment, the method includes the following steps:
1)在升高的温度下,将式I所示化合物的马来酸盐溶解于有机溶剂中,从而得到一热溶液;1) at elevated temperature, the maleate salt of the compound shown in formula I is dissolved in an organic solvent, thereby obtaining a hot solution;
2)将步骤1)所得的热溶液降温,从而析出固体;2) cooling the hot solution obtained in step 1) to precipitate solids;
3)收集并任选干燥步骤2)得到的固体。3) Collect and optionally dry the solid obtained in step 2).
在优选的实施方式中,所述有机溶剂是选自下组的一种或多种:C3-6酮(包括但不限于:丙酮、甲基乙基酮、甲基异丁基酮)。In a preferred embodiment, the organic solvent is one or more selected from the group consisting of C3-6 ketones (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone).
在具体的实施方式中,所述方法包括以下步骤:In a specific embodiment, the method includes the following steps:
1)将式I所示化合物的马来酸盐溶解于有机溶剂中,从而得到一溶液;1) dissolving the maleate of the compound shown in formula I in an organic solvent to obtain a solution;
2)将步骤1)所得的溶液挥发溶剂至干;2) volatilize the solvent from the solution obtained in step 1) to dryness;
3)收集并任选干燥步骤2)得到的固体。3) Collect and optionally dry the solid obtained in step 2).
在优选的实施方式中,步骤2)在25℃-50℃下进行。In a preferred embodiment, step 2) is carried out at 25°C-50°C.
在优选的实施方式中,式I所示化合物的马来酸盐与有机溶剂的质量体积比为20:1到5:1g/l;更优选10:1g/l。In a preferred embodiment, the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solvent is 20:1 to 5:1 g/l; more preferably 10:1 g/l.
在优选的实施方式中,所述有机溶剂是选自下组的一种或多种的混合溶剂,优选多种的混合溶剂:C1-6醇(包括但不限于:甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇)、C2-6腈(包括但不限于:乙腈)、C6-C8烷烃(包括但不限于己烷、庚烷)、C2-6醚(包括但不限于:甲醚、乙醚、甲基叔丁基醚)。In a preferred embodiment, the organic solvent is one or more mixed solvents selected from the following group, preferably a plurality of mixed solvents: C1-6 alcohol (including but not limited to: methanol, ethanol, propanol, Isopropanol, butanol, isobutanol), C2-6 nitriles (including but not limited to: acetonitrile), C6-C8 alkanes (including but not limited to hexane, heptane), C2-6 ethers (including but not limited to : methyl ether, diethyl ether, methyl tert-butyl ether).
在第六方面,本发明提供式I所示化合物的马来酸盐的无水A晶型,所述A晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:8.3±0.2、10.0±0.2、14.2±0.2和16.4±0.2°。In the sixth aspect, the present invention provides the anhydrous A crystal form of the maleate salt of the compound represented by formula I. The X-ray powder diffraction pattern of the A crystal form has characteristic peaks at the following 2θ angles: 8.3±0.2, 10.0 ±0.2, 14.2±0.2 and 16.4±0.2°.
在优选的实施方式中,所述A晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:8.3±0.2、10.0±0.2、12.0±0.2、13.6±0.2、14.2±0.2和16.4±0.2°。In a preferred embodiment, the X-ray powder diffraction pattern of the crystal form A has characteristic peaks at the following 2θ angles: 8.3±0.2, 10.0±0.2, 12.0±0.2, 13.6±0.2, 14.2±0.2 and 16.4±0.2 °.
在优选的实施方式中,所述A晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:8.3±0.2、10.0±0.2、12.0±0.2、13.6±0.2、14.2±0.2、16.4±0.2、16.7±0.2、18.3±0.2、20.1±0.2和26.0±0.2°。In a preferred embodiment, the X-ray powder diffraction pattern of the crystal form A has characteristic peaks at the following 2θ angles: 8.3±0.2, 10.0±0.2, 12.0±0.2, 13.6±0.2, 14.2±0.2, 16.4±0.2 , 16.7±0.2, 18.3±0.2, 20.1±0.2, and 26.0±0.2°.
在优选的实施方式中,所述A晶型的X-射线粉末衍射图谱如图2所示。In a preferred embodiment, the X-ray powder diffraction pattern of the crystal form A is shown in FIG. 2 .
在优选的实施方式中,所述A晶型的X射线粉末衍射图谱中,特征峰的峰位置及强度如下表所示:In a preferred embodiment, in the X-ray powder diffraction pattern of the A crystal form, the peak positions and intensities of the characteristic peaks are shown in the following table:


在优选的实施方式中,所述A晶型的X-射线粉末衍射图谱采用Au Kα射线衍射得到。In a preferred embodiment, the X-ray powder diffraction pattern of the A crystal form is obtained by Au Kα ray diffraction.
在优选的实施方式中,所述A晶型在差示扫描量热分析中显示在约188.2℃开始熔融。In a preferred embodiment, the crystalline Form A shows onset of melting at about 188.2°C in differential scanning calorimetry analysis.
在优选的实施方式中,所述A晶型在差示扫描量热分析中显示在190℃开始分解。In a preferred embodiment, said crystalline form A shows decomposition starting at 190° C. in differential scanning calorimetry analysis.
在优选的实施方式中,所述A晶型的差示扫描量热曲线如图4所示。In a preferred embodiment, the differential scanning calorimetry curve of the crystal form A is shown in FIG. 4 .
在优选的实施方式中,所述A晶型的热失重分析(TG)图如图3所示。In a preferred embodiment, the thermogravimetric analysis (TG) diagram of the crystal form A is shown in FIG. 3 .
在优选的实施方式中,所述A晶型的红外光谱图如图6所示。In a preferred embodiment, the infrared spectrum of the crystal form A is shown in FIG. 6 .
在优选的实施方式中,所述A晶型的拉曼光谱图如图7所示。In a preferred embodiment, the Raman spectrum of the crystal form A is shown in FIG. 7 .
在优选的实施方式中,所述A晶型的偏光照片如图1所示。In a preferred embodiment, the polarized photo of the crystal form A is shown in FIG. 1 .
在优选的实施方式中,所述A晶型的吸湿性分析(DVS)图如图5所示。In a preferred embodiment, the hygroscopicity analysis (DVS) diagram of the crystal form A is shown in FIG. 5 .
在优选的实施方式中,所述A晶型在80%湿度条件下引湿性为0.5%。In a preferred embodiment, the hygroscopicity of the crystal form A is 0.5% under the condition of 80% humidity.
在优选的实施方式中,所述A晶型是柱状晶体。In a preferred embodiment, the crystal form A is a columnar crystal.
在第七方面,本发明提供所述的A晶型的制备方法,所述方法包括以下步骤:In the seventh aspect, the present invention provides the preparation method of the A crystal form, the method comprising the following steps:
1)将式I所示化合物与马来酸进行成盐反应,其中有固体析出1) carry out salt-forming reaction with the compound shown in formula I and maleic acid, wherein solid is separated out
2)收集并任选干燥步骤1)中析出的固体。2) Collect and optionally dry the solid precipitated in step 1).
在具体的实施方式中,所述方法包括以下步骤:In a specific embodiment, the method includes the following steps:
1)将式I所示化合物的马来酸盐混悬于有机溶剂中,从而得到一混悬液;1) suspending the maleate salt of the compound shown in formula I in an organic solvent to obtain a suspension;
2)过滤步骤1)得到的混悬液,从而得到固体部分;2) filtering the suspension obtained in step 1) to obtain a solid portion;
3)收集并任选干燥得到的固体部分。3) Collect and optionally dry the resulting solid portion.
在优选的实施方式中,步骤1)将式I所示化合物的马来酸盐在25-50℃,优选50℃下 混悬于有机溶剂中,随后搅拌得到混悬液1-3天;更优选搅拌得到的混悬液3天。In a preferred embodiment, step 1) suspending the maleate salt of the compound represented by formula I in an organic solvent at 25-50°C, preferably 50°C, and then stirring to obtain a suspension for 1-3 days; more The resulting suspension is preferably stirred for 3 days.
在优选的实施方式中,式I所示化合物的马来酸盐与有机溶剂的质量体积比为40:1到5:1g/l;更优选15:1g/l。In a preferred embodiment, the mass volume ratio of the maleate salt of the compound represented by formula I to the organic solvent is 40:1 to 5:1 g/l; more preferably 15:1 g/l.
在优选的实施方式中,所述有机溶剂是选自下组的一种或多种:酯类(包括但不限于:乙酸乙酯、异丙基乙酯)。In a preferred embodiment, the organic solvent is one or more selected from the following group: esters (including but not limited to: ethyl acetate, isopropyl ethyl ester).
在具体的实施方式中,所述方法包括以下步骤:In a specific embodiment, the method includes the following steps:
1)在升高的温度下,将式I所示化合物的马来酸盐溶解于C3-6酮(包括但不限于:丙酮、甲基乙基酮、甲基异丁基酮)、甲基乙基酮或乙腈中,从而得到一热溶液;1) At elevated temperature, dissolve the maleate salt of the compound shown in formula I in C3-6 ketones (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone), methyl In ethyl ketone or acetonitrile, thereby obtaining a hot solution;
2)将步骤1)得到的热溶液降温,从而析出固体;2) cooling the hot solution obtained in step 1) to precipitate solids;
3)收集并任选干燥步骤2)得到的固体。3) Collect and optionally dry the solid obtained in step 2).
在优选的实施方式中,式I所示化合物的马来酸盐与C3-6酮(包括但不限于:丙酮、甲基乙基酮、甲基异丁基酮)、甲基乙基酮或乙腈的质量体积比为100:1到20:1g/l;更优选50:1g/l。In a preferred embodiment, the maleate of the compound shown in formula I is combined with C3-6 ketone (including but not limited to: acetone, methyl ethyl ketone, methyl isobutyl ketone), methyl ethyl ketone or The mass volume ratio of acetonitrile is 100:1 to 20:1 g/l; more preferably 50:1 g/l.
在第八方面,本发明提供一种药物组合物,所述药物组合物包含第一、二、四和六中任一方面所述的多晶型以及药学上可接受的载体或赋形剂。In the eighth aspect, the present invention provides a pharmaceutical composition comprising the polymorphic form of any one of the first, second, fourth and sixth aspects and a pharmaceutically acceptable carrier or excipient.
在第九方面,本发明提供第一、二、四和六中任一方面所述的多晶型在制备DPP4抑制剂中的用途。In the ninth aspect, the present invention provides the use of the polymorphic form described in any one of the first, second, fourth and sixth aspects in the preparation of a DPP4 inhibitor.
在优选的实施方式中,所述DPP4抑制剂是降血糖药物或糖尿病治疗药物。In a preferred embodiment, the DPP4 inhibitor is a hypoglycemic drug or a diabetes treatment drug.
在第十方面,本发明提供第一、二、四和六中任一方面所述的多晶型,用于制备DPP4抑制剂。In the tenth aspect, the present invention provides the polymorphic form of any one of the first, second, fourth and sixth aspects, which is used for preparing a DPP4 inhibitor.
在优选的实施方式中,所述DPP4抑制剂是降血糖药物或糖尿病治疗药物。In a preferred embodiment, the DPP4 inhibitor is a hypoglycemic drug or a diabetes treatment drug.
在第十一方面,本发明提供一种降血糖或治疗糖尿病的治疗方法,所述方法包括将治疗有效量的第一、二、四和六中任一方面所述的多晶型或第八方面所述的药物组合物给予有此需要的对象。In the eleventh aspect, the present invention provides a treatment method for lowering blood sugar or treating diabetes, the method comprising treating an effective amount of the polymorphic form described in any one of the first, second, fourth and sixth aspects or the eighth The pharmaceutical composition of the aspect is administered to a subject in need thereof.
在优选的实施方式中,所述对象是人。In a preferred embodiment, the subject is a human.
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。It should be understood that within the scope of the present invention, the above-mentioned technical features of the present invention and the technical features specifically described in the following (such as embodiments) can be combined with each other to form new or preferred technical solutions. Due to space limitations, we will not repeat them here.
图1显示了本发明的A晶型的偏光照片;Fig. 1 has shown the polarized photo of A crystal form of the present invention;
图2显示了本发明的A晶型的X-射线粉末衍射(XRPD)图;Figure 2 shows the X-ray powder diffraction (XRPD) pattern of the A crystal form of the present invention;
图3显示了本发明的A晶型的热失重分析(TG)图;Fig. 3 shows the thermogravimetric analysis (TG) figure of A crystal form of the present invention;
图4显示了本发明的A晶型的差示扫描量热分析(DSC)图;Fig. 4 has shown the differential scanning calorimetry (DSC) figure of A crystal form of the present invention;
图5显示了本发明的A晶型的吸湿性分析(DVS)图;Figure 5 shows the hygroscopicity analysis (DVS) figure of the A crystal form of the present invention;
图6显示了本发明的A晶型的红外光谱(IR)图;Figure 6 shows the infrared spectrum (IR) figure of the A crystal form of the present invention;
图7显示了本发明的A晶型的拉曼光谱(Raman)图;Figure 7 shows the Raman spectrum (Raman) figure of the A crystal form of the present invention;
图8显示了本发明的B晶型的偏光照片;Fig. 8 shows the polarized photo of B crystal form of the present invention;
图9显示了本发明的B晶型的X-射线粉末衍射(XRPD)图;Figure 9 shows an X-ray powder diffraction (XRPD) pattern of Form B of the present invention;
图10显示了本发明的B晶型的热失重分析(TG)图;Figure 10 shows the thermogravimetric analysis (TG) figure of B crystal form of the present invention;
图11显示了本发明的B晶型的差示扫描量热分析(DSC)图;Figure 11 shows the differential scanning calorimetry (DSC) diagram of the B crystal form of the present invention;
图12显示了本发明的B晶型的吸湿性分析(DVS)图;Figure 12 shows the hygroscopicity analysis (DVS) figure of B crystal form of the present invention;
图13显示了本发明的B晶型的红外光谱(IR)图;Figure 13 shows the infrared spectrum (IR) figure of B crystal form of the present invention;
图14显示了本发明的B晶型的拉曼光谱(Raman)图;Figure 14 shows the Raman spectrum (Raman) figure of B crystal form of the present invention;
图15显示了本发明的C晶型的偏光照片;Fig. 15 has shown the polarized photo of C crystal form of the present invention;
图16显示了本发明的C晶型的X-射线粉末衍射(XRPD)图;Figure 16 shows the X-ray powder diffraction (XRPD) pattern of the C crystal form of the present invention;
图17显示了本发明的C晶型的热失重分析(TG)图;Figure 17 shows the thermogravimetric analysis (TG) diagram of the C crystal form of the present invention;
图18显示了本发明的C晶型的差示扫描量热分析(DSC)图;Figure 18 shows the differential scanning calorimetry (DSC) diagram of the C crystal form of the present invention;
图19显示了本发明的C晶型的吸湿性分析(DVS)图;Figure 19 shows the hygroscopicity analysis (DVS) diagram of the C crystal form of the present invention;
图20显示了本发明的C晶型的红外光谱(IR)图;Figure 20 shows the infrared spectrum (IR) diagram of the C crystal form of the present invention;
图21显示了本发明的C晶型的拉曼光谱(Raman)图;Figure 21 shows the Raman spectrum (Raman) figure of C crystal form of the present invention;
图22显示了本发明的A晶型、B晶型和C晶型的X-射线粉末衍射(XRPD)叠加比较;Figure 22 shows the X-ray powder diffraction (XRPD) overlay comparison of the A crystal form, the B crystal form and the C crystal form of the present invention;
图23显示了本发明的A晶型、B晶型和C晶型的热失重分析(TG)图谱叠加比较;Figure 23 shows the thermogravimetric analysis (TG) spectrum overlay comparison of A crystal form, B crystal form and C crystal form of the present invention;
图24显示了本发明的A晶型、B晶型和C晶型的差示扫描量热分析(DSC)叠加比较;Figure 24 shows the differential scanning calorimetry (DSC) overlay comparison of Form A, Form B and Form C of the present invention;
图25显示了本发明的A晶型、B晶型和C晶型的引湿性分许(DVS)叠加比较;Figure 25 shows the overlay comparison of hygroscopicity analysis (DVS) of A crystal form, B crystal form and C crystal form of the present invention;
图26显示了本发明的A晶型、B晶型和C晶型的Ramam谱图叠加比较;Figure 26 shows the overlay comparison of the Ramam spectrum of the A crystal form, the B crystal form and the C crystal form of the present invention;
图27显示了本发明的A晶型、B晶型和C晶型的IR谱图叠加比较;和Figure 27 shows the overlay comparison of the IR spectra of Form A, Form B and Form C of the present invention; and
图28显示了B晶型的结构。Figure 28 shows the structure of Form B.
发明人经过广泛而深入的研究,针对式I所示化合物的马来酸盐的多晶型问题,采用不同的结晶条件和实验手段,系统地筛选了可能的晶型。最终发现式I所示化合物的马来酸盐存在3种不同的固体形态,分别为无水晶型Form A、一水合物Form B以及一水合物Form C。这些新晶型的结晶度较高、吸湿性较低以及稳定性优异,同时这些晶型的制备工艺简便,从而有利于药物的工艺处理和物化性能的改善,并提高成药性能。在此基础上完成了本发明。After extensive and in-depth research, the inventors have systematically screened possible crystal forms by using different crystallization conditions and experimental means in view of the polymorphic form of the maleate salt of the compound represented by formula I. Finally, it was found that the maleate salt of the compound represented by formula I exists in three different solid forms, which are anhydrous Form A, monohydrate Form B, and monohydrate Form C. These new crystal forms have high crystallinity, low hygroscopicity and excellent stability, and at the same time, the preparation process of these crystal forms is simple, which is beneficial to the process treatment of drugs and the improvement of physical and chemical properties, and improves the drug-making performance. The present invention has been accomplished on this basis.

多晶型Polymorph
晶型研究包括晶体发现和晶型优选的两个阶段,在晶体发现阶段,主要采用多种结晶手段,如熔融结晶,溶液挥发,快速冷却和混悬法的结晶方法,通过改变结晶条件,溶剂,温度,速度和混悬溶剂比例等影响药物结晶的外部因素。采用高通量样品制备平台,同时制备数百次结晶试验,运用微量样品制备技术和分析测试手段。制备和发现新的晶型。在晶型优选阶段,要对于新的晶型晶型工艺放大和制备条件摸索,采用多种固体表征手段,如x-射线衍射,固体核磁共振,拉曼光谱,红外光谱等手段晶型晶体表征,另外,要采用DSC、TGA、DVS、HPLC等对晶型进行物化性能研究,比较不同晶型的吸湿性、化学稳定、物理状态稳定性、可加工性等进行研究。最后选择最为优选的固体形态进行开发。Crystal form research includes two stages of crystal discovery and crystal form optimization. In the crystal discovery stage, various crystallization methods are mainly used, such as melt crystallization, solution volatilization, rapid cooling and suspension crystallization methods. By changing crystallization conditions, solvents , external factors such as temperature, speed and suspension solvent ratio affect drug crystallization. A high-throughput sample preparation platform is used to prepare hundreds of crystallization tests at the same time, using micro-sample preparation technology and analysis and testing methods. Preparation and discovery of new crystal forms. In the crystal form optimization stage, it is necessary to explore new crystal form process amplification and preparation conditions, and use various solid characterization methods, such as x-ray diffraction, solid state nuclear magnetic resonance, Raman spectroscopy, infrared spectroscopy and other means of crystal form crystal characterization In addition, DSC, TGA, DVS, HPLC, etc. should be used to study the physical and chemical properties of the crystal form, and to compare the hygroscopicity, chemical stability, physical state stability, and processability of different crystal forms. Finally, the most preferred solid form is selected for development.
针对式I所示化合物的马来酸盐,本发明提供了三种稳定的新晶型。For the maleate salt of the compound represented by formula I, the present invention provides three stable new crystal forms.
在具体的实施方式中,本发明提供式I所示化合物的马来酸盐的一水合物B晶型,所述B晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:8.5±0.2、10.0±0.2、10.3±0.2、13.8±0.2、14.5±0.2、17.2±0.2、17.7±0.2、18.3±0.2、22.2±0.2和25.9±0.2°。In a specific embodiment, the present invention provides the monohydrate B crystal form of the maleate salt of the compound shown in formula I, and the X-ray powder diffraction pattern of the B crystal form has a characteristic peak at the following 2θ angle: 8.5± 0.2, 10.0±0.2, 10.3±0.2, 13.8±0.2, 14.5±0.2, 17.2±0.2, 17.7±0.2, 18.3±0.2, 22.2±0.2, and 25.9±0.2°.
通过对该晶型进一步的表征,本发明人发现所述B晶型是一水合物;无色柱状晶体;空间群为P 212121;晶胞参数(a,b,c,α,β,γ)为

在具体的实施方式中,本发明提供式I所示化合物的马来酸盐的一水合物C晶型,所述C晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:4.7±0.2、6.7±0.2、9.5±0.2、13.0±0.2、13.6±0.2、14.3±0.2、14.7±0.2、15.8±0.2、18.0±0.2和19.3±0.2°。In a specific embodiment, the present invention provides the monohydrate C crystal form of the maleate salt of the compound shown in formula I, and the X-ray powder diffraction pattern of the C crystal form has a characteristic peak at the following 2θ angle: 4.7± 0.2, 6.7±0.2, 9.5±0.2, 13.0±0.2, 13.6±0.2, 14.3±0.2, 14.7±0.2, 15.8±0.2, 18.0±0.2, and 19.3±0.2°.
通过对该晶型进一步的表征,本发明人发现该晶型是柱状晶体,在差示扫描量热分析中显示在熔融前(155℃)分解;并且在80%湿度条件下引湿性为0.7%。Through further characterization of the crystal form, the inventors found that the crystal form is a columnar crystal, which is decomposed before melting (155° C.) in differential scanning calorimetry analysis; and has a hygroscopicity of 0.7% under 80% humidity conditions .
在具体的实施方式中,本发明提供式I所示化合物的马来酸盐的无水A晶型,所述A晶型的X-射线粉末衍射图谱在以下2θ角存在特征峰:8.3±0.2、10.0±0.2、12.0±0.2、13.6±0.2、14.2±0.2、16.4±0.2、16.7±0.2、18.3±0.2、20.1±0.2和26.0±0.2°。In a specific embodiment, the present invention provides the anhydrous A crystal form of the maleate salt of the compound represented by formula I, and the X-ray powder diffraction pattern of the A crystal form has a characteristic peak at the following 2θ angle: 8.3±0.2 , 10.0±0.2, 12.0±0.2, 13.6±0.2, 14.2±0.2, 16.4±0.2, 16.7±0.2, 18.3±0.2, 20.1±0.2, and 26.0±0.2°.
通过对该晶型进一步的表征,本发明人发现该晶型是柱状晶体,在差示扫描量热分析中显示在约188.2℃开始熔融,在190℃开始分解;并且在80%湿度条件下引湿性为0.5%。Through further characterization of the crystal form, the inventors found that the crystal form is a columnar crystal, which starts to melt at about 188.2°C and decomposes at 190°C in differential scanning calorimetry analysis; Moisture is 0.5%.
本发明的各种晶型的XRPD图谱的2θ角以及相对强度如下表所示:The 2θ angle and relative intensity of the XRPD collection of various crystal forms of the present invention are shown in the following table:


本发明的药物组合物以及施用方式The pharmaceutical composition and administration method of the present invention
在本发明的多晶型的基础上,本发明进一步提供了包含所述多晶型的药物组合物。由于本发明的多晶型具有结晶度高、吸湿性小,并形成规整的晶体型态等优点,本发明的药物组合物由此具备优异的成药性能。On the basis of the polymorphic form of the present invention, the present invention further provides a pharmaceutical composition comprising the polymorphic form. Since the polymorphic form of the present invention has the advantages of high crystallinity, low hygroscopicity, and regular crystal form, the pharmaceutical composition of the present invention has excellent drug-making properties.
本发明的药物组合物因为其中的式I所示化合物而具备抑制DPP4的能力,从而能够用作DPP4抑制剂。在具体的实施方式中,本发明的药物组合物可以用作降血糖药物或糖尿病治疗药物The pharmaceutical composition of the present invention can be used as a DPP4 inhibitor because the compound represented by formula I has the ability to inhibit DPP4. In a specific embodiment, the pharmaceutical composition of the present invention can be used as a hypoglycemic drug or a diabetes treatment drug
本发明的药物组合物还包含任选的药学上可接受的载体。在本文中,术语“组合物”旨在涵盖包含特定量的特定成分的产品,以及直接或间接地以特定量的特定成分的组合产生的任何产品;而药学上可接受的载体是指对有机体不引起明显的刺激性并且不干扰所给予化合物的生物活性和性质的载体、稀释剂或赋形剂;即,所述载体、稀释剂或赋形剂必须与制剂的 其它成分相容并且对其接受者无害。The pharmaceutical composition of the present invention also includes an optional pharmaceutically acceptable carrier. As used herein, the term "composition" is intended to cover a product comprising the specified ingredients in the specified amounts, and any product resulting directly or indirectly from the combination of the specified ingredients in the specified amounts; A carrier, diluent, or excipient that does not cause significant irritation and does not interfere with the biological activity and properties of the administered compound; that is, the carrier, diluent, or excipient must be compatible with the other ingredients of the formulation and The recipient is harmless.
可以采用本领域技术人员公知的方法制备本发明的药物组合物。例如,可以将本发明的化合物与药学上可接受的载体、稀释剂或赋形剂混合来制备相应的药物组合物。进一步地,本领域技术人员可以将本发明的化合物或药物组合物制成各种合适的剂型。根据所需的剂型,本领域技术人员也可以选择相应的药学上可接受的载体、稀释剂或赋形剂。The pharmaceutical composition of the present invention can be prepared by methods known to those skilled in the art. For example, the compound of the present invention can be mixed with a pharmaceutically acceptable carrier, diluent or excipient to prepare the corresponding pharmaceutical composition. Furthermore, those skilled in the art can prepare the compound or pharmaceutical composition of the present invention into various suitable dosage forms. According to the desired dosage form, those skilled in the art can also select corresponding pharmaceutically acceptable carriers, diluents or excipients.
本发明的药物组合物中可以包含安全有效量的所述多晶型。所述“安全有效量”指的是:化合物(或晶型)的量足以明显改善病情,而不至于产生严重的副作用。A safe and effective amount of the polymorphic form may be included in the pharmaceutical composition of the present invention. The "safe and effective amount" refers to: the amount of the compound (or crystal form) is sufficient to significantly improve the condition without causing serious side effects.
“药学上可以接受的载体”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。“相容性”在此指的是组合物中各组份能和本发明的活性成分以及它们之间相互掺和,而不明显降低活性成分的药效。药学上可以接受的载体部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂(如吐温
本发明的多晶型物或药物组合物的施用方式没有特别限制,代表性的施用方式包括(但并不限于):口服、直肠、肠胃外(静脉内、肌肉内或皮下)、和局部给药。The mode of administration of the polymorph or pharmaceutical composition of the present invention is not particularly limited, and representative modes of administration include (but are not limited to): oral, rectal, parenteral (intravenous, intramuscular or subcutaneous), and topical administration medicine.
用于口服给药的固体剂型包括胶囊剂、片剂、丸剂、散剂和颗粒剂。在这些固体剂型中,活性成分与至少一种常规惰性赋形剂(或载体)混合,如柠檬酸钠或磷酸二钙,或与下述成分混合:(a)填料或增容剂,例如,淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸;(b)粘合剂,例如,羟甲基纤维素、藻酸盐、明胶、聚乙烯基吡咯烷酮、蔗糖和阿拉伯胶;(c)保湿剂,例如,甘油;(d)崩解剂,例如,琼脂、碳酸钙、马铃薯淀粉或木薯淀粉、藻酸、某些复合硅酸盐、和碳酸钠;(e)缓溶剂,例如石蜡;(f)吸收加速剂,例如,季胺化合物;(g)润湿剂,例如鲸蜡醇和单硬脂酸甘油酯;(h)吸附剂,例如,高岭土;和(i)润滑剂,例如,滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠,或其混合物。胶囊剂、片剂和丸剂中,剂型也可包含缓冲剂。Solid dosage forms for oral administration include capsules, tablets, pills, powders and granules. In these solid dosage forms, the active ingredient is admixed with at least one conventional inert excipient (or carrier), such as sodium citrate or dicalcium phosphate, or with: (a) fillers or extenders, for example, Starch, lactose, sucrose, glucose, mannitol and silicic acid; (b) binders such as hydroxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose and acacia; (c) humectants, For example, glycerol; (d) disintegrants, such as agar, calcium carbonate, potato starch or tapioca starch, alginic acid, certain complex silicates, and sodium carbonate; (e) slow agents, such as paraffin; (f) Absorption accelerators such as quaternary ammonium compounds; (g) wetting agents such as cetyl alcohol and glyceryl monostearate; (h) adsorbents such as kaolin; and (i) lubricants such as talc, hard Calcium stearate, magnesium stearate, solid polyethylene glycol, sodium lauryl sulfate, or mixtures thereof. In capsules, tablets and pills, the dosage form may also contain buffering agents.
固体剂型如片剂、糖丸、胶囊剂、丸剂和颗粒剂可采用包衣和壳材制备,如肠衣和其它本领域公知的材料。它们可包含不透明剂,并且,这种组合物中活性成分的释放可以延迟的方式在消化道内的某一部分中释放。可采用的包埋组分的实例是聚合物质和蜡类物质。必要时,活性成分也可与上述赋形剂中的一种或多种形成微胶囊形式。Solid dosage forms such as tablets, dragees, capsules, pills, and granules can be prepared with coatings and shell materials, such as enteric coatings and others well known in the art. They may contain opacifying agents and the release of the active ingredient from such compositions may be in a certain part of the alimentary canal in a delayed manner. Examples of usable embedding components are polymeric substances and waxy substances. The active ingredient can also be in the form of microcapsules with one or more of the above-mentioned excipients, if necessary.
用于口服给药的液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆或酊剂。除了活性成分外,液体剂型可包含本领域中常规采用的惰性稀释剂,如水或其它溶剂,增溶剂和乳化剂,例知,乙醇、异丙醇、碳酸乙酯、乙酸乙酯、丙二醇、1,3-丁二醇、二甲基甲酰胺以及油,特别是棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油或这些物质的混合物等。Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups or tinctures. In addition to the active ingredient, liquid dosage forms may contain inert diluents conventionally used in the art, such as water or other solvents, solubilizers and emulsifiers, for example, ethanol, isopropanol, ethyl carbonate, ethyl acetate, propylene glycol, 1 , 3-butanediol, dimethylformamide and oils, especially cottonseed oil, peanut oil, corn germ oil, olive oil, castor oil and sesame oil or mixtures of these substances, etc.
除了这些惰性稀释剂外,组合物也可包含助剂,如润湿剂、乳化剂和悬浮剂、甜味剂、 矫味剂和香料。Besides such inert diluents, the compositions can also contain adjuvants, such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
除了活性成分外,悬浮液可包含悬浮剂,例如,乙氧基化异十八烷醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、甲醇铝和琼脂或这些物质的混合物等。Suspensions, in addition to the active ingredient, may contain suspending agents, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum methoxide and agar, mixtures of these substances, and the like.
用于肠胃外注射的组合物可包含生理上可接受的无菌含水或无水溶液、分散液、悬浮液或乳液,和用于重新溶解成无菌的可注射溶液或分散液的无菌粉末。适宜的含水和非水载体、稀释剂、溶剂或赋形剂包括水、乙醇、多元醇及其适宜的混合物。Compositions for parenteral injection may comprise physiologically acceptable sterile aqueous or anhydrous solutions, dispersions, suspensions or emulsions, and sterile powders for reconstitution into sterile injectable solutions or dispersions. Suitable aqueous and non-aqueous carriers, diluents, solvents or vehicles include water, ethanol, polyols, and suitable mixtures thereof.
用于局部给药的本发明的多晶型物的剂型包括软膏剂、散剂、贴剂、喷射剂和吸入剂。活性成分在无菌条件下与生理上可接受的载体及任何防腐剂、缓冲剂,或必要时可能需要的推进剂一起混合。Dosage forms of the polymorphs of the invention for topical administration include ointments, powders, patches, sprays and inhalants. The active ingredient is mixed under sterile conditions with a physiologically acceptable carrier and any preservatives, buffers, or propellants which may be required, if necessary.
疾病的预防和治疗方法Disease prevention and treatment methods
如上所述,鉴于本发明的多晶型能够作为DPP4抑制剂,本领域技术人员能够理解,本发明的多晶型或药物组合物可以用于降低对象的血糖或者治疗对象的糖尿病。As mentioned above, considering that the polymorphic form of the present invention can be used as a DPP4 inhibitor, those skilled in the art can understand that the polymorphic form or pharmaceutical composition of the present invention can be used to lower blood sugar or treat diabetes in a subject.
本发明的降低对象的血糖或者治疗对象的糖尿病的方法包括将治疗有效量的所述多晶型或物组合物给予有此需要的对象。所述对象包括但不限于人。The method of the present invention for lowering blood sugar or treating diabetes in a subject comprises administering to a subject in need thereof a therapeutically effective amount of said polymorphic form or composition of matter. Such subjects include, but are not limited to, humans.
本发明的优点Advantages of the invention
1.本发明的多晶型结晶度较高、吸湿性较低以及稳定性优异;1. The polymorphic form of the present invention has higher crystallinity, lower hygroscopicity and excellent stability;
2.本发明的多晶型的制备工艺简便,从而有利于药物的工艺处理和物化性能的改善;2. The preparation process of the polymorphic form of the present invention is simple and convenient, thereby being conducive to the improvement of the process treatment and physical and chemical properties of the medicine;
3.本发明的多晶型的成药性能提高。3. The druggability of the polymorphic form of the present invention is improved.
以下结合具体实施案例对本发明的技术方案进一步描述,但以下实施案例不构成对本发明的限制,所有依据本发明的原理和技术手段采用的各种施用方法,均属于本发明范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。The technical solutions of the present invention are further described below in conjunction with specific examples of implementation, but the following examples of implementation do not constitute limitations to the present invention, and all various application methods adopted according to the principles and technical means of the present invention belong to the scope of the present invention. For the experimental methods without specific conditions indicated in the following examples, the conventional conditions or the conditions suggested by the manufacturer are usually followed. Percentages and parts are by weight unless otherwise indicated.
实施例Example
材料与方法Materials and Methods
化合物(2S,3R)-2-氨基-9-甲氧基-3-(2,4,5-三氟苯基)-2,3-二氢-1H-苯并[f]色满-8-腈(HL012)参考CN 105566276A中所述合成。Compound (2S,3R)-2-amino-9-methoxy-3-(2,4,5-trifluorophenyl)-2,3-dihydro-1H-benzo[f]chroman-8 -Nitrile (HL012) refers to the synthesis described in CN 105566276A.
本发明涉及的仪器及所得晶型的检测方法如下所述:The detection method of instrument involved in the present invention and gained crystal form is as follows:
TGA方法TGA method
仪器型号:Netzsch TG 209F3,温度范围:30-400℃,扫描速率:10K/min,吹扫气:25mL/min,保护气:15mL/min;Instrument model: Netzsch TG 209F3, temperature range: 30-400°C, scan rate: 10K/min, purge gas: 25mL/min, protective gas: 15mL/min;
DSC方法DSC method
仪器号:Perkin Elmer DSC 1200,温度范围:-40-400℃,扫描速率:10℃/min,氮 气流速:50mL/min;Instrument number: Perkin Elmer DSC 1200, temperature range: -40-400°C, scan rate: 10°C/min, nitrogen flow rate: 50mL/min;
XRPD方法XRPD method
仪器型号:Bruker D8 advance,靶:Cu Kα(40kV,40mA),样品到检测器距离:30cm,扫描范围:3°-40°(2theta值),扫描步径:0.05sInstrument model: Bruker D8 advance, target: Cu Kα (40kV, 40mA), distance from sample to detector: 30cm, scanning range: 3°-40° (2theta value), scanning step: 0.05s
DVS方法DVS method
仪器型号:SMS DVS Intrinsic,0~95%RH,温度:25℃Instrument model: SMS DVS Intrinsic, 0~95%RH, temperature: 25℃
实施例1.化合物(2S,3R)-2-氨基-9-甲氧基-3-(2,4,5-三氟苯基)-2,3-二氢-1H-苯并[f]色满-8-腈的马来酸盐的制备Example 1. Compound (2S,3R)-2-amino-9-methoxy-3-(2,4,5-trifluorophenyl)-2,3-dihydro-1H-benzo[f] Preparation of the maleate salt of chroman-8-carbonitrile
称取100mg API于且茄型瓶中,加入定量的溶剂将API配置成0.01M的溶液,而后加入0.02M配置好的马来酸溶液。将反应相在70℃条件下混悬1小时后冷却至室温静置12小时。将析出的固体过滤,且获得的固体用少量乙腈洗涤3次。之后将获得的固体在是室温干燥即得标题化合物。Weigh 100mg of API into an eggplant-shaped bottle, add a certain amount of solvent to make the API a 0.01M solution, and then add a 0.02M maleic acid solution. The reaction phase was suspended at 70° C. for 1 hour, then cooled to room temperature and allowed to stand for 12 hours. The precipitated solid was filtered, and the obtained solid was washed 3 times with a small amount of acetonitrile. The obtained solid was then dried at room temperature to obtain the title compound.
实施例2.Example 2.
20mg HL012-MA置于锥形瓶中,加入乙酸乙酯溶剂1mL。加入磁力搅拌子在50度条件下搅拌,形成混悬液。转速为200rpm,保持在室温条件下搅拌3天。混悬液过滤,用少量乙酸乙酯溶剂洗涤后,于室温减压干燥。得到结晶性粉末(A型)16.6mg,产率为83%。20mg of HL012-MA was placed in a conical flask, and 1mL of ethyl acetate solvent was added. Add a magnetic stirring bar and stir at 50°C to form a suspension. The rotation speed was 200 rpm, and the stirring was kept at room temperature for 3 days. The suspension was filtered, washed with a small amount of ethyl acetate solvent, and dried under reduced pressure at room temperature. 16.6 mg of a crystalline powder (type A) was obtained with a yield of 83%.
实施例3.Example 3.
10mg HL012-MA溶解在乙腈/甲基异丁基酮(体积比1:1)混合溶剂1mL中。在50度条件下完全挥干。得到结晶性粉末(B型)9.5mg,产率为95%。10mg of HL012-MA was dissolved in 1mL of acetonitrile/methyl isobutyl ketone (volume ratio 1:1) mixed solvent. Evaporate completely at 50 degrees. 9.5 mg of crystalline powder (type B) was obtained, and the yield was 95%.
实施例4.Example 4.
50mg HL012-MA在50度条件下溶解在1mL的甲基乙基酮中,缓慢的冷却至室温,保持在室温条件下3天。过滤,所得的固体于室温减压干燥。得到结晶性粉末(A型)30mg,产率为60%。50mg of HL012-MA was dissolved in 1mL of methyl ethyl ketone at 50 degrees, slowly cooled to room temperature, and kept at room temperature for 3 days. After filtration, the resulting solid was dried under reduced pressure at room temperature. 30 mg of a crystalline powder (type A) was obtained, and the yield was 60%.
实施例5.Example 5.
20mg HL012-MA置于锥形瓶中,加入异丙醇溶剂1mL。加入磁力搅拌子搅拌,形成混悬液。转速为200rpm,保持在室温条件下搅拌3天。混悬液过滤,用少量异丙醇溶剂洗涤后,于室温减压干燥。得到结晶性粉末(B型)19mg,产率为95%。20mg of HL012-MA was placed in a conical flask, and 1mL of isopropanol solvent was added. Add a magnetic stirrer and stir to form a suspension. The rotation speed was 200 rpm, and the stirring was kept at room temperature for 3 days. The suspension was filtered, washed with a small amount of isopropanol solvent, and dried under reduced pressure at room temperature. 19 mg of crystalline powder (type B) was obtained, and the yield was 95%.
实施例6.Example 6.
20mg HL012-MA置于锥形瓶中,加入丙酮溶剂1mL。加入磁力搅拌子搅拌,形成混悬液。转速为200rpm,保持在室温条件下搅拌3天。混悬液过滤,用少量丙酮溶剂洗涤后, 于室温减压干燥。得到结晶性粉末(B型)18mg,产率为90%。20mg of HL012-MA was placed in a conical flask, and 1mL of acetone solvent was added. Add a magnetic stirrer and stir to form a suspension. The rotation speed was 200 rpm, and the stirring was kept at room temperature for 3 days. The suspension was filtered, washed with a small amount of acetone solvent, and dried under reduced pressure at room temperature. 18 mg of crystalline powder (type B) was obtained, and the yield was 90%.
实施例7.Example 7.
20mg HL012-MA置于锥形瓶中,加入正己烷溶剂1mL。加入磁力搅拌子搅拌,形成混悬液。转速为200rpm,保持在室温条件下搅拌3天。混悬液过滤,用少量正己烷溶剂洗涤后,于室温减压干燥。得到结晶性粉末(B型)18mg,产率为90%。20mg of HL012-MA was placed in a conical flask, and 1mL of n-hexane solvent was added. Add a magnetic stirrer and stir to form a suspension. The rotation speed was 200 rpm, and the stirring was kept at room temperature for 3 days. The suspension was filtered, washed with a small amount of n-hexane solvent, and dried under reduced pressure at room temperature. 18 mg of crystalline powder (type B) was obtained, and the yield was 90%.
实施例8.Example 8.
50mg HL012-MA在50度条件下溶解在1mL的甲醇中,缓慢的冷却至室温,保持在室温条件下3天。过滤,所得的固体于室温减压干燥。得到结晶性粉末(B型)26mg,产率为52%。50mg of HL012-MA was dissolved in 1mL of methanol at 50 degrees, slowly cooled to room temperature, and kept at room temperature for 3 days. After filtration, the resulting solid was dried under reduced pressure at room temperature. 26 mg of crystalline powder (type B) was obtained, and the yield was 52%.
实施例9.Example 9.
将2g HL012-MA加入丙酮/水(V/V=3:1),升温至回流(55±5℃),保温回流搅拌2h,缓慢降温至0-5度,过滤,滤饼用预先冷至5±5℃的丙酮淋洗。收集滤饼,60±5℃烘料,得到产品HL012MA的晶体,所得晶型为B。收率:92%。Add 2g of HL012-MA into acetone/water (V/V=3:1), heat up to reflux (55±5°C), keep warm and reflux and stir for 2 hours, slowly cool down to 0-5°C, filter, and filter the cake with pre-cooled Rinse with acetone at 5±5°C. Collect the filter cake, bake the material at 60±5°C, and obtain the crystal of the product HL012MA, and the obtained crystal form is B. Yield: 92%.
实施例10.Example 10.
10mg HL012-MA溶解在乙腈溶剂1mL中。在25度条件下完全挥干,得到结晶性粉末(B型)9mg,产率为90%。10mg of HL012-MA was dissolved in 1mL of acetonitrile solvent. Completely evaporated to dryness at 25°C to obtain 9 mg of crystalline powder (Type B) with a yield of 90%.
实施例11.Example 11.
10mg HL012-MA溶解在四氢呋喃溶剂1mL中。在25度条件下完全挥干,得到结晶性粉末(B型)9mg,产率为90%。10mg of HL012-MA was dissolved in 1mL of tetrahydrofuran solvent. Completely evaporated to dryness at 25°C to obtain 9 mg of crystalline powder (Type B) with a yield of 90%.
实施例12.Example 12.
20mg HL012-MA置于锥形瓶中,加入乙酸乙酯剂1mL。加入磁力搅拌子搅拌,形成混悬液。转速为200rpm,保持在室温条件下搅拌3天。混悬液过滤,用少量乙酸乙酯溶剂洗涤后,于室温减压干燥。得到结晶性粉末(C型)18mg,产率为90%。20mg of HL012-MA was placed in a conical flask, and 1mL of ethyl acetate was added. Add a magnetic stirrer and stir to form a suspension. The rotation speed was 200 rpm, and the stirring was kept at room temperature for 3 days. The suspension was filtered, washed with a small amount of ethyl acetate solvent, and dried under reduced pressure at room temperature. 18 mg of crystalline powder (Type C) was obtained, and the yield was 90%.
实施例13.Example 13.
20mg HL012-MA置于锥形瓶中,加入异丙醇/甲苯(体积比1:1)剂1mL。加入磁力搅拌子搅拌,形成混悬液。转速为200rpm,保持在室温条件下搅拌3天。混悬液过滤,用少量异乙酸乙酯溶剂洗涤后,于室温减压干燥。得到结晶性粉末(C型)16mg,产率为80%。20mg of HL012-MA was placed in a conical flask, and 1mL of isopropanol/toluene (volume ratio 1:1) was added. Add a magnetic stirrer and stir to form a suspension. The rotation speed was 200 rpm, and the stirring was kept at room temperature for 3 days. The suspension was filtered, washed with a small amount of ethyl isoacetate solvent, and dried under reduced pressure at room temperature. 16 mg of crystalline powder (Type C) was obtained, and the yield was 80%.
实施例14.Example 14.
50mg HL012-MA在50度条件下溶解在1mL的丙酮中,缓慢的冷却至室温,保持在室温条件下3天。过滤,所得的固体于室温减压干燥。得到结晶性粉末(C型)20mg,产率为40%。50mg of HL012-MA was dissolved in 1mL of acetone at 50°C, cooled slowly to room temperature, and kept at room temperature for 3 days. After filtration, the resulting solid was dried under reduced pressure at room temperature. 20 mg of crystalline powder (Type C) was obtained, and the yield was 40%.
实施例15.Example 15.
10mg HL012-MA溶解在甲醇/甲基叔丁基醚(体积比1:1)混合溶剂1mL中。在50度条件下完全挥干。得到结晶性粉末(C型)10mg,产率为100%。10mg of HL012-MA was dissolved in 1mL of methanol/methyl tert-butyl ether (volume ratio 1:1) mixed solvent. Evaporate completely at 50 degrees. 10 mg of crystalline powder (Type C) was obtained, and the yield was 100%.
实施例16.Example 16.
10mg HL012-MA溶解在乙腈/正己烷(体积比1:1)混合溶剂1mL中。在50度条件下完全挥干。得到结晶性粉末(C型)10mg,产率为100%。10mg of HL012-MA was dissolved in 1mL of acetonitrile/n-hexane (volume ratio 1:1) mixed solvent. Evaporate completely at 50 degrees. 10 mg of crystalline powder (Type C) was obtained, and the yield was 100%.
实施例17.晶型B的单晶解析Example 17. Single Crystal Analysis of Form B
单晶解析Single crystal analysis
制备方法:于室温,在乙腈溶液中缓慢挥发可得到晶型B单晶。晶型B的单晶数据信息如下表所:Preparation method: Slowly volatilize in acetonitrile solution at room temperature to obtain Form B single crystal. The single crystal data information of Form B is shown in the following table:
晶体基本信息-单晶数据Crystal Basic Information - Single Crystal Data

晶体基本信息-测定数据Crystal Basic Information - Determination Data
晶体结构如图28所示。The crystal structure is shown in Figure 28.
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。All documents mentioned in this application are incorporated by reference in this application as if each were individually incorporated by reference. In addition, it should be understood that after reading the above teaching content of the present invention, those skilled in the art can make various changes or modifications to the present invention, and these equivalent forms also fall within the scope defined by the appended claims of the present application.
Claims (15)
Translated fromChinese
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110803022.1ACN115611843A (en) | 2021-07-15 | 2021-07-15 | Polymorphism of DPP4 inhibitory active compound and preparation method thereof |
| CN202110803022.1 | 2021-07-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023284872A1true WO2023284872A1 (en) | 2023-01-19 |
Family
ID=84856189
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2022/106082CeasedWO2023284872A1 (en) | 2021-07-15 | 2022-07-15 | Polymorph of compound having dpp4-inhibitory activity and preparation method therefor |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN115611843A (en) |
| WO (1) | WO2023284872A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105566276A (en)* | 2014-10-31 | 2016-05-11 | 华东理工大学 | Benzo-hexatomic ring derivative used as DPP-4 inhibitor and application of benzo-hexatomic ring derivative |
| CN110240586A (en)* | 2018-03-09 | 2019-09-17 | 山东百极地长制药有限公司 | Preparation method of 2,3-dihydro-1H-benzo[f]chroman-2-amine derivative |
| CN110237068A (en)* | 2018-03-09 | 2019-09-17 | 山东百极地长制药有限公司 | Application of benzo six-membered ring derivatives as long-acting DPP-4 inhibitors |
- 2021
- 2021-07-15CNCN202110803022.1Apatent/CN115611843A/enactivePending
- 2022
- 2022-07-15WOPCT/CN2022/106082patent/WO2023284872A1/ennot_activeCeased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105566276A (en)* | 2014-10-31 | 2016-05-11 | 华东理工大学 | Benzo-hexatomic ring derivative used as DPP-4 inhibitor and application of benzo-hexatomic ring derivative |
| CN110240586A (en)* | 2018-03-09 | 2019-09-17 | 山东百极地长制药有限公司 | Preparation method of 2,3-dihydro-1H-benzo[f]chroman-2-amine derivative |
| CN110237068A (en)* | 2018-03-09 | 2019-09-17 | 山东百极地长制药有限公司 | Application of benzo six-membered ring derivatives as long-acting DPP-4 inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| LI SHILIANG, QIN CHUN, CUI SHICHAO, XU HONGLING, WU FANGSHU, WANG JIAWEI, SU MINGBO, FANG XIAOYU, LI DAN, JIAO QIAN, ZHANG MING, X: "Discovery of a Natural-Product-Derived Preclinical Candidate for Once-Weekly Treatment of Type 2 Diabetes", JOURNAL OF MEDICINAL CHEMISTRY, vol. 62, no. 5, 14 March 2019 (2019-03-14), US , pages 2348 - 2361, XP055878577, ISSN: 0022-2623, DOI: 10.1021/acs.jmedchem.8b01491* |
Also Published As
| Publication number | Publication date |
|---|---|
| CN115611843A (en) | 2023-01-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN109311832B (en) | Pamoic acid salt of vortioxetine and crystal forms thereof | |
| CN103930419A (en) | Crystal form of azilsartan and preparation method thereof | |
| US7759481B2 (en) | Solid state forms of 5-azacytidine and processes for preparation thereof | |
| EP2646457B1 (en) | Optimized synthesis of pure, non-polymorphic, crystalline bile acids with defined particle size | |
| JP2015508090A (en) | Solid form dabigatran etexilate mesylate and process for its preparation | |
| JP7682923B2 (en) | Crystalline forms of gepotidacin | |
| WO2018000250A1 (en) | New ibrutinib crystal form and preparation method therefor | |
| JP2018529769A (en) | Crystal form A of inositol nicotinate and process for producing the same | |
| WO2022052925A1 (en) | Repotrectinib crystal form and preparation method therefor | |
| CN104039779B (en) | Crystal form of azilsartan medoxomil potassium and its preparation method and use | |
| WO2023284872A1 (en) | Polymorph of compound having dpp4-inhibitory activity and preparation method therefor | |
| CN102952138B (en) | The salt of a kind of pyrazolopyrimidinone compound, polymorph and pharmaceutical composition, preparation method and application | |
| JP2018199718A (en) | Pharmaceutical formulations containing 3-(4-cinnamyl-1-piperazinyl) amino derivatives of 3-formylrifamycin sv and 3-formylrifamycin s and process for their preparation | |
| WO2022166774A1 (en) | Crystal form of 3-hydroxy-5-pregnane-20-one derivative, and preparation method therefor and use thereof | |
| WO2023284871A1 (en) | Polymorphic form of compound having dpp4 inhibitory activity and preparation method therefor | |
| CN115073518A (en) | Diedaravone phosphate compound, its preparation method and use | |
| WO2011163469A1 (en) | Hydrated form of anti-inflammatory roflumilast-n-oxide | |
| CN114349809A (en) | Beta-nicotinamide mononucleotide crystal form A and preparation method thereof | |
| WO2016197980A1 (en) | Baicalin crystal form a, and preparation method and application thereof | |
| WO2021104257A1 (en) | Crystalline form of acetylcholinesterase inhibitor and preparation method therefor and application thereof | |
| CN106478616B (en) | Crystalline form of GPR40 agonist and preparation method thereof | |
| CN114391016B (en) | Salt of nucleoside analogue, crystal form, pharmaceutical composition and application thereof | |
| CN105308043B (en) | Crystal formation of hepatitis C medicine and preparation method thereof, its pharmaceutical composition and purposes | |
| EP1674468A1 (en) | Polymorphs of clopidogrel hydrobromide | |
| US6596774B1 (en) | Synthesis, lipid peroxidation and cytotoxic evaluation of 10-substituted 1,5-dichloro-9(10H)-anthracenone derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:22841505 Country of ref document:EP Kind code of ref document:A1 | |
| NENP | Non-entry into the national phase | Ref country code:DE | |
| 122 | Ep: pct application non-entry in european phase | Ref document number:22841505 Country of ref document:EP Kind code of ref document:A1 |