WO2020177012A1 - Nucleic acid sequence for direct rna library construction, method for directly constructing sequencing library based on rna samples, and use thereof - Google Patents
Nucleic acid sequence for direct rna library construction, method for directly constructing sequencing library based on rna samples, and use thereofDownload PDFInfo
- Publication number
- WO2020177012A1 WO2020177012A1PCT/CN2019/076697CN2019076697WWO2020177012A1WO 2020177012 A1WO2020177012 A1WO 2020177012A1CN 2019076697 WCN2019076697 WCN 2019076697WWO 2020177012 A1WO2020177012 A1WO 2020177012A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nucleic acid
- acid sequence
- rna
- sequencing
- sequence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images

Classifications
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
Definitions
- the present inventionrelates to the field of gene sequencing, in particular to a nucleic acid sequence used for direct RNA library construction, a method and application for directly constructing a sequencing library based on RNA samples.
- Pacbio's third-generation sequencingis based on the principle of sequencing while synthesizing. It uses SMRT (Single Molecule Real-Time Fluorescence Sequencing Technology) chips as a carrier to perform sequencing reactions. During sequencing, genomic DNA is broken into many small fragments, which are then made into droplets. Disperse into different ZMW nanopores. When the polymerization reaction occurs at the bottom of the ZMW well, the nucleotides labeled with different fluorescence will be retained by the polymerase in the fluorescence detection area of the small hole. According to the type of fluorescence and the duration of fluorescence, the type of template DNA base composition can be determined.
- SMRTSingle Molecule Real-Time Fluorescence Sequencing Technology
- the transcriptome of a cellcontains a wealth of information, including gene structure, transcript expression level and antisense transcription. The best way to capture this information is to accurately and quantitatively reveal the presence and identity of bases without knowing the sequence in advance. Of course, this method is best to generate a continuous sequence across the splice junction.
- the most commonly used RNA-seq strategyis to use poly(dT) primers or random hexamer primers for cDNA synthesis. Then PCR is used to amplify these cDNA strands, but this may reduce the complexity of the cDNA library, affect the relative abundance of cDNA, and cause certain types of RNA to be lost. In addition, during the PCR amplification process, all modifications of RNA are lost.
- the present inventionaims to solve one of the technical problems in related technologies at least to a certain extent, and provides a method and kit for directly performing library building and sequencing on RNA.
- the method for directly constructing a library and sequencing RNA provided by the present inventiondoes not require PCR amplification during the sequencing process of the library, and there is no bias of PCR; in the whole process, no interruption processing is performed, and the original RNA can be maintained
- the lengthis particularly useful for the study of splice variants, and the data obtained by sequencing is chain-specific.
- RNA sequencing RNAfirst add Illumina adapters to the obtained mRNA with a poly(A) structure, and after the library is quantified, a bridge PCR amplification process is performed to amplify the signal.
- Illumina methodis a PCR-FREE process in the process of library construction, Illumina is based on the principle of SBS sequencing, and the signal is still amplified by bridge PCR amplification during the sequencing process. Therefore, this direct RNA sequencing method based on the Illumina platform is not a complete PCR-FREE, nor can it directly read the methylation and other modification information on the RNA.
- the Pacbio sequencing platformuses the principle of full-length transcriptome sequencing for RNA sequencing.
- the starting sampleis total RNA extracted from tissue samples, and cDNA synthesized by reverse transcription and PCR amplification is repaired by damage to the end. Repair, connect with known adapters, digest by enzyme reaction, and finally obtain a dumbbell-shaped library, which is tested by Agilent 2100 and Qubit HS and then sequenced on Pacbio.
- RNAcan be reverse-transcribed into an RNA-DNA hybrid strand, and then library construction and sequencing can be performed, thereby overcoming the need for reverse transcription into cDNA for conventional RNA sequencing. Double-stranded, and then PCR amplification method to build a library to carry out the process of sequencing. Taking mRNA as an example, this method is especially suitable for sequencing mRNAs with different splicing bodies.
- the present inventionprovides the following technical solutions:
- the present inventionprovides a nucleic acid sequence for direct RNA library construction, comprising: a first nucleic acid sequence and a second nucleic acid sequence, the first nucleic acid sequence and the second nucleic acid sequence are connected
- the first nucleic acid sequenceis close to the 5'end of the nucleic acid sequence
- the second nucleic acid sequenceis close to the 3'end of the nucleic acid sequence
- the first nucleic acid sequenceincludes 1-40 bases
- the dinucleotide sequenceincludes a polyT sequence composed of at least 20 bases.
- the second nucleic acid sequencecontains a polyT sequence, and the polyT sequence can bind to polyA on the RNA.
- the nucleic acid sequencealso contains a first nucleic acid sequence composed of any A, T, C, and G.
- the first nucleic acid sequence and the second nucleic acid sequenceare connected, which can reduce the difficulty of separately synthesizing the second nucleic acid sequence and also reduce the nucleic acid sequence Difficulty of purification.
- the first nucleic acid sequencecan be composed of four bases: A, T, C, and G, and can be 1-40 bases in length, for example, 5-40 bases, 10-40 bases, 20-40 bases. Bases, 15-35 bases, 20-35 bases, etc., the number corresponding to the four bases A, T, C, and G is not particularly limited.
- the mRNA informationcan be effectively reverse-transcribed, so as to realize the precise reverse transcription of the synthetic DNA strand using RNA as a template.
- the aforementioned nucleic acid sequencemay further include the following technical features:
- the first nucleic acid sequenceincludes 25 bases.
- the polyT sequenceconsists of 30 bases.
- Most of the RNA samplesare mRNA. Take mRNA as an example. Usually most of the mRNA has 20-30 consecutive A bases at the 3'end.
- the polyT sequence in the nucleic acid sequenceis limited to 30 bases, which can satisfy the absolute requirement. Reverse transcription of most mRNA; at the same time, the fixed 30 base setting can be used to find the transcription start and stop sites during subsequent data analysis. For example, a sequence of 30 consecutive A bases obtained by sequencing is the termination site. If it is significantly less than 30 consecutive A bases, it may be the characteristics of the insert itself.
- the second nucleic acid sequencefurther includes:
- Degenerate base Vsaid degenerate base V is connected to said polyT sequence, said degenerate base V is located at the 3'end of said polyT sequence, and said degenerate base V is base A, G Or C.
- the second nucleic acid sequencefurther includes a degenerate base N, the degenerate base N is connected to the degenerate base V, and the degenerate base N is located in the The 3'end of the degenerate base V, the degenerate base N is the base A, T, G or C.
- RNA samplesmay have poly(A) sequences of different lengths at the 3'end. Taking mRNA as an example, the length of the 3'end poly(A) varies greatly among different species, even in different parts of the same species. The difference is also large.
- one or two degenerate basescan be added to the end of the poly(T) sequence of the nucleic acid sequence.
- any three consecutive base sequences on the first nucleic acid sequenceare different.
- the number corresponding to the four bases of A, T, C, and G in the first nucleic acid sequenceis not particularly limited, as long as there are no three consecutive identical bases. For example, if there are 3 consecutive A bases, it may cause abnormal base recognition during sequencing.
- nucleic acid sequenceis shown in SEQ ID NO:1.
- the present inventionprovides a partially double-stranded RNA-DNA hybrid strand, comprising: a first strand and a second strand, the first strand is an RNA strand, the The 3'end of the first strand is a polyA sequence; the second strand is the nucleic acid sequence according to the first aspect of the present invention; at least part of the polyA sequence at the 3'end of the first strand and the second strand At least part of the polyT sequence of the strand is complementary to form a double strand.
- the present inventionprovides a method for constructing a sequencing library based on an RNA sample, which includes: combining a nucleic acid sequence containing a polyT sequence with a polyA sequence on the RNA sample to obtain a partial double-strand
- the RNA-DNA hybrid strand, the partial double strandis formed by complementary at least part of the polyA sequence and at least part of the polyT sequence, and the nucleic acid sequence is the nucleic acid sequence according to any one of the embodiments of the first aspect of the present invention; based on the The RNA-DNA hybrid strands containing partial double-strands are synthesized by reverse transcription to synthesize the DNA strands complementary to the RNA sample to obtain products containing the RNA-DNA hybrid double strands for damage repair and end repair, and the sequencing adapter is connected to construct Obtain sequencing library.
- the nucleic acid sequence containing the polyT sequenceis combined with the polyA sequence on the RNA sample to obtain an RNA-DNA hybrid strand containing at least a part of the polyA sequence and the part of the double strand formed by the complementary polyT sequence. Then use the RNA sample as a template, reverse transcription to obtain a DNA strand complementary to the RNA sample, thereby obtaining a product containing an RNA-DNA hybrid double-strand; performing damage repair on the obtained product containing an RNA-DNA hybrid double-strand And end repair, connect the sequencing adapter to construct a sequencing library.
- RNA-DNA hybrid strand structurethe RNA can be calibrated with the sequence of cDNA complementary to the RNA, thereby improving data quality.
- the obtained RNA-DNA secondary structureis stable, and the library is not easily degraded during storage and transportation, thereby improving sequencing quality and yield.
- the above method for constructing a sequencing library based on RNA samplesmay further include the following technical features:
- the RNA sampleis an mRNA sample.
- the 3'end of the mRNAusually contains a polyA tail, which can be combined with a nucleic acid sequence containing a polyT sequence by the above-mentioned method to obtain an RNA-DNA hybrid duplex.
- the mRNA samplecan be sequenced.
- RNA-DNA hybrid double-strandsinto blunt ends by exonuclease digestion based on the product containing the RNA-DNA hybrid duplex, and then Then proceed to the damage repair and end repair.
- the obtained RNA-DNA hybrid double-stranded productmay contain some unreacted residual single-stranded RNA and exposed single-stranded nucleic acid sequences, which can be digested and removed by exonuclease, and then damage repair and end repair are performed. Thereby reducing the influence of single-stranded RNA and single-stranded nucleic acid sequences on the sequencing results.
- the exonucleaseis ExoVII.
- the exonucleasecan effectively digest the single-stranded RNA or single-stranded DNA fragments exposed by the RNA-DNA hybrid double-stranded ends.
- the exonucleaseis used to react for 15-20 minutes at 37 degrees Celsius to perform the digestion treatment. Controlling the reaction time to about 15-20 minutes can make the digestion complete without affecting the quality of sequencing. When the digestion time is too short, the digestion will be incomplete and the adapters cannot be connected; while the time is too long to cause damage to the DNA-RNA and form gaps, resulting in poor sequencing quality.
- the methodfurther includes: performing damage repair and end repair based on the product containing the RNA-DNA hybrid duplex, after connecting the sequencing adapter, using exonuclease ExoIII and exonuclease ExoVII is digested and then constructed to obtain the sequencing library.
- exonuclease ExoVIIis to further digest the fragments at the ends of the RNA-DNA hybrid double-stranded strands
- exonuclease ExoIIIis to digest the gapped RNA-DNA hybrid double-strands.
- the exonuclease ExoIII and ExoVIIare used for digestion.
- the main functionis to digest the invalid library and improve the efficiency of sequencing.
- the exonuclease ExoIII and ExoVIIare used to react at 37 degrees Celsius for 50 to 70 minutes. Under these conditions, the digestion can be completed without affecting the quality of sequencing.
- the present inventionprovides a sequencing library obtained by the method described in the third aspect of the present invention.
- the present inventionprovides a method for sequencing an RNA sample, including: constructing a sequencing library based on the RNA sample according to any one of the embodiments of the second aspect of the present invention;
- the sequencing libraryuses the Pacbio sequencing platform to sequence the RNA samples.
- the Pacbio sequencing platform mentioned hererefers to all sequencing platforms launched by Pacbio, such as the RSII and Sequel platforms, and the Sequel II platform that will be launched in the future.
- the present inventionprovides a kit including the nucleic acid sequence according to any one of the embodiments of the first aspect of the present invention.
- the beneficial effect achieved by the present inventionis at least manifested as: using the nucleic acid sequence provided by the present invention as primers can realize the direct library building and sequencing of RNA samples, so that the original length of RNA can be maintained without the need for conventional second-generation sequencing. RNA is broken into small fragments and then sequenced, making it suitable for the study of mRNA variable shearing. Moreover, because the sequencing data is chain-specific, the directionality of the sequencing data can be distinguished, and it can be identified whether the sequencing data is the sense strand mRNA or the complementary strand of the mRNA, thereby realizing high-precision analysis of the sequencing data.
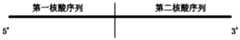
- Fig. 1is a schematic structural diagram of a nucleic acid sequence for RNA library construction according to an embodiment of the present invention.
- Fig. 2is a schematic diagram of constructing an RNA-DNA hybrid duplex based on mRNA according to an embodiment of the present invention, and performing restriction digestion.
- first and secondare only used for descriptive purposes, and cannot be understood as indicating or implying relative importance or implicitly indicating the number of indicated technical features. Therefore, the features defined with “first” and “second” may explicitly or implicitly include at least one of the features. In the description of the present invention, "a plurality of” means at least two, such as two, three, etc., unless otherwise specifically defined.
- RNA-DNA hybrid strands containing partial double-strandsrefers to: formed by RNA strands and DNA strands, and the formed RNA-DNA hybrid strands have only part of the double strands, and the rest are Single-stranded RNA and single-stranded DNA.
- RNA-DNA hybrid double strandrefers to the RNA-DNA double strand formed by the RNA strand and the DNA strand. Compared with the aforementioned RNA-DNA hybrid strands containing partial double strands, the RNA-DNA hybrid double strands contain no or almost no single-stranded fragments. Of course, even if it contains a few single-stranded fragments, these single-stranded fragments are not part of the sequencing data or part of the sequencing library, and can be digested and removed by exonuclease.
- reverse transcription and reverse transcriptionhave the same meaning, and both refer to the process of synthesizing DNA using RNA as a template.
- the present inventionprovides a nucleic acid sequence that can be used for RNA library construction, as shown in FIG. 1, comprising: a first nucleic acid sequence and a second nucleic acid sequence, the first nucleic acid sequence and the second nucleic acid sequence are connected, so The first nucleic acid sequence is close to the 5'end of the nucleic acid sequence, and the second nucleic acid sequence is close to the 3'end of the nucleic acid sequence; the first nucleic acid sequence includes 1-40 bases; the second nucleic acid sequence The sequence includes a polyT sequence consisting of at least 20 bases.
- the nucleic acid sequencecan be used as a fusion primer for the reverse transcription reaction of RNA.
- the present inventionalso provides a method for constructing a sequencing library based on an RNA sample, which includes: combining a nucleic acid sequence containing a polyT sequence with a polyA sequence on the RNA sample to obtain an RNA-DNA hybrid strand containing a partial double strand
- the partially double-strandedis formed by complementation of at least part of the polyA sequence and at least part of the polyT sequence, and the nucleic acid sequence is the nucleic acid sequence according to any one of the embodiments of the first aspect of the present invention; based on the RNA-containing partially double-stranded RNA- DNA hybrid strands, reverse transcription to synthesize a DNA strand complementary to the RNA sample to obtain products containing RNA-DNA hybrid double strands, perform damage repair and end repair, connect sequencing adapters, and construct a sequencing library.
- the RNA sampleis mixed with the polyT-containing nucleic acid sequence, reacted at 72 degrees Celsius for 3 to 5 minutes, and then reduced to 42 degrees Celsius at a rate of 0.1 to 0.5 degrees Celsius/sec, and React for 2 to 5 minutes at 42 degrees Celsius to obtain a partially double-stranded RNA-DNA hybrid strand.
- the temperatureis set the temperature to 72 degrees Celsius or lower than 72 degrees Celsius to prevent RNA from being degraded due to excessive temperature; at the same time, consider the simplicity of the 3'end of nucleic acid sequences containing polyT.
- Combining basesusing gradually reduced temperatures, can effectively increase the binding efficiency of polyT-containing nucleic acid sequences and RNA samples, for example, reduce to 42 degrees Celsius at a rate of 0.1 to 0.5 degrees Celsius/sec; and react at 42 degrees Celsius for 2-5 minutes to promote the reaction full.
- DTT and dNTPare added and mixed, reacted at 42 degrees Celsius for 80-100 minutes, and then reacted at 70-75 degrees Celsius. ⁇ 15 minutes to synthesize a DNA strand complementary to the RNA sample by reverse transcription.
- DNA and dNTPare added and reacted at 37 degrees Celsius for 50-70 minutes to repair the damage. In some embodiments of the present invention, DNA is added and reacted at 25-30 degrees Celsius for 10-20 minutes to perform the end repair.
- a ligase and a linkerare added, and the reaction is carried out at 25-30 degrees Celsius for 12-16 hours to connect the sequencing linker.
- the experimental group sequenced mRNAaccording to the following method.
- the starting sample usedis the total RNA extracted from tissue samples, using oligo d(T) magnetic beads to extract mRNA, using specific primers, reverse transcription to transform the mRNA into an RNA-DNA hybrid strand, and then pass the enzyme VII digestion, damage repair, end repair, ligation with known adapters, enzyme reaction digestion, BluePippin sorting, and finally a dumbbell-shaped library, which is detected and synthesized by Agilent2100 and Qubit HS, and then sequenced on the Pacbio computer. After the data is off the computer, CCS is performed Data correction.
- specific operationsrefer to 1, and the text description is as follows:
- oligo d (T) magnetic beadsto transfer mRNA in total RNA.
- the magnetic beads usedare purchased from Novozan VAHTSTM DNA Clean Beads (lot: N411).
- reaction conditions72°C for 3 minutes, slow ramp to slowly decrease to 42°C at a rate of 0.1°C/s, and then keep at 42°C for 2 minutes.
- the reaction systemis as follows:
- sequence of the fusion primer usedis (SEQ ID NO:1):
- Vrepresents a degenerate base, which is A, G, or C
- Nrepresents a degenerate base, which is A, T, G, or C. That is, the fusion primer forms a different sequence according to the type of the two bases at the 3'end (V can be A, G or C, and N can be A, T, G or C) to form a composite primer for mRNA The reverse transcription reaction.
- reaction systemreverse transcription to synthesize the complementary strand cDNA of mRNA
- the reaction conditionsare: 42°C90min, 70°C10min, 4°Chold
- the reaction systemis:
- the reagents used in this stepare all from the TAKARA company, the kit number is 634926 (SMARTer PCR cDNA Synthesis Kit).
- Use enzyme VII to digest the residual single-stranded RNA in the system, as well as the exposed primer sequences, and the reaction conditionsare: digestion at 37°C for 15 minutes.
- the reaction systemis:
- reaction systemwas prepared and reacted for 10 minutes at 25 degrees Celsius for end repair.
- reaction systemwas prepared and digested with ExoIII and ExoVII enzymes for 60 minutes at 37 degrees Celsius.
- reaction systemwas prepared and reacted at 20 degrees Celsius for 60 minutes.
- Template Prep Kit 1.0 Reagent(PACIFIC BIOSCIENCES lot: 100-991-900).
- the control group sequenced mRNAaccording to the following method.
- the starting sample usedis the total RNA extracted from the tissue sample (the starting sample is the same as the experimental group).
- the double-stranded DNAis then digested with enzyme VII, damage repair, end repair, ligation with known adapters, enzyme reaction digestion, BluePippin sorting, and finally a dumbbell-shaped library, which is detected and synthesized by Agilent2100 and Qubit HS, and then Pacbio is put on the machine Sequencing, the text description is as follows:
- reaction conditionsare: 42°C90min, 70°C10min, 4°Chold, the reaction system is:
- reaction conditionsare: 95°C, 2min; 98°C, 20S, 65°C, 15S, 72°C, 5min; 72°C, 10min, 4°Chold.
- the reaction systemis:
- reaction systemwas prepared and digested with ExoIII and ExoVII enzymes for 60 minutes at 37 degrees Celsius.
- a (%), T (%), G (%), and C (%) in Table 1respectively refer to the proportions of bases A, T, G, and C in the effective data on the entire chip; Take base A as an example.
- a (%)is the percentage of the content of A bases on the entire chip to the content of A bases + T bases + C bases + G bases on the entire chip.
- the experimental groupis reduced to 12.02%, and the available data is improved. It shows that using the experimental group method can significantly reduce the repetitive sequence ratio, thereby improving the utilization of data.
- the experimental group methodcan be used to build a database and sequence samples with different GC content.
- the method provided by the present inventionis used to build and sequence mRNAs. There is no PCR process in the library building process, which reduces the generation of duplicate sequences and can effectively improve the data utilization rate. Save sequencing costs. And because there is no PCR process, the GC bias caused by PCR is reduced.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Physics & Mathematics (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Analytical Chemistry (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description
Translated fromChinese优先权信息Priority information
无。no.
本发明涉及基因测序领域,具体涉及一种用于RNA直接建库的核酸序列、基于RNA样本直接构建测序文库的方法及应用。The present invention relates to the field of gene sequencing, in particular to a nucleic acid sequence used for direct RNA library construction, a method and application for directly constructing a sequencing library based on RNA samples.
Pacbio第三代测序基于边合成边测序的原理,以SMRT(单分子实时荧光测序技术)芯片为载体进行测序反应,测序时将基因组DNA打断成许多小的片段,制成液滴后将其分散到不同的ZMW纳米孔中。当ZMW孔底部聚合反应发生时,被不同荧光标记的核苷酸会在小孔的荧光探测区域中被聚合酶滞留,根据荧光的种类和荧光持续时间就可以判定模板DNA碱基组成的种类。Pacbio's third-generation sequencing is based on the principle of sequencing while synthesizing. It uses SMRT (Single Molecule Real-Time Fluorescence Sequencing Technology) chips as a carrier to perform sequencing reactions. During sequencing, genomic DNA is broken into many small fragments, which are then made into droplets. Disperse into different ZMW nanopores. When the polymerization reaction occurs at the bottom of the ZMW well, the nucleotides labeled with different fluorescence will be retained by the polymerase in the fluorescence detection area of the small hole. According to the type of fluorescence and the duration of fluorescence, the type of template DNA base composition can be determined.
细胞的转录组蕴含了丰富的信息,包括基因的结构、转录本的表达水平和反义转录。捕获这些信息的最好方法是准确、定量地揭示碱基的存在和身份,而不需要预先知道序列。当然,这种方法最好能生成跨越剪接点的连续序列。目前最常用的RNA-seq策略是利用poly(dT)引物或随机六聚体引物进行cDNA合成。之后再通过PCR来扩增这些cDNA链,不过这可能会降低cDNA文库的复杂性,影响cDNA的相对丰度,并造成某些种类的RNA丢失。此外,在PCR扩增过程中,RNA的所有修饰都会丢失。The transcriptome of a cell contains a wealth of information, including gene structure, transcript expression level and antisense transcription. The best way to capture this information is to accurately and quantitatively reveal the presence and identity of bases without knowing the sequence in advance. Of course, this method is best to generate a continuous sequence across the splice junction. At present, the most commonly used RNA-seq strategy is to use poly(dT) primers or random hexamer primers for cDNA synthesis. Then PCR is used to amplify these cDNA strands, but this may reduce the complexity of the cDNA library, affect the relative abundance of cDNA, and cause certain types of RNA to be lost. In addition, during the PCR amplification process, all modifications of RNA are lost.
针对RNA的碱基信息,如何实现RNA信息的测定,还需要进一步改进。For the base information of RNA, how to realize the measurement of RNA information needs further improvement.
发明内容Summary of the invention
本发明旨在至少在一定程度上解决相关技术中的技术问题之一,提供了一种对RNA直接进行建库和测序的方法以及试剂盒。The present invention aims to solve one of the technical problems in related technologies at least to a certain extent, and provides a method and kit for directly performing library building and sequencing on RNA.
本发明所提供的对于RNA进行直接建库测序的方法,在建库测序过程中无需PCR扩增,不会存在PCR的偏向性;在整个过程中,没有进行打断处理,可以保持RNA的原始长度,对剪接变体的研究特别有用,而且测序获得的数据是链特异性的。The method for directly constructing a library and sequencing RNA provided by the present invention does not require PCR amplification during the sequencing process of the library, and there is no bias of PCR; in the whole process, no interruption processing is performed, and the original RNA can be maintained The length is particularly useful for the study of splice variants, and the data obtained by sequencing is chain-specific.
本发明的发明人在研究过程中发现:针对RNA的测序,不同测序平台会采取不同的测序手段。以Illumina平台为例,在对RNA进行测序的过程中,首先对获取到带poly(A)结构的mRNA加上Illumina的接头,文库定量之后,进行桥式PCR扩增放大信号的过程。Illumina的 方法,虽然在建库的过程中是PCR-FREE的过程,但是Illumina基于SBS的测序原理,在测序的过程中依然要通过桥式PCR扩增来放大信号。所以这种基于Illumina平台的RNA直接测序方法,并不是完全的PCR-FREE,也不能直接读取到RNA上面的甲基化等修饰信息。现有的Illumina RNA直接测序技术中,测序过程中有桥式PCR扩增,必然会导致同一个插入片段有较多的重复,造成数据浪费。以Nanopore平台为例,其在poly(T)的接头上面连接上一个分子马达蛋白(可以引导核酸分子进去测序孔),用T4DNA连接酶将带poly(A)的mRNA和引物连接后,loading到Nanopore纳米测序孔。在建库测序都是完全的PCR-FREE过程,也可以直接读取到RNA上面的碱基修饰信息。但是单链状态的RNA极其不稳定,极易降解,造成测序的质量和产量越来越差。The inventor of the present invention discovered during the research process that for RNA sequencing, different sequencing platforms will adopt different sequencing methods. Taking the Illumina platform as an example, in the process of sequencing RNA, first add Illumina adapters to the obtained mRNA with a poly(A) structure, and after the library is quantified, a bridge PCR amplification process is performed to amplify the signal. Although the Illumina method is a PCR-FREE process in the process of library construction, Illumina is based on the principle of SBS sequencing, and the signal is still amplified by bridge PCR amplification during the sequencing process. Therefore, this direct RNA sequencing method based on the Illumina platform is not a complete PCR-FREE, nor can it directly read the methylation and other modification information on the RNA. In the existing Illumina direct RNA sequencing technology, there is bridge PCR amplification during the sequencing process, which will inevitably lead to more repetitions of the same insert and cause data waste. Take the Nanopore platform as an example. A molecular motor protein (which can guide nucleic acid molecules into the sequencing hole) is connected to the poly(T) linker. After connecting the poly(A) mRNA and primers with T4DNA ligase, load it to Nanopore nano sequencing hole. Sequencing during library construction is a complete PCR-FREE process, and the base modification information on RNA can also be read directly. However, single-stranded RNA is extremely unstable and easily degraded, resulting in poorer sequencing quality and yield.
而以Pacbio测序平台为例,其对RNA测序采用全长转录组测序过程原理,例如起始样品为组织样本提取得到的总RNA,经逆转录和PCR扩增合成的cDNA,经损伤修复,末端修复,与已知接头连接,酶反应消化,最终得到哑铃形的文库,经过Agilent2100和Qubit HS检测合格后进行Pacbio上机测序。由于利用Pacbio测序平台进行全长转录组测序,其中含有PCR的过程,必然会导致从一个片段有较多的重复,造成数据的浪费,从而导致成本增加。而且PCR反应,也会导致原始mRNA上甲基化等修饰的丢失。Taking the Pacbio sequencing platform as an example, it uses the principle of full-length transcriptome sequencing for RNA sequencing. For example, the starting sample is total RNA extracted from tissue samples, and cDNA synthesized by reverse transcription and PCR amplification is repaired by damage to the end. Repair, connect with known adapters, digest by enzyme reaction, and finally obtain a dumbbell-shaped library, which is tested by Agilent 2100 and Qubit HS and then sequenced on Pacbio. Due to the use of Pacbio sequencing platform for full-length transcriptome sequencing, which includes a PCR process, it will inevitably lead to more repetitions from one fragment, resulting in waste of data and increasing costs. Moreover, the PCR reaction will also lead to the loss of modifications such as methylation on the original mRNA.
由此,对于RNA来说,如何能够通过测序获得完整可靠的RNA上的生物信息,至关重要。本发明的发明人在研究过程中发现,可以通过引入引物序列,将RNA反转录成为RNA-DNA的杂合链,然后进行建库和测序,从而可以克服常规RNA测序需要反转录成cDNA双链,然后进行PCR扩增建库的方法来进行测序的过程。以mRNA为例,该方法尤其适用于具备不同剪切体的mRNA的测序。Therefore, for RNA, how to obtain complete and reliable biological information on RNA through sequencing is very important. The inventor of the present invention discovered in the research process that by introducing a primer sequence, RNA can be reverse-transcribed into an RNA-DNA hybrid strand, and then library construction and sequencing can be performed, thereby overcoming the need for reverse transcription into cDNA for conventional RNA sequencing. Double-stranded, and then PCR amplification method to build a library to carry out the process of sequencing. Taking mRNA as an example, this method is especially suitable for sequencing mRNAs with different splicing bodies.
具体而言,本发明提供了如下技术方案:Specifically, the present invention provides the following technical solutions:
根据本发明的第一方面,本发明提供了一种用于RNA直接建库的核酸序列,包括:第一核酸序列和第二核酸序列,所述第一核酸序列和所述第二核酸序列相连,所述第一核酸序列靠近所述核酸序列的5’端,所述第二核酸序列靠近所述核酸序列的3’端;所述第一核酸序列包括1~40个碱基;所述第二核酸序列包括至少20个碱基组成的polyT序列。第二核酸序列中含有polyT序列,polyT序列能够与RNA上的polyA结合。同时该核酸序列中还含有由任意A、T、C和G组成的第一核酸序列,该第一核酸序列和第二核酸序列相连,可以降低单独合成第二核酸序列的难度,也降低核酸序列纯化的难度。第一核酸序列可以由A、T、C、G四种碱基组成,长度可以为1~40个碱基,例如可以为5~40个碱基,10~40个碱基,20~40个碱基,15~35个碱基,20~35个碱基等等,A、T、C、G四种碱基对应的个数不做特殊限制。利用这段第一核酸序列,可以有效反转录出mRNA的信息,从而实现以RNA为模板,合成DNA链的精准的反转录。According to the first aspect of the present invention, the present invention provides a nucleic acid sequence for direct RNA library construction, comprising: a first nucleic acid sequence and a second nucleic acid sequence, the first nucleic acid sequence and the second nucleic acid sequence are connected The first nucleic acid sequence is close to the 5'end of the nucleic acid sequence, and the second nucleic acid sequence is close to the 3'end of the nucleic acid sequence; the first nucleic acid sequence includes 1-40 bases; The dinucleotide sequence includes a polyT sequence composed of at least 20 bases. The second nucleic acid sequence contains a polyT sequence, and the polyT sequence can bind to polyA on the RNA. At the same time, the nucleic acid sequence also contains a first nucleic acid sequence composed of any A, T, C, and G. The first nucleic acid sequence and the second nucleic acid sequence are connected, which can reduce the difficulty of separately synthesizing the second nucleic acid sequence and also reduce the nucleic acid sequence Difficulty of purification. The first nucleic acid sequence can be composed of four bases: A, T, C, and G, and can be 1-40 bases in length, for example, 5-40 bases, 10-40 bases, 20-40 bases. Bases, 15-35 bases, 20-35 bases, etc., the number corresponding to the four bases A, T, C, and G is not particularly limited. Using this first nucleic acid sequence, the mRNA information can be effectively reverse-transcribed, so as to realize the precise reverse transcription of the synthetic DNA strand using RNA as a template.
根据本发明的实施例,以上所述的核酸序列可以进一步包括如下技术特征:According to an embodiment of the present invention, the aforementioned nucleic acid sequence may further include the following technical features:
在本发明的一些实施例中,所述第一核酸序列包括25个碱基。In some embodiments of the present invention, the first nucleic acid sequence includes 25 bases.
在本发明的一些实施例中,所述polyT序列由30个碱基组成。RNA样本中大多数均是mRNA,以mRNA为例,通常大部分的mRNA 3’端的具有20-30个连续的A碱基,将核酸序列中的polyT序列限定为30个碱基,可以满足绝大部分mRNA的反转录;同时,固定的30个碱基的设定可以为了后续数据分析的时候,找到转录起始和终止位点。例如测序获得连续30个A碱基的序列即为终止位点,如果明显小于30个连续A碱基,则可能为插入片段本身的特质。In some embodiments of the present invention, the polyT sequence consists of 30 bases. Most of the RNA samples are mRNA. Take mRNA as an example. Usually most of the mRNA has 20-30 consecutive A bases at the 3'end. The polyT sequence in the nucleic acid sequence is limited to 30 bases, which can satisfy the absolute requirement. Reverse transcription of most mRNA; at the same time, the fixed 30 base setting can be used to find the transcription start and stop sites during subsequent data analysis. For example, a sequence of 30 consecutive A bases obtained by sequencing is the termination site. If it is significantly less than 30 consecutive A bases, it may be the characteristics of the insert itself.
在本发明的一些实施例中,所述第二核酸序列进一步包括:In some embodiments of the present invention, the second nucleic acid sequence further includes:
简并碱基V,所述简并碱基V与所述polyT序列相连,所述简并碱基V位于所述polyT序列的3’端,所述简并碱基V为碱基A、G或者C。Degenerate base V, said degenerate base V is connected to said polyT sequence, said degenerate base V is located at the 3'end of said polyT sequence, and said degenerate base V is base A, G Or C.
在本发明的一些实施例中,所述第二核酸序列进一步包括简并碱基N,所述简并碱基N与所述简并碱基V相连,所述简并碱基N位于所述简并碱基V的3’端,所述简并碱基N为碱基A、T、G或者C。RNA样本3’末端可能有不同长度的poly(A)序列,以mRNA为例,其3’末端poly(A)长度在不同物种中差异较大,甚至在同一个物种不同部位poly(A)长度差异也较大。为了能够更好的使本发明提供的核酸序列锚定到mRNA的3’末端,可以在核酸序列的poly(T)序列末端添加一个或者两个简并碱基。In some embodiments of the present invention, the second nucleic acid sequence further includes a degenerate base N, the degenerate base N is connected to the degenerate base V, and the degenerate base N is located in the The 3'end of the degenerate base V, the degenerate base N is the base A, T, G or C. RNA samples may have poly(A) sequences of different lengths at the 3'end. Taking mRNA as an example, the length of the 3'end poly(A) varies greatly among different species, even in different parts of the same species. The difference is also large. In order to better anchor the nucleic acid sequence provided by the present invention to the 3'end of the mRNA, one or two degenerate bases can be added to the end of the poly(T) sequence of the nucleic acid sequence.
在本发明的一些实施例中,所述第一核酸序列上任意连续3个碱基序列不同。第一核酸序列中A、T、C、G四种碱基对应的个数不做特殊限制,只要没有连续三个相同的碱基即可。例如,若存在连续3个A碱基,可能会造成测序时碱基识别异常。In some embodiments of the present invention, any three consecutive base sequences on the first nucleic acid sequence are different. The number corresponding to the four bases of A, T, C, and G in the first nucleic acid sequence is not particularly limited, as long as there are no three consecutive identical bases. For example, if there are 3 consecutive A bases, it may cause abnormal base recognition during sequencing.
在本发明的一些实施例中,所述核酸序列如SEQ ID NO:1所示。In some embodiments of the present invention, the nucleic acid sequence is shown in SEQ ID NO:1.
根据本发明的第二方面,本发明提供了一种含有部分双链的RNA-DNA杂合链,包括:第一条链和第二条链,所述第一条链为RNA链,所述第一条链的3’末端为polyA序列;所述第二条链为本发明第一方面所述的核酸序列;所述第一条链3’末端的至少部分polyA序列和所述第二条链的至少部分polyT序列互补形成双链。According to the second aspect of the present invention, the present invention provides a partially double-stranded RNA-DNA hybrid strand, comprising: a first strand and a second strand, the first strand is an RNA strand, the The 3'end of the first strand is a polyA sequence; the second strand is the nucleic acid sequence according to the first aspect of the present invention; at least part of the polyA sequence at the 3'end of the first strand and the second strand At least part of the polyT sequence of the strand is complementary to form a double strand.
根据本发明的第三方面,本发明提供了一种基于RNA样本构建测序文库的方法,包括:基于含有polyT序列的核酸序列,与所述RNA样本上的polyA序列结合,以便获得含有部分双链的RNA-DNA杂合链,所述部分双链由至少部分polyA序列和至少部分polyT序列互补形成,所述核酸序列为根据本发明第一方面任一实施例所述的核酸序列;基于所述含有部分双链的RNA-DNA杂合链,逆转录合成与所述RNA样本互补的DNA链,以便获得含有RNA-DNA杂合双链的产物,进行损伤修复和末端修复,连接测序接头,构建获得测序文库。According to the third aspect of the present invention, the present invention provides a method for constructing a sequencing library based on an RNA sample, which includes: combining a nucleic acid sequence containing a polyT sequence with a polyA sequence on the RNA sample to obtain a partial double-strand The RNA-DNA hybrid strand, the partial double strand is formed by complementary at least part of the polyA sequence and at least part of the polyT sequence, and the nucleic acid sequence is the nucleic acid sequence according to any one of the embodiments of the first aspect of the present invention; based on the The RNA-DNA hybrid strands containing partial double-strands are synthesized by reverse transcription to synthesize the DNA strands complementary to the RNA sample to obtain products containing the RNA-DNA hybrid double strands for damage repair and end repair, and the sequencing adapter is connected to construct Obtain sequencing library.
利用含有polyT序列的核酸序列,与RNA样本上的polyA序列结合,从而获得含有至少部分polyA序列和polyT序列互补所形成的部分双链的RNA-DNA杂合链。然后以RNA样本为模板,逆转录获得与所述RNA样本互补的DNA链,从而获得含有RNA-DNA杂合双链的产物;对所获得的含有RNA-DNA杂合双链的产物进行损伤修复和末端修复,连接测序接头,构建获得测序文库。The nucleic acid sequence containing the polyT sequence is combined with the polyA sequence on the RNA sample to obtain an RNA-DNA hybrid strand containing at least a part of the polyA sequence and the part of the double strand formed by the complementary polyT sequence. Then use the RNA sample as a template, reverse transcription to obtain a DNA strand complementary to the RNA sample, thereby obtaining a product containing an RNA-DNA hybrid double-strand; performing damage repair on the obtained product containing an RNA-DNA hybrid double-strand And end repair, connect the sequencing adapter to construct a sequencing library.
利用本发明提供的方法,对RNA样本构建测序文库,在建库过程中没有PCR扩增的过程,减少了重复序列(duplicate)和嵌合体的产生,可以有效提高数据利用率,从而节约测序成本。而且没有PCR,可以直接读取RNA上面原始碱基修饰(甲基化、乙酰化等)信息。而且由于没有PCR的过程,减少了PCR造成的GC偏向性,对不同GC的样本都可以进行建库和测序。由于收构建的测序文库是基于RNA-DNA杂合链结构,可以用与RNA互补的cDNA的序列校准RNA,从而提高数据质量。而且,所获得的RNA-DNA二级结构稳定,文库储存和运输过程不易被降解,从而可以提高测序质量和产量。Utilizing the method provided by the present invention to construct a sequencing library for RNA samples, there is no PCR amplification process during the library construction process, which reduces the generation of duplicate sequences (duplicates) and chimeras, can effectively improve data utilization, and save sequencing costs . And without PCR, you can directly read the original base modification (methylation, acetylation, etc.) information on the RNA. And because there is no PCR process, the GC bias caused by PCR is reduced, and the library and sequencing can be performed on samples of different GCs. Since the constructed sequencing library is based on the RNA-DNA hybrid strand structure, the RNA can be calibrated with the sequence of cDNA complementary to the RNA, thereby improving data quality. Moreover, the obtained RNA-DNA secondary structure is stable, and the library is not easily degraded during storage and transportation, thereby improving sequencing quality and yield.
根据本发明的实施例,以上基于RNA样本构建测序文库的方法,可以进一步包括如下技术特征:According to an embodiment of the present invention, the above method for constructing a sequencing library based on RNA samples may further include the following technical features:
在本发明的一些实施例中,所述RNA样本为mRNA样本。mRNA的3’端通常含有polyA尾巴,可以通过上述方法,与含有polyT序列的核酸序列结合,获得RNA-DNA杂合双链,通过对杂合双链构建文库,来实现mRNA样本的测序。In some embodiments of the present invention, the RNA sample is an mRNA sample. The 3'end of the mRNA usually contains a polyA tail, which can be combined with a nucleic acid sequence containing a polyT sequence by the above-mentioned method to obtain an RNA-DNA hybrid duplex. By constructing a library of the hybrid duplex, the mRNA sample can be sequenced.
在本发明的一些实施例中,进一步包括:基于所述含有RNA-DNA杂合双链的产物,利用外切酶消化将所述RNA-DNA杂合双链的两端消化为平末端,然后再进行所述损伤修复和末端修复。所获得的含有RNA-DNA杂合双链的产物中可能含有一些未反应的残留的单链RNA以及暴露出来的单链核酸序列,可以利用外切酶消化去除,然后进行损伤修复和末端修复,从而减少单链RNA以及单链核酸序列对于测序结果的影响。In some embodiments of the present invention, it further includes: digesting the two ends of the RNA-DNA hybrid double-strands into blunt ends by exonuclease digestion based on the product containing the RNA-DNA hybrid duplex, and then Then proceed to the damage repair and end repair. The obtained RNA-DNA hybrid double-stranded product may contain some unreacted residual single-stranded RNA and exposed single-stranded nucleic acid sequences, which can be digested and removed by exonuclease, and then damage repair and end repair are performed. Thereby reducing the influence of single-stranded RNA and single-stranded nucleic acid sequences on the sequencing results.
在本发明的一些实施例中,所述外切酶为ExoVII。利用该外切酶可以有效实现RNA-DNA杂合双链末端所暴露出来的单链RNA或者单链DNA片段的消化。In some embodiments of the present invention, the exonuclease is ExoVII. The exonuclease can effectively digest the single-stranded RNA or single-stranded DNA fragments exposed by the RNA-DNA hybrid double-stranded ends.
在本发明的一些实施例中,利用所述外切酶在37摄氏度条件下反应15~20分钟进行所述消化处理。将反应时间控制在15-20分钟左右,可以使得消化完全,且不会影响测序质量。当消化时间过短时,会导致消化不完全,接头无法连接;而时间过长对导致DNA-RNA造成损伤,形成缺口,从而导致测序质量变差。In some embodiments of the present invention, the exonuclease is used to react for 15-20 minutes at 37 degrees Celsius to perform the digestion treatment. Controlling the reaction time to about 15-20 minutes can make the digestion complete without affecting the quality of sequencing. When the digestion time is too short, the digestion will be incomplete and the adapters cannot be connected; while the time is too long to cause damage to the DNA-RNA and form gaps, resulting in poor sequencing quality.
在本发明的一些实施例中,所述方法进一步包括:基于所述含有RNA-DNA杂合双链的产物,进行损伤修复和末端修复,连接测序接头后,利用外切酶ExoIII和外切酶ExoVII进行消化处理,再构建获得所述测序文库。外切酶ExoVII的作用在于进一步消化掉RNA-DNA杂合双链末端处于单链状态的片段,外切酶ExoIII的作用在于消化掉有缺口的 RNA-DNA杂合双链。连接测序接头后利用外切酶ExoIII和外切酶ExoVII进行消化处理,主要作用是为了消化掉无效的文库,提高测序的效率。In some embodiments of the present invention, the method further includes: performing damage repair and end repair based on the product containing the RNA-DNA hybrid duplex, after connecting the sequencing adapter, using exonuclease ExoIII and exonuclease ExoVII is digested and then constructed to obtain the sequencing library. The role of exonuclease ExoVII is to further digest the fragments at the ends of the RNA-DNA hybrid double-stranded strands, and the role of exonuclease ExoIII is to digest the gapped RNA-DNA hybrid double-strands. After connecting the sequencing adapter, the exonuclease ExoIII and ExoVII are used for digestion. The main function is to digest the invalid library and improve the efficiency of sequencing.
在本发明的一些实施例中,利用外切酶ExoIII和外切酶ExoVII在37摄氏度条件下反应50~70分钟。在该条件下可以使得消化完全,且不会影响测序质量。In some embodiments of the present invention, the exonuclease ExoIII and ExoVII are used to react at 37 degrees Celsius for 50 to 70 minutes. Under these conditions, the digestion can be completed without affecting the quality of sequencing.
根据本发明的第四方面,本发明提供了一种测序文库,所述测序文库通过本发明第三方面所述的方法获得。According to the fourth aspect of the present invention, the present invention provides a sequencing library obtained by the method described in the third aspect of the present invention.
根据本发明的第五方面,本发明提供了一种对RNA样本进行测序的方法,包括:基于所述RNA样本,根据本发明第二方面任一实施例所述的方法构建测序文库;基于所述测序文库,利用Pacbio测序平台对所述RNA样本进行测序。这里所述的Pacbio测序平台,指的是Pacbio公司推出的所有测序平台,例如包括RSII和Sequel平台,还有未来即将推出市场的Sequel II平台等等。According to the fifth aspect of the present invention, the present invention provides a method for sequencing an RNA sample, including: constructing a sequencing library based on the RNA sample according to any one of the embodiments of the second aspect of the present invention; The sequencing library uses the Pacbio sequencing platform to sequence the RNA samples. The Pacbio sequencing platform mentioned here refers to all sequencing platforms launched by Pacbio, such as the RSII and Sequel platforms, and the Sequel II platform that will be launched in the future.
根据本发明的第四方面,本发明提供了一种试剂盒,所述试剂盒包括本发明第一方面任一实施例所述的核酸序列。According to the fourth aspect of the present invention, the present invention provides a kit including the nucleic acid sequence according to any one of the embodiments of the first aspect of the present invention.
本发明所取得的有益效果至少表现为:利用本发明提供的核酸序列作为引物,可以实现RNA样本的直接建库和测序,从而可以保持RNA的原始长度,而不必如常规二代测序一样,将RNA打断成小片段后再进行测序,从而使得适用于mRNA可变剪切的研究。而且由于测序的数据是链特异性的,从而可以区分测序数据的方向性,鉴定出测序数据是正义链mRNA,还是mRNA的互补链,从而实现测序数据的高精度分析。The beneficial effect achieved by the present invention is at least manifested as: using the nucleic acid sequence provided by the present invention as primers can realize the direct library building and sequencing of RNA samples, so that the original length of RNA can be maintained without the need for conventional second-generation sequencing. RNA is broken into small fragments and then sequenced, making it suitable for the study of mRNA variable shearing. Moreover, because the sequencing data is chain-specific, the directionality of the sequencing data can be distinguished, and it can be identified whether the sequencing data is the sense strand mRNA or the complementary strand of the mRNA, thereby realizing high-precision analysis of the sequencing data.
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:The above and/or additional aspects and advantages of the present invention will become obvious and easy to understand from the description of the embodiments in conjunction with the following drawings, in which:
图1是根据本发明的一个实施例提供的用于RNA建库的核酸序列的结构示意图。Fig. 1 is a schematic structural diagram of a nucleic acid sequence for RNA library construction according to an embodiment of the present invention.
图2是根据本发明的一个实施例提供的基于mRNA构建RNA-DNA杂合双链,并进行酶切消化的示意图。Fig. 2 is a schematic diagram of constructing an RNA-DNA hybrid duplex based on mRNA according to an embodiment of the present invention, and performing restriction digestion.
下面详细描述本发明的实施例,所述实施例的示例在附图中示出。下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。同时,对本发明中出现的术语进行解释,以方便本领域技术人员的理解。需要说明的是,这些解释仅用来方便对本发明进行理解,而不应看做是对本发明保护范围的限制。The embodiments of the present invention are described in detail below, and examples of the embodiments are shown in the accompanying drawings. The embodiments described below with reference to the accompanying drawings are exemplary, and are intended to explain the present invention, but should not be construed as limiting the present invention. At the same time, the terms appearing in the present invention are explained to facilitate the understanding of those skilled in the art. It should be noted that these explanations are only used to facilitate the understanding of the present invention, and should not be regarded as limiting the protection scope of the present invention.
本文中,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性 或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在本发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。In this article, the terms "first" and "second" are only used for descriptive purposes, and cannot be understood as indicating or implying relative importance or implicitly indicating the number of indicated technical features. Therefore, the features defined with "first" and "second" may explicitly or implicitly include at least one of the features. In the description of the present invention, "a plurality of" means at least two, such as two, three, etc., unless otherwise specifically defined.
本文中,“含有部分双链的RNA-DNA杂合链”指的是:由RNA链和DNA链所形成的,且所形成的RNA-DNA杂合链中仅具有部分双链,其余部分为单链RNA和单链DNA。In this article, "RNA-DNA hybrid strands containing partial double-strands" refers to: formed by RNA strands and DNA strands, and the formed RNA-DNA hybrid strands have only part of the double strands, and the rest are Single-stranded RNA and single-stranded DNA.
本文中,“RNA-DNA杂合双链”指的是RNA链和DNA链所形成的RNA-DNA双链。该RNA-DNA杂合双链与上述含有部分双链的RNA-DNA杂合链相比,RNA-DNA杂合双链中不含有或者几乎不含有单链片段。当然,即便是含有些许单链片段,这些单链片段也不作为测序数据的一部分或者测序文库的一部分,可以利用外切酶消化去除。Herein, "RNA-DNA hybrid double strand" refers to the RNA-DNA double strand formed by the RNA strand and the DNA strand. Compared with the aforementioned RNA-DNA hybrid strands containing partial double strands, the RNA-DNA hybrid double strands contain no or almost no single-stranded fragments. Of course, even if it contains a few single-stranded fragments, these single-stranded fragments are not part of the sequencing data or part of the sequencing library, and can be digested and removed by exonuclease.
本文中,反转录和逆转录具有相同的含义,均是指以RNA为模板合成DNA的过程。In this article, reverse transcription and reverse transcription have the same meaning, and both refer to the process of synthesizing DNA using RNA as a template.
本发明提供了一种能够用于RNA建库的核酸序列,如图1所示,包括:第一核酸序列和第二核酸序列,所述第一核酸序列和所述第二核酸序列相连,所述第一核酸序列靠近所述核酸序列的5’端,所述第二核酸序列靠近所述核酸序列的3’端;所述第一核酸序列包括1~40个碱基;所述第二核酸序列包括至少20个碱基组成的polyT序列。该核酸序列可以作为融合引物,用于RNA的逆转录反应。The present invention provides a nucleic acid sequence that can be used for RNA library construction, as shown in FIG. 1, comprising: a first nucleic acid sequence and a second nucleic acid sequence, the first nucleic acid sequence and the second nucleic acid sequence are connected, so The first nucleic acid sequence is close to the 5'end of the nucleic acid sequence, and the second nucleic acid sequence is close to the 3'end of the nucleic acid sequence; the first nucleic acid sequence includes 1-40 bases; the second nucleic acid sequence The sequence includes a polyT sequence consisting of at least 20 bases. The nucleic acid sequence can be used as a fusion primer for the reverse transcription reaction of RNA.
本发明还提供了一种基于RNA样本构建测序文库的方法,包括:基于含有polyT序列的核酸序列,与所述RNA样本上的polyA序列结合,以便获得含有部分双链的RNA-DNA杂合链,所述部分双链由至少部分polyA序列和至少部分polyT序列互补形成,所述核酸序列为根据本发明第一方面任一实施例所述的核酸序列;基于所述含有部分双链的RNA-DNA杂合链,逆转录合成与所述RNA样本互补的DNA链,以便获得含有RNA-DNA杂合双链的产物,进行损伤修复和末端修复,连接测序接头,构建获得测序文库。The present invention also provides a method for constructing a sequencing library based on an RNA sample, which includes: combining a nucleic acid sequence containing a polyT sequence with a polyA sequence on the RNA sample to obtain an RNA-DNA hybrid strand containing a partial double strand The partially double-stranded is formed by complementation of at least part of the polyA sequence and at least part of the polyT sequence, and the nucleic acid sequence is the nucleic acid sequence according to any one of the embodiments of the first aspect of the present invention; based on the RNA-containing partially double-stranded RNA- DNA hybrid strands, reverse transcription to synthesize a DNA strand complementary to the RNA sample to obtain products containing RNA-DNA hybrid double strands, perform damage repair and end repair, connect sequencing adapters, and construct a sequencing library.
在本发明的一些实施例中,将所述RNA样本与所述含有polyT的核酸序列混合,在72摄氏度反应3~5分钟,然后以0.1~0.5摄氏度/秒的速度降低至42摄氏度,并在42摄氏度下反应2~5分钟,从而获得含有部分双链的RNA-DNA杂合链。在将RNA样本和含有polyT的核酸序列混合反应时,将温度设定为72摄氏度,或者低于72℃,为了防止温度过高RNA被降解;同时考虑到含有polyT的核酸序列3’末端的简并碱基,采用逐渐减低的温度,可以有效增加含有polyT的核酸序列与RNA样本的结合效率,例如以0.1~0.5摄氏度/秒的速度降低至42摄氏度;且在42℃反应2-5min促进反应充分。In some embodiments of the present invention, the RNA sample is mixed with the polyT-containing nucleic acid sequence, reacted at 72 degrees Celsius for 3 to 5 minutes, and then reduced to 42 degrees Celsius at a rate of 0.1 to 0.5 degrees Celsius/sec, and React for 2 to 5 minutes at 42 degrees Celsius to obtain a partially double-stranded RNA-DNA hybrid strand. When mixing RNA samples with nucleic acid sequences containing polyT, set the temperature to 72 degrees Celsius or lower than 72 degrees Celsius to prevent RNA from being degraded due to excessive temperature; at the same time, consider the simplicity of the 3'end of nucleic acid sequences containing polyT. Combining bases, using gradually reduced temperatures, can effectively increase the binding efficiency of polyT-containing nucleic acid sequences and RNA samples, for example, reduce to 42 degrees Celsius at a rate of 0.1 to 0.5 degrees Celsius/sec; and react at 42 degrees Celsius for 2-5 minutes to promote the reaction full.
在本发明的一些实施例中,基于所述含有部分双链的RNA-DNA杂合链,加入DTT和dNTP混合,在42摄氏度条件下反应80~100分钟,然后在70~75摄氏度下反应10~15分钟,从而逆转录合成与所述RNA样本互补的DNA链。In some embodiments of the present invention, based on the partially double-stranded RNA-DNA hybrid strands, DTT and dNTP are added and mixed, reacted at 42 degrees Celsius for 80-100 minutes, and then reacted at 70-75 degrees Celsius. ~15 minutes to synthesize a DNA strand complementary to the RNA sample by reverse transcription.
在本发明的一些实施例中,加入DNA和dNTP在37摄氏度条件下反应50~70分钟, 进行所述损伤修复。在本发明的一些实施例中,加入DNA在25~30摄氏度条件下反应10~20分钟,进行所述末端修复。In some embodiments of the present invention, DNA and dNTP are added and reacted at 37 degrees Celsius for 50-70 minutes to repair the damage. In some embodiments of the present invention, DNA is added and reacted at 25-30 degrees Celsius for 10-20 minutes to perform the end repair.
在本发明的一些实施例中,加入连接酶和连接接头,在25~30摄氏度条件下反应12~16小时,以便连接所述测序接头。In some embodiments of the present invention, a ligase and a linker are added, and the reaction is carried out at 25-30 degrees Celsius for 12-16 hours to connect the sequencing linker.
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。The solution of the present invention will be explained below in conjunction with examples. Those skilled in the art will understand that the following embodiments are only used to illustrate the present invention and should not be regarded as limiting the scope of the present invention. Where specific techniques or conditions are not indicated in the examples, the procedures shall be carried out in accordance with the techniques or conditions described in the literature in the field or in accordance with the product specification. The reagents or instruments used without the manufacturer's indication are all conventional products that are commercially available.
实施例Example
实验组test group
实验组按照如下方法对mRNA进行测序。所用到的起始样品为组织样本提取得到的总RNA,使用oligo d(T)的磁珠调取mRNA,使用特定引物,经逆转录将mRNA变成一条RNA-DNA杂合链,然后经过酶VII消化,损伤修复,末端修复,与已知接头连接,酶反应消化,BluePippin分选,最终得到哑铃形的文库,经过Agilent2100和Qubit HS检测合成后进行Pacbio上机测序,数据下机后进行CCS数据矫正。具体操作参照如1,文字描述如下:The experimental group sequenced mRNA according to the following method. The starting sample used is the total RNA extracted from tissue samples, using oligo d(T) magnetic beads to extract mRNA, using specific primers, reverse transcription to transform the mRNA into an RNA-DNA hybrid strand, and then pass the enzyme VII digestion, damage repair, end repair, ligation with known adapters, enzyme reaction digestion, BluePippin sorting, and finally a dumbbell-shaped library, which is detected and synthesized by Agilent2100 and Qubit HS, and then sequenced on the Pacbio computer. After the data is off the computer, CCS is performed Data correction. For specific operations, refer to 1, and the text description is as follows:
(1)使用oligo d(T)的磁珠调取total RNA中的mRNA。其中所用到的磁珠购自于诺唯赞VAHTSTM DNA Clean Beads(lot:N411)。(1) Use oligo d (T) magnetic beads to transfer mRNA in total RNA. The magnetic beads used are purchased from Novozan VAHTSTM DNA Clean Beads (lot: N411).
(2)使用融合引物结合到mRNA poly(A)上,反应条件为:72℃3min,slow ramp to以0.1℃/s的速率缓慢降低为42℃,然后在42摄氏度保持2min,反应体系如下:(2) Use fusion primers to bind to mRNA poly(A). The reaction conditions are: 72°C for 3 minutes, slow ramp to slowly decrease to 42°C at a rate of 0.1°C/s, and then keep at 42°C for 2 minutes. The reaction system is as follows:

其中,所用到的融合引物的序列为(SEQ ID NO:1):Among them, the sequence of the fusion primer used is (SEQ ID NO:1):
5'-AAGCAGTGGTATCAACGCAGAGTAC TTT TTT TTT TTT TTT TTT TTT TTT TTT TTTVN-3',其中V代表简并碱基,为A、G或者C;N代表简并碱基,为A、T、G或者C。即该融合引物依据3’末端的两个碱基的类型(V可以为A、G活C,N可以为A、T、G或C),形成不同的序列,从而组成复合引物,用于mRNA的逆转录反应。5'-AAGCAGTGGTATCAACGCAGAGTAC TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT N-3', where V represents a degenerate base, which is A, G, or C; N represents a degenerate base, which is A, T, G, or C. That is, the fusion primer forms a different sequence according to the type of the two bases at the 3'end (V can be A, G or C, and N can be A, T, G or C) to form a composite primer for mRNA The reverse transcription reaction.
(3)按照如下反应体系,逆转录合成mRNA的互补链cDNA,反应条件为:42℃90min,70℃10min,4℃hold,反应体系为:(3) According to the following reaction system, reverse transcription to synthesize the complementary strand cDNA of mRNA, the reaction conditions are: 42℃90min, 70℃10min, 4℃hold, the reaction system is:

此步骤所用的试剂均来自TAKARA公司,货号为634926的试剂盒(SMARTer PCR cDNA Synthesis Kit)。The reagents used in this step are all from the TAKARA company, the kit number is 634926 (SMARTer PCR cDNA Synthesis Kit).
(4)利用酶VII消化体系中残留的单链RNA,还有暴露出来的引物序列,反应条件为:37℃温度下,消化15min。反应体系为:(4) Use enzyme VII to digest the residual single-stranded RNA in the system, as well as the exposed primer sequences, and the reaction conditions are: digestion at 37°C for 15 minutes. The reaction system is:

(5)建库:损伤修复+末端修复+加接头+片段分选。具体如下:(5) Library building: damage repair + end repair + linker addition + fragment sorting. details as follows:
A、损伤修复反应A. Damage repair response
制备如下反应体系,在37摄氏度条件下反应60分钟,进行损伤修复。Prepare the following reaction system and react at 37 degrees Celsius for 60 minutes to repair damage.

B、末端修复反应(25℃,10min)B. End repair reaction (25℃, 10min)
制备如下反应体系,在25摄氏度条件下反应10分钟,进行末端修复。The following reaction system was prepared and reacted for 10 minutes at 25 degrees Celsius for end repair.

C、加接头C, add connector
制备如下反应体系,在25摄氏度条件下反应12~16小时左右,连接接头。Prepare the following reaction system, react at 25 degrees Celsius for about 12-16 hours, and connect the joints.

D、双酶消化D. Double enzyme digestion
制备如下反应体系,在37摄氏度条件下,利用ExoIII和ExoVII酶消化60分钟。The following reaction system was prepared and digested with ExoIII and ExoVII enzymes for 60 minutes at 37 degrees Celsius.

(6)制备上机:加primer+加测序聚合酶。(6) Preparation on the machine: adding primer+ and sequencing polymerase.
A、测序引物连接A. Sequencing primer connection
制备如下反应体系,在20摄氏度下反应60分钟。The following reaction system was prepared and reacted at 20 degrees Celsius for 60 minutes.

B、测序聚合酶连接B. Sequencing polymerase connection
制备如下反应体系,在30摄氏度下反应60分钟。The following reaction system was prepared and reacted at 30 degrees Celsius for 60 minutes.


其中,所用到的商业建库试剂盒名称为Template Prep Kit 1.0 Reagent(PACIFIC BIOSCIENCES lot:100-991-900)。Among them, the name of the commercial library building kit used is Template Prep Kit 1.0 Reagent (PACIFIC BIOSCIENCES lot: 100-991-900).
对照组Control group
对照组按照如下方法对mRNA进行测序。所用到的起始样品为组织样本提取得到的总RNA(起始样品与实验组相同),使用oligo d(T)的磁珠调取mRNA,逆转录合成mRNA的互补链,PCR扩增出大量的DNA双链,然后经过酶VII消化,损伤修复,末端修复,与已知接头连接,酶反应消化,BluePippin分选,最终得到哑铃形的文库,经过Agilent2100和Qubit HS检测合成后进行Pacbio上机测序,文字描述如下:The control group sequenced mRNA according to the following method. The starting sample used is the total RNA extracted from the tissue sample (the starting sample is the same as the experimental group). Use oligo d (T) magnetic beads to extract mRNA, reverse transcription to synthesize the complementary strand of mRNA, and a large amount of PCR amplification The double-stranded DNA is then digested with enzyme VII, damage repair, end repair, ligation with known adapters, enzyme reaction digestion, BluePippin sorting, and finally a dumbbell-shaped library, which is detected and synthesized by Agilent2100 and Qubit HS, and then Pacbio is put on the machine Sequencing, the text description is as follows:
(1)逆转录合成mRNA的互补链,反应条件为:42℃90min,70℃10min,4℃hold,反应体系为:(1) Reverse transcription to synthesize the complementary strand of mRNA, the reaction conditions are: 42℃90min, 70℃10min, 4℃hold, the reaction system is:

(2)PCR扩增,反应条件为:95℃2min;98℃,20S,65℃,15S,72℃,5min;72℃,10min,4℃hold。反应体系为:(2) PCR amplification, the reaction conditions are: 95℃, 2min; 98℃, 20S, 65℃, 15S, 72℃, 5min; 72℃, 10min, 4℃hold. The reaction system is:


(3)建库:损伤修复+末端修复+加接头+片段分选。具体如下:(3) Library building: damage repair + end repair + linker addition + fragment sorting. details as follows:
A、损伤修复反应A. Damage repair response
制备如下反应体系,在37摄氏度下反应60分钟,进行损伤修复。Prepare the following reaction system and react at 37 degrees Celsius for 60 minutes to repair damage.

B、末端修复反应B. End repair reaction
制备如下反应体系,在25摄氏度下反应10分钟,进行末端修复。Prepare the following reaction system and react at 25 degrees Celsius for 10 minutes to perform end repair.

C、加接头C, add connector
制备如下反应体系,在25摄氏度下反应12~16小时左右,连接接头。Prepare the following reaction system, react at 25 degrees Celsius for about 12-16 hours, and connect the joints.

D、双酶消化D. Double enzyme digestion
制备如下反应体系,在37摄氏度条件下,利用ExoIII和ExoVII酶进行消化处理60分钟。The following reaction system was prepared and digested with ExoIII and ExoVII enzymes for 60 minutes at 37 degrees Celsius.

(4)制备上机:加primer+加测序聚合酶。(4) Preparation on the machine: adding primer+ and sequencing polymerase.
A、测序引物连接A. Sequencing primer connection
制备如下反应体系,在20摄氏度反应60分钟,连接测序引物。Prepare the following reaction system, react at 20 degrees Celsius for 60 minutes, and connect the sequencing primers.

B、测序聚合酶连接(30℃,60min)B. Sequencing polymerase connection (30℃, 60min)
制备如下反应体系,在30摄氏度下反应60分钟,连接测序聚合酶。Prepare the following reaction system, react at 30 degrees Celsius for 60 minutes, and connect the sequencing polymerase.

将实验组和对照组处理得到的溶液分别利用Pacbio测序平台进行上机测序,获得测序数据如下表所示:The solutions processed by the experimental group and the control group were respectively used for sequencing on the Pacbio sequencing platform, and the sequencing data obtained are shown in the following table:
表1 实验组和对照组的测序结果Table 1 Sequencing results of experimental group and control group


其中,表1中A(%)、T(%)、G(%)、C(%)分别指的是整张芯片上有效数据中碱基A、T、G和C的占比;以碱基A为例,A(%)是整张芯片上A碱基的含量与整张芯片上A碱基+T碱基+C碱基+G碱基的含量的百分比。理论正常情况下,根据碱基互补配对原则和碱基平衡原理,碱基含量A=T,G=C。Among them, A (%), T (%), G (%), and C (%) in Table 1 respectively refer to the proportions of bases A, T, G, and C in the effective data on the entire chip; Take base A as an example. A (%) is the percentage of the content of A bases on the entire chip to the content of A bases + T bases + C bases + G bases on the entire chip. Theory Normally, according to the principle of base complementary pairing and the principle of base balance, base content A=T, G=C.
从表1的结果可以看出,无论是实验组还是对照组,数据产量均大于5Gb,酶读长均大于10Kb。就数据产量来说,无论是按照实验组还是对照组的方法,均未对测序结果造成不利的影响。It can be seen from the results in Table 1, whether it is the experimental group or the control group, the data output is greater than 5Gb, and the enzyme read length is greater than 10Kb. In terms of data output, no matter whether it was in accordance with the method of the experimental group or the control group, it did not adversely affect the sequencing results.
关于重复序列比对到基因组上的比例Duplicate rate on genome(%),相较于对照组的29.94%,实验组降低至12.02%,可利用数据提高。说明采用实验组的方法可以明显的降低重复序列比,从而提高数据的利用率。Regarding the Duplicate rate on genome (%) of the repetitive sequence alignment to the genome, compared to 29.94% in the control group, the experimental group is reduced to 12.02%, and the available data is improved. It shows that using the experimental group method can significantly reduce the repetitive sequence ratio, thereby improving the utilization of data.
进一步比较,可以看出,相较于对照组的测序数据,实验组所获得的A和T、G和C碱基的偏离程度明显减少(理论正常情况下,根据碱基互补配对原则和碱基平衡原理,A=T,G=C)。说明采用实验组的方法,由于没有PCR过程的存在,可以显著减少PCR造成的GC偏向性,利用实验组的方法可以对不同GC含量的样本进行建库和测序。Further comparison, it can be seen that compared with the control group's sequencing data, the degree of deviation of the A and T, G and C bases obtained by the experimental group is significantly reduced (in the theoretical normal situation, according to the principle of base complementary pairing and base Principle of balance, A=T, G=C). It shows that the experimental group method can significantly reduce the GC bias caused by PCR because there is no PCR process. The experimental group method can be used to build a database and sequence samples with different GC content.
从以上实施例可以看出,采用本发明所提供的方法对mRNA进行建库和测序,建库过程中没有PCR的过程,减少了重复序列(duplicate)的产生,可以有效提高数据利用率,从而节约测序成本。而且由于没有PCR的过程,减少了PCR造成的GC偏向性。It can be seen from the above examples that the method provided by the present invention is used to build and sequence mRNAs. There is no PCR process in the library building process, which reduces the generation of duplicate sequences and can effectively improve the data utilization rate. Save sequencing costs. And because there is no PCR process, the GC bias caused by PCR is reduced.
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。In the description of this specification, descriptions with reference to the terms "one embodiment", "some embodiments", "examples", "specific examples", or "some examples" etc. mean specific features described in conjunction with the embodiment or example , Structure, materials or features are included in at least one embodiment or example of the present invention. In this specification, the schematic representations of the above terms do not necessarily refer to the same embodiment or example. Moreover, the described specific features, structures, materials or characteristics can be combined in any one or more embodiments or examples in a suitable manner. In addition, those skilled in the art can combine and combine the different embodiments or examples and the characteristics of the different embodiments or examples described in this specification without contradicting each other.
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。Although the embodiments of the present invention have been shown and described above, it can be understood that the above-mentioned embodiments are exemplary and should not be construed as limiting the present invention. Those of ordinary skill in the art can comment on the foregoing within the scope of the present invention. The embodiment undergoes changes, modifications, substitutions and modifications.
Claims (18)
Translated fromChinesePriority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2019/076697WO2020177012A1 (en) | 2019-03-01 | 2019-03-01 | Nucleic acid sequence for direct rna library construction, method for directly constructing sequencing library based on rna samples, and use thereof |
| CN201980087580.3ACN113574181A (en) | 2019-03-01 | 2019-03-01 | Nucleic acid sequence for direct RNA library construction, method and application for direct construction of sequencing library based on RNA samples |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2019/076697WO2020177012A1 (en) | 2019-03-01 | 2019-03-01 | Nucleic acid sequence for direct rna library construction, method for directly constructing sequencing library based on rna samples, and use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020177012A1true WO2020177012A1 (en) | 2020-09-10 |
Family
ID=72336866
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2019/076697CeasedWO2020177012A1 (en) | 2019-03-01 | 2019-03-01 | Nucleic acid sequence for direct rna library construction, method for directly constructing sequencing library based on rna samples, and use thereof |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN113574181A (en) |
| WO (1) | WO2020177012A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113718023A (en)* | 2021-08-23 | 2021-11-30 | 普瑞基准生物医药(苏州)有限公司 | Prokaryotic ribonucleic acid direct sequencing method based on bovine nanopore sequencing technology |
| CN114410744A (en)* | 2022-01-27 | 2022-04-29 | 深圳安吉康尔医学检验实验室 | Method for processing sample, nucleic acid extraction method and library thereof |
| CN114507728A (en)* | 2022-03-03 | 2022-05-17 | 苏州贝康医疗器械有限公司 | Capture primer and application thereof |
| CN119776345A (en)* | 2024-12-26 | 2025-04-08 | 北京普译生物科技有限公司 | Connecting bridge and library construction method of RNA library |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114582419B (en)* | 2022-01-29 | 2023-02-10 | 苏州大学 | Sliding window based gene sequence poly A tail extraction method |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009148617A2 (en)* | 2008-06-04 | 2009-12-10 | The Salk Institute For Biological Studies | Grepseq: an almost inexhaustible, cost-effective, high-throughput protocol for the generation of selector sequences |
| CN103649335A (en)* | 2011-05-04 | 2014-03-19 | Htg分子诊断有限公司 | Quantitative nuclease protection assay (QNPA) and sequencing (QNPS) improvements |
| CN104160040A (en)* | 2012-03-06 | 2014-11-19 | 伊鲁米纳剑桥有限公司 | Improved methods of nucleic acid sequencing |
| CN105985945A (en)* | 2015-01-30 | 2016-10-05 | 深圳华大基因研究院 | mRNA fragmentation method and method for constructing sequencing library based on same |
| CN107636163A (en)* | 2015-04-29 | 2018-01-26 | 加利福尼亚大学董事会 | Compositions and methods for constructing strand-specific cDNA libraries |
- 2019
- 2019-03-01WOPCT/CN2019/076697patent/WO2020177012A1/ennot_activeCeased
- 2019-03-01CNCN201980087580.3Apatent/CN113574181A/enactivePending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009148617A2 (en)* | 2008-06-04 | 2009-12-10 | The Salk Institute For Biological Studies | Grepseq: an almost inexhaustible, cost-effective, high-throughput protocol for the generation of selector sequences |
| CN103649335A (en)* | 2011-05-04 | 2014-03-19 | Htg分子诊断有限公司 | Quantitative nuclease protection assay (QNPA) and sequencing (QNPS) improvements |
| CN104160040A (en)* | 2012-03-06 | 2014-11-19 | 伊鲁米纳剑桥有限公司 | Improved methods of nucleic acid sequencing |
| CN105985945A (en)* | 2015-01-30 | 2016-10-05 | 深圳华大基因研究院 | mRNA fragmentation method and method for constructing sequencing library based on same |
| CN107636163A (en)* | 2015-04-29 | 2018-01-26 | 加利福尼亚大学董事会 | Compositions and methods for constructing strand-specific cDNA libraries |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113718023A (en)* | 2021-08-23 | 2021-11-30 | 普瑞基准生物医药(苏州)有限公司 | Prokaryotic ribonucleic acid direct sequencing method based on bovine nanopore sequencing technology |
| CN114410744A (en)* | 2022-01-27 | 2022-04-29 | 深圳安吉康尔医学检验实验室 | Method for processing sample, nucleic acid extraction method and library thereof |
| CN114507728A (en)* | 2022-03-03 | 2022-05-17 | 苏州贝康医疗器械有限公司 | Capture primer and application thereof |
| CN114507728B (en)* | 2022-03-03 | 2024-03-22 | 苏州贝康医疗器械有限公司 | Capturing primer and application thereof |
| CN119776345A (en)* | 2024-12-26 | 2025-04-08 | 北京普译生物科技有限公司 | Connecting bridge and library construction method of RNA library |
Also Published As
| Publication number | Publication date |
|---|---|
| CN113574181A (en) | 2021-10-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20240352507A1 (en) | Method for increasing throughput of single molecule sequencing by concatenating short dna fragments | |
| WO2020177012A1 (en) | Nucleic acid sequence for direct rna library construction, method for directly constructing sequencing library based on rna samples, and use thereof | |
| TWI742059B (en) | DNA amplification method | |
| TWI837127B (en) | Single tube bead-based dna co-barcoding for accurate and cost-effective sequencing, haplotyping, and assembly | |
| CN106795514B (en) | Bubble linker and its application in nucleic acid library construction and sequencing | |
| CN108220392A (en) | Methods for Enrichment and Determination of Target Nucleotide Sequences | |
| US20220033811A1 (en) | Method and kit for preparing complementary dna | |
| JP7641118B2 (en) | Probes and methods for enriching target regions using same for high-throughput sequencing | |
| US20150087556A1 (en) | COMPOSITIONS AND METHODS FOR MAKING cDNA LIBRARIES FROM SMALL RNAs | |
| CN112359093B (en) | Method and kit for preparing and expressing and quantifying free miRNA library in blood | |
| CN105602939A (en) | DNA amplification method | |
| WO2020252720A1 (en) | Method for constructing library on basis of rna samples, and use thereof | |
| WO2021253372A1 (en) | High-compatibility pcr-free library building and sequencing method | |
| CN102559864B (en) | Particle capable of performing mono-molecular nucleic acid amplification and preparation method as well as application thereof | |
| CN117106873A (en) | Single-cell multi-omics parallel sequencing method and its application based on third-generation sequencing platform | |
| CN109750086B (en) | Construction method of single-chain circular library | |
| CN115874291A (en) | Method for marking and simultaneously detecting DNA (deoxyribonucleic acid) and RNA (ribonucleic acid) molecules in sample | |
| WO2025000136A1 (en) | Method for preparing strand-specific library for rapid detection of various types of rnas, and high-throughput sequencing technique | |
| US20200370108A1 (en) | Methods and compositions for selecting and amplifying dna targets in a single reaction mixture | |
| WO2014086037A1 (en) | Method for constructing nucleic acid sequencing library and applications thereof | |
| US20090305288A1 (en) | Methods for amplifying nucleic acids and for analyzing nucleic acids therewith | |
| KR20210079309A (en) | Barcoding of Nucleic Acids | |
| Su et al. | MASTR-seq: Multiplexed Analysis of Short Tandem Repeats with sequencing | |
| CN113355389B (en) | Method and application of targeted enrichment nucleic acid target region using CRISPR/Cas12a system | |
| CN117757895A (en) | Single-stranded DNA library construction kit and application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:19918033 Country of ref document:EP Kind code of ref document:A1 | |
| NENP | Non-entry into the national phase | Ref country code:DE | |
| 122 | Ep: pct application non-entry in european phase | Ref document number:19918033 Country of ref document:EP Kind code of ref document:A1 |