WO2019042470A1 - BLOCKER OF CD47/SIRPα AND APPLICATION THEREOF - Google Patents
BLOCKER OF CD47/SIRPα AND APPLICATION THEREOFDownload PDFInfo
- Publication number
- WO2019042470A1 WO2019042470A1PCT/CN2018/103974CN2018103974WWO2019042470A1WO 2019042470 A1WO2019042470 A1WO 2019042470A1CN 2018103974 WCN2018103974 WCN 2018103974WWO 2019042470 A1WO2019042470 A1WO 2019042470A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- group
- cancer
- alkyl
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0*C(CNc1c(*)c(*)c2*)*(C=C3*)c1c2C3=OChemical compound*C(CNc1c(*)c(*)c2*)*(C=C3*)c1c2C3=O0.000description6
- UEHLEGXZIBQJPB-UHFFFAOYSA-NCC(C1)NCCN1NChemical compoundCC(C1)NCCN1NUEHLEGXZIBQJPB-UHFFFAOYSA-N0.000description1
- YXFDUTUDVQNZIZ-UHFFFAOYSA-NCCOC(C(C(c1c2)=O)=CN(c(cc3)ccc3F)c1cc(-c1ccccc1)c2F)=OChemical compoundCCOC(C(C(c1c2)=O)=CN(c(cc3)ccc3F)c1cc(-c1ccccc1)c2F)=OYXFDUTUDVQNZIZ-UHFFFAOYSA-N0.000description1
- PWNNUBNYZAKFGX-UHFFFAOYSA-NCCOC(CC(c(cc(c(N)c1)N)c1N)=O)=OChemical compoundCCOC(CC(c(cc(c(N)c1)N)c1N)=O)=OPWNNUBNYZAKFGX-UHFFFAOYSA-N0.000description1
- IFNWESYYDINUHV-OLQVQODUSA-NC[C@H]1N[C@@H](C)CNC1Chemical compoundC[C@H]1N[C@@H](C)CNC1IFNWESYYDINUHV-OLQVQODUSA-N0.000description1
- LJKWSFFLEICUTH-UHFFFAOYSA-NCc(c(F)c1)cc(N(C=C2C(OCc3cccc(B(O)O)c3)=O)c(cc3)ccc3F)c1C2=OChemical compoundCc(c(F)c1)cc(N(C=C2C(OCc3cccc(B(O)O)c3)=O)c(cc3)ccc3F)c1C2=OLJKWSFFLEICUTH-UHFFFAOYSA-N0.000description1
- IYPZRUYMFDWKSS-UHFFFAOYSA-NNN1CCNCC1Chemical compoundNN1CCNCC1IYPZRUYMFDWKSS-UHFFFAOYSA-N0.000description1
- VZXRDKPYISFRMU-UHFFFAOYSA-NOC(C(C(c1c2)=O)=CN(c(cc3)ccc3F)c1cc(-c1ccccc1)c2F)=OChemical compoundOC(C(C(c1c2)=O)=CN(c(cc3)ccc3F)c1cc(-c1ccccc1)c2F)=OVZXRDKPYISFRMU-UHFFFAOYSA-N0.000description1
Images

















Classifications
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4741—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having oxygen as a ring hetero atom, e.g. tubocuraran derivatives, noscapine, bicuculline
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D215/54—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3
- C07D215/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3 with oxygen atoms in position 4
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D455/00—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine
- C07D455/03—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing quinolizine ring systems directly condensed with at least one six-membered carbocyclic ring, e.g. protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine
- C07D455/04—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing quinolizine ring systems directly condensed with at least one six-membered carbocyclic ring, e.g. protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing a quinolizine ring system condensed with only one six-membered carbocyclic ring, e.g. julolidine
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/056—Ortho-condensed systems with two or more oxygen atoms as ring hetero atoms in the oxygen-containing ring
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/06—Peri-condensed systems
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present inventionrelates to the field of medicinal chemistry; in particular, the present invention relates to compounds which block the interaction of SIRP ⁇ protein with CD47 and uses thereof.
- SIRP ⁇Signal regulatory protein ⁇
- SIRP ⁇is a widely expressed glycoprotein molecule, also known as a protein tyrosine phosphatase substrate containing the SHP-2 domain, and belongs to the transmembrane protein of the immunoglobulin superfamily.
- SIRP ⁇contains an Immune-receptor tyrosin-based inhibitory motif (ITIM). When cells are stimulated by growth factors, SIRP ⁇ can inhibit the growth factor activity by phosphorylation of ITIM. SIRP ⁇ can be widely expressed on the surface of myeloid cells such as macrophages and dendritic cells.
- CD47also known as Integrin-associated protein (IAP), belongs to the membrane protein of the immunoglobulin superfamily and is highly expressed on various tumor cell membranes. SIRP ⁇ is an important surface receptor of CD47. The CD47-SIRP ⁇ signal produced by the combination of the two has a negative regulatory effect in the immune system and is of great significance in the process of macrophage phagocytosis.
- SIRP ⁇When SIRP ⁇ binds to CD47, it leads to aggregation of receptor molecules leading to tyrosine phosphorylation and activation and inhibition of macrophage synaptophysin accumulation, in which SIRP ⁇ with phosphorylated ITIM can recruit and activate tyrosine
- the phosphorylases SHP-1 and SHP-2which transmit an inhibitory signal, thereby inhibiting the phagocytosis of macrophages, ultimately leading to immune escape of tumor cells. Therefore, blocking the binding of SIRP ⁇ to CD47 can restore the related functions of macrophages, and finally achieve the effect of treating tumors.
- the present inventionprovides the use of a compound of Formula I, or a pharmaceutically acceptable salt, prodrug, or solvate thereof, for the manufacture of a medicament for blocking the interaction of a SIRP ⁇ protein with CD47,
- Yis selected from N or C, wherein when Y is N, R 3 is absent;
- R 1is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 alkyl, halogen, optionally substituted C 1 -C 10 alkoxy, optionally substituted C 6 -C 10 aryl, optionally substituted Benzyl, nitro, CN;
- R 2is selected from the group consisting of hydrogen, C 1 -C 10 alkyl or cycloalkyl, halogen, C 1 -C 10 alkoxy, optionally substituted C 6 -C 10 aryl, NR a R b , Wherein R a and R b are independently selected from the group consisting of: hydrogen, optionally substituted C 1 -C 6 alkyl;
- R 1 and R 2may be joined to form Wherein n is an integer from 1 to 3;
- R 3is selected from the group consisting of hydrogen, halogen, optionally substituted C 1 -C 10 alkoxy, optionally substituted alkylthio;
- R 4is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 alkyl, optionally substituted C 3 -C 10 cycloalkyl, optionally substituted C 6 -C 10 aryl, optionally substituted benzyl ;
- R 3 and R 4form an optionally substituted six-membered ring, or an optionally substituted oxygen-containing or sulfur-containing six-membered heterocyclic ring;
- R 5is selected from the group consisting of: hydrogen, optionally substituted C 1 -C 10 alkyl, halogen;
- R 6is selected from the group consisting of: H, R c —COOH, R c —COOR d , R c —CONR 8 R 9 , optionally substituted hydroxy C 1 -C 3 alkyl;
- R cis absent or optionally substituted -(CH 2 ) m -, m is an integer selected from 1 to 3;
- R dis an optionally substituted C 1 -C 3 alkyl group;
- R 8 and R 9Independently selected from: H, optionally substituted C 1 -C 10 alkyl, optionally substituted C 6 -C 10 aryl;
- R 7is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 alkyl, halogen, amino.
- the compoundis as shown in Formula II,
- R 1is selected from the group consisting of: hydrogen, optionally substituted C 1 -C 10 alkyl, halogen, optionally substituted C 6 -C 10 aryl;
- R 2is selected from the group consisting of hydrogen, C 1 -C 3 alkyl, An optionally substituted C 6 -C 10 aryl group, NR a R b , wherein R a and R b are as defined above;
- R 3is selected from the group consisting of: hydrogen, halogen, optionally substituted C 1 -C 10 alkoxy;
- R 4is selected from the group consisting of: an optionally substituted cyclopropyl, an optionally substituted C 1 -C 6 alkyl group, an optionally substituted phenyl group, an optionally substituted benzyl group;
- R 3 and R 4form an optionally substituted six-membered ring
- R 7is selected from the group consisting of hydrogen and amino groups
- Yis selected from: C or N, wherein when Y is N, R 3 is absent.
- the compoundis as shown in Formula III,
- R 1is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 alkyl, halogen;
- R 2is selected from the group consisting of hydrogen
- R 7is selected from the group consisting of hydrogen and amino groups
- R 10is selected from the group consisting of: hydrogen, optionally substituted C 1 -C 10 alkyl;
- Zis selected from: C, O, S;
- R 6is as described above.
- R 6is R c -COOH or R c -COOR d ; wherein R c is absent or optionally substituted -(CH 2 ) m -, m is an integer selected from 1 to 3 R d is an optionally substituted C 1 -C 3 alkyl group; more preferably, R 6 is -COOH.
- the compoundis selected from the group consisting of:
- the compoundis selected from the group consisting of:
- the medicamentis for inhibiting a tumor, or preventing or treating an infection caused by a bacterium, a virus or a fungus, or treating an inflammatory disease.
- the tumorincludes, but is not limited to, melanoma, lung cancer (preferably non-small cell lung cancer), kidney cancer, ovarian cancer, prostate cancer, breast cancer, colon cancer, bone cancer, pancreatic cancer, skin cancer , head and neck cancer, uterine cancer, rectal cancer, anal cancer, stomach cancer, testicular cancer, fallopian tube cancer, endometrial cancer, cervical cancer, vaginal cancer, vulvar cancer, Hodgkin's disease, non-Hodgkin's lymphoma, esophagus Cancer, small intestine cancer, endocrine system cancer, thyroid cancer, parathyroid carcinoma, adrenal cancer, soft tissue sarcoma, urethral cancer, penile cancer, acute myeloid leukemia, chronic myelogenous leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, pediatric Solid tumor, lymphocytic lymphoma, bladder cancer, renal or ureteral cancer, renal pelvic cancer, central nervous system
- lung cancer
- the virusesinclude, but are not limited to, hepatitis viruses (types A, B, and C), herpes viruses, influenza viruses, adenoviruses, coronaviruses, measles viruses, dengue viruses, polioviruses, rabies viruses;
- the bacteriainclude, but are not limited to, Chlamydia, Rickettsia, Mycobacterium, Staphylococcus, Pneumococci, Vibrio cholerae, Clostridium tetanus;
- the fungusincludes, but is not limited to, Candida, Aspergillus, dermatitis;
- the inflammatory diseasesinclude, but are not limited to, ankylosing spondylitis, autoimmune hemolytic anemia, arthritis, myasthenia gravis, systemic lupus erythematosus, rheumatoid arthritis, pernicious anemia, polymyositis.
- the present inventionprovides a compound of the formula: or a pharmaceutically acceptable salt, prodrug or solvate thereof,
- R 1is selected from the group consisting of hydrogen, halogen (preferably F or Cl), optionally substituted C 1 -C 10 alkyl (preferably C 1 -C 6 alkyl, more preferably C 1 -C 3 alkyl), nitro, Optionally substituted C 6 -C 10 aryl (preferably phenyl), cyano;
- R 2is selected from the group consisting of: an optionally substituted substituted phenyl, butyl, morpholinyl, piperidinyl, piperazinyl, NR a R b ; R a and R b are independently selected from: hydrogen, optionally substituted C 1 -C 6 alkyl;
- R 4is selected from the group consisting of: an optionally substituted C 3 -C 8 cycloalkyl group (preferably an optionally substituted cyclohexyl group), an optionally substituted C 1 -C 6 alkyl group (for example, an optionally substituted butyl group), (CH 2 ) o - optionally substituted phenyl (preferable ), optionally replaced o is an integer selected from 0-2
- R 6is selected from the group consisting of: COOH, C(O)-OC 1 -C 4 alkyl (preferably COOCH 3 or COOCH 2 CH 3 ), CONR 8 R 9 , CONHC 6 H 5 , CH 2 OH;
- R 8 and R 9are independently selected from: H, optionally substituted C 1 -C 10 alkyl (preferably optionally substituted C 1 -C 6 alkyl), optionally substituted C 6 -C 10 aryl (preferably Optionally substituted phenyl).
- the inventionprovides a compound selected from the group consisting of:
- the present inventionprovides a pharmaceutical composition
- a pharmaceutical compositioncomprising the compound of the second or third aspect of the present invention, or a pharmaceutically acceptable salt, prodrug, solvate thereof, and optionally A pharmaceutically acceptable carrier or excipient.
- the pharmaceutical compositionis for inhibiting tumors, or preventing or treating infections caused by bacteria, viruses or fungi or treating inflammatory diseases.
- the tumorincludes, but is not limited to, melanoma, lung cancer (preferably non-small cell lung cancer), kidney cancer, ovarian cancer, prostate cancer, breast cancer, colon cancer, bone cancer, pancreatic cancer, skin cancer , head and neck cancer, uterine cancer, rectal cancer, anal cancer, stomach cancer, testicular cancer, fallopian tube cancer, endometrial cancer, cervical cancer, vaginal cancer, vulvar cancer, Hodgkin's disease, non-Hodgkin's lymphoma, esophagus Cancer, small intestine cancer, endocrine system cancer, thyroid cancer, parathyroid carcinoma, adrenal cancer, soft tissue sarcoma, urethral cancer, penile cancer, acute myeloid leukemia, chronic myelogenous leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, pediatric Solid tumor, lymphocytic lymphoma, bladder cancer, renal or ureteral cancer, renal pelvic cancer, central nervous system
- lung cancer
- the virusesinclude, but are not limited to, hepatitis viruses (types A, B, and C), herpes viruses, influenza viruses, adenoviruses, coronaviruses, measles viruses, dengue viruses, polioviruses, rabies viruses;
- the bacteriainclude, but are not limited to, Chlamydia, Rickettsia, Mycobacterium, Staphylococcus, Pneumococci, Vibrio cholerae, Clostridium tetanus;
- the fungusincludes, but is not limited to, Candida, Aspergillus, dermatitis;
- the inflammatory diseasesinclude, but are not limited to, ankylosing spondylitis, autoimmune hemolytic anemia, arthritis, myasthenia gravis, systemic lupus erythematosus, rheumatoid arthritis, pernicious anemia, polymyositis.
- the present inventionprovides a method of blocking binding of SIRP ⁇ to CD47, comprising blocking SIRP ⁇ by using the compound of the first, second or third aspect of the invention or the pharmaceutical composition of the fourth aspect The step of CD47 binding.
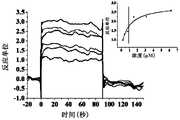
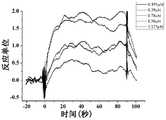
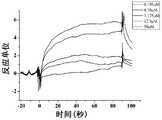
- Figure 1shows a SPR diagram of Compound 8 of the present invention and human SIRP ⁇ protein
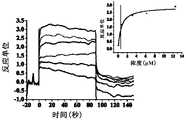
- Figure 2shows a SPR diagram of Compound 9 of the present invention and human SIRP ⁇ protein
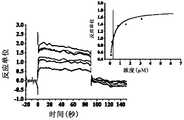
- Figure 3shows a SPR diagram of Compound 11 of the present invention and human SIRP ⁇ protein
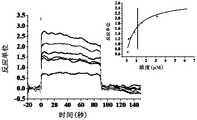
- Figure 4shows a SPR diagram of Compound 13 of the present invention and human SIRP ⁇ protein
- Figure 5shows a SPR diagram of Compound 14 of the present invention and human SIRP ⁇ protein
- Figure 6shows a SPR diagram of Compound 15 of the present invention and human SIRP ⁇ protein
- Figure 7shows a SPR map of Compound 16 of the present invention and human SIRP ⁇ protein
- Figure 8shows a SPR diagram of Compound 17 of the present invention and human SIRP ⁇ protein
- Figure 9shows a SPR diagram of Compound 18 of the present invention and human SIRP ⁇ protein
- Figure 10shows a SPR diagram of Compound 19 of the present invention and human SIRP ⁇ protein
- Figure 11shows a SPR diagram of Compound 20 of the present invention and human SIRP ⁇ protein
- Figure 12shows a SPR diagram of Compound 23 of the present invention and human SIRP ⁇ protein
- Figure 13shows a SPR map of Compound 24 of the present invention and human SIRP ⁇ protein
- Figure 14shows a SPR diagram of Compound 25 of the present invention and human SIRP ⁇ protein
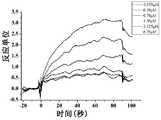
- Figure 15shows a SPR diagram of the compound D2 of the present invention and a human SIRP ⁇ protein
- Figure 16shows an SPR diagram of the compound D3 of the present invention and a human SIRP ⁇ protein
- Figure 17shows a SPR diagram of the compound D4 of the present invention and a human SIRP ⁇ protein
- Figure 18shows an SPR diagram of the compound D7 of the present invention and a human SIRP ⁇ protein
- Figure 19shows an SPR diagram of the compound D8 of the present invention and a human SIRP ⁇ protein
- Figure 20shows an SPR diagram of the compound D9 of the present invention and a human SIRP ⁇ protein
- Figure 21shows a SPR diagram of the compound D11 of the present invention and a human SIRP ⁇ protein
- Figure 22shows a SPR diagram of the compound D15 of the present invention and a human SIRP ⁇ protein
- Figure 23shows a SPR diagram of the compound D17 of the present invention and a human SIRP ⁇ protein
- Figure 24shows the SPR map of the compound D21 of the present invention and the human SIRP ⁇ protein.
- SIRP ⁇ proteinrefers to Signal Regulatory Protein ⁇ (SIRP ⁇ ), which is a broad class of glycoprotein molecules.
- CD47also known as integrin-related protein, is also a membrane protein belonging to the immunoglobulin superfamily. SIRP ⁇ is an important surface receptor for CD47, and the signals produced by the combination of the two have a negative regulatory effect in the immune system.
- alkylrefers to a saturated branched or straight chain alkyl group having a carbon chain length of from 1 to 10 carbon atoms, preferably an alkyl group of from 1 to 6 carbon atoms, more preferably from 1 to 3 carbon atoms. Alkyl. Examples of alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, heptyl, and the like.
- cycloalkylrefers to a saturated cyclic alkyl group having a carbon chain length of from 3 to 10 carbon atoms, preferably a cycloalkyl group having a cycloalkyl group of from 3 to 6 carbon atoms; for example but not limited to ) cyclopropyl.
- an alkyl or cycloalkyl groupcan be substituted, for example by a halogen.
- a halogen-substituted C 1 -C 3 alkyl groupis preferred.
- alkoxyrefers to an oxy group substituted with an alkyl group.
- Preferred alkoxy groupsare alkoxy groups of 1 to 10 carbon atoms, preferably 1 to 4 carbon atoms, more preferably 1 to 3 carbon atoms. Examples of alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, and the like.
- the alkoxy groupcan be substituted, for example by a halogen.
- a halogen-substituted C 1 -C 3 alkoxy groupis preferred.
- heterocyclylincludes, but is not limited to, a 5- or 6-membered heterocyclic group containing from 1 to 3 heteroatoms selected from O, S or N, including but not limited to furyl, thienyl, pyrrolyl, Pyrrolidinyl, pyrazolyl, imidazolyl, triazolyl, oxazolyl, pyranyl, pyridyl, pyrimidinyl, pyrazinyl, piperidinyl, morpholinyl, isodecyl and the like.
- a heterocyclic groupcan be substituted, for example, by a halogen.
- aminorefers to a group of the formula "NR a R b ", wherein R a and R b may be independently selected from H or an optionally substituted C 1 -C 6 alkyl group, for example, C 1 -C 6 haloalkyl. In specific embodiments herein the “amino” may be -NH 2.
- halogenrefers to fluoro, chloro, bromo and iodo. In a preferred embodiment, the halogen is chlorine or fluorine.
- Carboxyor “carboxylic acid group” refers to a group of the formula “R c -COOH” wherein R c is optionally substituted C 1 -C 3 alkyl. In a specific embodiment, “carboxy” or “carboxylic acid group” means "-COOH”.
- ester groupor “carboxylate group” refers to a group of the formula “R c -COOR d ", wherein R c is as defined above; R d is optionally substituted C 1 -C 3 alkyl.
- substituents which may be substituted for other groupsinclude, but are not limited to, hydroxyl, halogen, C 1 -C 6 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 6 alkoxy , phenyl, amino, nitro.
- the present inventionprovides a compound having a common parent core which is capable of binding SIRP ⁇ , thereby blocking the interaction of SIRP ⁇ with CD47. These compounds are low in toxicity and safe.
- the inventionprovides a compound of Formula I, or a pharmaceutically acceptable salt, prodrug, solvate thereof,
- Yis selected from N or C, wherein when Y is N, R 3 is absent;
- R 1is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 alkyl, halogen, optionally substituted C 1 -C 10 alkoxy, optionally substituted C 6 -C 10 aryl, optionally substituted Benzyl, nitro, CN;
- R 2is selected from the group consisting of hydrogen, C 1 -C 10 alkyl or cycloalkyl, halogen, C 1 -C 10 alkoxy, optionally substituted C 6 -C 10 aryl, NR a R b , Wherein R a and R b are independently selected from the group consisting of: hydrogen, optionally substituted C 1 -C 6 alkyl;
- R 1 and R 2may be joined to form Wherein n is an integer from 1 to 3;
- R 3is selected from the group consisting of hydrogen, halogen, optionally substituted C 1 -C 10 alkoxy, optionally substituted alkylthio;
- R 4is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 alkyl, optionally substituted C 3 -C 10 cycloalkyl, optionally substituted C 6 -C 10 aryl, optionally substituted benzyl ;
- R 3 and R 4form an optionally substituted six-membered ring, or an optionally substituted oxygen-containing or sulfur-containing six-membered heterocyclic ring;
- R 5is selected from the group consisting of: hydrogen, optionally substituted C 1 -C 10 alkyl, halogen;
- R 6is selected from the group consisting of: H, R c —COOH, R c —COOR d , R c —CONR 8 R 9 , optionally substituted hydroxy C 1 -C 3 alkyl;
- R cis absent or optionally substituted -(CH 2 ) m -, m is an integer selected from 1 to 3;
- R dis an optionally substituted C 1 -C 3 alkyl group;
- R 8 and R 9Independently selected from: H, optionally substituted C 1 -C 10 alkyl, optionally substituted C 6 -C 10 aryl;
- R 7is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 alkyl, halogen, amino.
- the compound of the inventionis as shown in Formula II,
- R 1is selected from the group consisting of: hydrogen, optionally substituted C 1 -C 10 alkyl, halogen, optionally substituted C 6 -C 10 aryl;
- R 2is selected from the group consisting of hydrogen, C 1 -C 3 alkyl, An optionally substituted C 6 -C 10 aryl group, NR a R b , wherein R a and R b are as defined above;
- R 3is selected from the group consisting of: hydrogen, halogen, optionally substituted C 1 -C 10 alkoxy;
- R 4is selected from the group consisting of: an optionally substituted cyclopropyl, an optionally substituted C 1 -C 6 alkyl group, an optionally substituted phenyl group, an optionally substituted benzyl group;
- R 3 and R 4form an optionally substituted six-membered ring
- R 7is selected from the group consisting of hydrogen and amino groups
- Yis selected from: C or N, wherein when Y is N, R 3 is absent.
- R 3 and R 4may form a ring to form a compound of the following formula III.
- R 1is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 alkyl, halogen;
- R 2is selected from the group consisting of hydrogen
- R 7is selected from the group consisting of hydrogen and amino groups
- R 10is selected from the group consisting of: hydrogen, optionally substituted C 1 -C 10 alkyl;
- Zis selected from: C, O, S;
- R 6is as defined above.
- the compound of the inventionis a compound of formula IV, or a pharmaceutically acceptable salt, prodrug, solvate thereof,
- R 1is selected from the group consisting of hydrogen, halogen (preferably F or Cl), optionally substituted C 1 -C 10 alkyl (preferably C 1 -C 6 alkyl, more preferably C 1 -C 3 alkyl), nitro, Optionally substituted C 6 -C 10 aryl (preferably phenyl), cyano;
- R 2is selected from the group consisting of: an optionally substituted substituted phenyl, butyl, morpholinyl, piperidinyl, piperazinyl, NR a R b ; R a and R b are independently selected from: hydrogen, optionally substituted C 1 -C 6 alkyl;
- R 4is selected from the group consisting of: an optionally substituted C 3 -C 8 cycloalkyl group (preferably an optionally substituted cyclohexyl group), an optionally substituted C 1 -C 6 alkyl group (for example, an optionally substituted butyl group), (CH 2 ) o - optionally substituted phenyl (preferable ), optionally replaced o is an integer selected from 0-2;
- R 6is selected from the group consisting of: COOH, C(O)-OC 1 -C 4 alkyl (preferably COOCH 3 or COOCH 2 CH 3 ), CONR 8 R 9 , CONHC 6 H 5 , CH 2 OH;
- R 8 and R 9are independently selected from: H, optionally substituted C 1 -C 10 alkyl (preferably optionally substituted C 1 -C 6 alkyl), optionally substituted C 6 -C 10 aryl (preferably Optionally substituted phenyl).
- the formula of the present inventionalso includes a compound in which a specific substituent in the compound specifically disclosed by the present invention is selected from other substituents in the formula; It will also be understood by those skilled in the art that such compounds are capable of obtaining and capable of having the same or similar activities as the compounds specifically disclosed in the Examples. Thus, in particular embodiments, all of the substituents in the formula of the invention may each be a corresponding group in any of the compounds specifically disclosed herein.
- the present inventionfurther provides a pharmaceutical composition
- a pharmaceutical compositioncomprising a therapeutically effective amount of a compound of the present invention or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
- Examples of pharmaceutically acceptable salts of the compounds of the inventioninclude, but are not limited to, inorganic and organic acid salts such as the hydrochloride, hydrobromide, sulfate, citrate, lactate, tartrate, maleate salts. , fumarate, mandelate and oxalate; and inorganic and formed with bases such as sodium hydroxy, tris(hydroxymethyl)aminomethane (TRIS, tromethamine) and N-methylglucamine Organic base salt. While each person's needs vary, one skilled in the art can determine the preferred dosage for every 10 active ingredients in the pharmaceutical compositions of the present invention.
- inorganic and organic acid saltssuch as the hydrochloride, hydrobromide, sulfate, citrate, lactate, tartrate, maleate salts. , fumarate, mandelate and oxalate
- basessuch as sodium hydroxy, tris(hydroxymethyl)aminomethane (TRIS, tromethamine) and N-methylglucamine Organic base
- a unit oral dosecan include from about 0.01 to 50 mg, preferably from about 0.1 to 10 mg, of a compound of the invention.
- the unit dosemay be administered one or more times per day in one or more tablets, each tablet containing from about 0.1 to 50 mg, conveniently from about 0.25 to 10 mg of the compound of the invention or a solvate thereof.
- the pharmaceutical composition of the present inventioncan be formulated into a form suitable for various administration routes, including but not limited to being formulated for parenteral, subcutaneous, intravenous, intramuscular, intraperitoneal, transdermal, oral, intrathecal, intracranial A form of administration, intranasal or topical, for the treatment of tumors and other diseases.
- the amount administeredis an amount effective to ameliorate or eliminate one or more conditions.
- an effective amountis an amount sufficient to ameliorate or in some way alleviate the symptoms associated with the disease.
- Such dosescan be administered as a single dose or can be administered according to an effective therapeutic regimen.
- the amount administeredmay cure the disease, but administration is usually to improve the symptoms of the disease.
- the dosage of the drugwill be determined by the age of the patient, the health and weight, the type of concurrent treatment, the frequency of treatment, and the desired therapeutic benefit.
- the pharmaceutical preparation of the present inventioncan be administered to any mammal as long as they can obtain the therapeutic effect of the compound of the present invention. Important among these mammals is humans.
- the compounds of the invention or pharmaceutical compositions thereofare useful for treating diseases mediated by the interaction of SIRP ⁇ with CD47.
- the disease mediated by the interaction of SIRP ⁇ and CD47is various cancers.
- the cancerincludes, but is not limited to, melanoma, lung cancer (preferably non-small cell lung cancer), kidney cancer, ovarian cancer, prostate cancer, breast cancer, colon cancer, bone cancer, pancreatic cancer, skin cancer, head and neck cancer, uterine cancer, Rectal cancer, anal cancer, gastric cancer, testicular cancer, fallopian tube cancer, endometrial cancer, cervical cancer, vaginal cancer, vulvar cancer, Hodgkin's disease, non-Hodgkin's lymphoma, esophageal cancer, small intestine cancer, endocrine system Cancer, thyroid cancer, parathyroid carcinoma, adrenal cancer, soft tissue sarcoma, urethral cancer, penile cancer, acute myeloid leukemia, chronic myelogenous leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, pediatric solid tumor, lymphocytic lymph Tumor, bladder cancer, kidney or ureteral cancer, renal pelvic cancer, central nervous system (CNS) tumor,
- the pharmaceutical or pharmaceutical preparation of the inventioncan be produced in a known manner. For example, it is manufactured by a conventional mixing, granulating, tableting, dissolving, or freeze drying process. In the manufacture of oral formulations, the mixture can be selectively milled by combining the solid adjuvant with the active compound. If necessary or necessary, after adding an appropriate amount of auxiliary agent, the mixture of particles is processed to obtain a tablet or tablet core.
- Suitable excipientsare, in particular, fillers, such as sugars such as lactose or sucrose, mannitol or sorbitol; cellulose preparations or calcium phosphates, such as tricalcium phosphate or calcium hydrogen phosphate; and binders, such as starch pastes, including corn starch.
- a disintegrating agentsuch as the above-mentioned starch, and carboxymethyl starch, crosslinked polyvinylpyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate may be added.
- Adjuvantsare especially flow regulators and lubricants, for example, silica, talc, stearates such as calcium magnesium stearate, stearic acid or polyethylene glycol.
- the tablet corecan be provided with a suitable coating that is resistant to gastric juice.
- a concentrated sugar solutioncan be applied.
- This solutionmay contain gum arabic, talc, polyvinylpyrrolidone, polyethylene glycol and/or titanium dioxide, a lacquer solution and a suitable organic solvent or solvent mixture.
- a suitable cellulose solutionsuch as cellulose acetate phthalic acid or hydroxypropyl methylcellulose phthalic acid can be used.
- a dye or pigmentcan be added to the coating of the tablet or tablet core. For example, a combination for identifying or for characterizing the dosage of an active ingredient.
- the pharmaceutical compositions of the present inventionare in a dosage form suitable for oral administration, including, but not limited to, tablets, solutions, suspensions, capsules, granules, powders.
- the inventionfurther provides a method of treating a disease mediated by the interaction of SIRP ⁇ with CD47, the method comprising administering to a subject in need thereof a compound or pharmaceutical composition of the invention.
- Methods of administrationinclude, but are not limited to, various methods of administration well known in the art, which can be determined based on the actual circumstances of the patient. These methods include, but are not limited to, parenteral, subcutaneous, intravenous, intramuscular, intraperitoneal, transdermal, buccal, intrathecal, intracranial, nasal or topical routes of administration.
- the inventionalso encompasses the use of a compound of the invention in the manufacture of a medicament for the prophylaxis or treatment of a disease mediated by the interaction of SIRP ⁇ with CD47.
- the present inventorshave for the first time discovered that a series of compounds can bind to SIRP ⁇ , thereby blocking the interaction of SIRP ⁇ with CD47;
- the compound of the present inventionis a highly effective and low-toxic SIRP ⁇ blocker, and thus has important academic value and practical significance.
- the product of the previous step(65 mg, 0.18 mmol) and anhydrous piperazine (62 mg, 0.72 mmol) were added to a 25 mL reaction flask, dissolved in pyridine, and slowly refluxed under stirring. After reacting for 8 hours, the pyridine was distilled off under reduced pressure to obtain The crude product was heated to reflux with 1 mL of ethanol, and after 30 minutes, it was cooled to room temperature, and the solid was precipitated, suction filtered, washed with a small amount of ethanol, and dried. 20 mg of a pale yellow solid was obtained in a yield of 27%. The yield was low, and the product was also obtained by esterification with the purchased bulk drug, with a yield of 66.3%.
- the ethyl ester product(100 mg, 0.26 mmol) was placed in a 25 mL reaction flask, 5% NaOH solution was added, and refluxed at 100 ° C. After 4 hours, the reaction was completed, pH was adjusted to 2 to 3, solids were precipitated, suction filtered, washed with water, and dried. Dry to give a pale yellow solid (yield: 55 mg).
- the ethyl ester product(0.315 mg, 0.87 mmol), cesium fluoride (0.143 mg, 0.87 mmol), phenylboronic acid (0.184 mg, 1.13 mmol) was placed in a 50 mL three-necked flask, dissolved in toluene, and deoxygenated for 15 minutes. The amount of palladium catalyst was deoxygenated for 15 minutes, and heated under reflux with argon.
- the ethyl ester product(500 mg, 1.37 mmol) was placed in a 100 mL reaction flask, and a 5% NaOH solution was added thereto, and refluxed at 100 ° C. After 4 hours, the reaction was completed, pH was adjusted to 2 to 3, solids were precipitated, suction filtered, washed with water, and dried. Dry to give 296 mg of a white solid (yield: 64.14%).
- the acetate (80 mg, 0.19 mmol) of the above-mentioned drug substancewas dissolved in methanol, and a methanol solution containing NaBH 4 (29 mg, 0.76 mmol) was added dropwise thereto, and the mixture was stirred for 1 hour, and a catalytic amount of p-toluenesulfonic acid was added thereto, followed by heating to reflux.
- the binding constant of the compound of the present invention to SIRP ⁇was determined by surface plasmon resonance.
- the following compounds 1-24 used in this examplewere purchased from Aladdin Corporation.
- the surface plasmon resonance (SPR) experimentwas performed using a Biacore T200 to determine the binding constant between the compound of the present invention and the human SIRP ⁇ protein.
- the specific experimental stepsare as follows: First, the purchased SIRP ⁇ protein (Beijing Yiqiao Shenzhou Biotechnology Co., Ltd.) was diluted to 50 ⁇ g/ml with sodium acetate pH 4.5, and the protein was coupled to the CM7 chip using an amino coupling kit to 10 ⁇ l. The flow rate of /min is combined for 600 s and the final coupling amount is approximately 15000 RU.
- the CM7 chipwas equilibrated to a steady state with a buffer (1.05 x PBS, 0.05% P20).
- the compoundwas then diluted with running buffer (1.05 x PBS, 0.05% P20, 1% DMSO) to a range of different concentrations, with running buffer flowing through the surface of the chip at a flow rate of 30 ⁇ L/min, a binding time of 90 s, and a dissociation time of 120 s.
- running buffer(1.05 x PBS, 0.05% P20, 1% DMSO)
- This exampleinvestigates the binding affinities of the 13 compounds of the invention to the SIRP ⁇ protein.
- Biacore T200CM5 sensor chip, maintenance chip, Biacore maintenance kit type 2, dimethyl sulfoxide (DMSO), filter head (0.22 ⁇ M), 10X HBS, amino coupling kit.
- DMSOdimethyl sulfoxide
- the proteinis coupled to the surface of the CM5 chip, and when the analyte flows through the surface with the solution, binding to the coupled protein produces a response signal, the response signal and the amount of analyte bound to the sensor chip. In direct proportion.
- the K D valuewas obtained by steady state fitting.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Virology (AREA)
- Oncology (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biotechnology (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Neurology (AREA)
- Diabetes (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Translated fromChineseDescription
Translated fromChinese本发明涉及药物化学领域;具体地说,本发明涉及阻断SIRPα蛋白与CD47相互作用的化合物及其应用。The present invention relates to the field of medicinal chemistry; in particular, the present invention relates to compounds which block the interaction of SIRPα protein with CD47 and uses thereof.
信号调节蛋白α(Signal regulatory proteinα,SIRPα)是一类广泛表达的糖蛋白分子,又称包含SHP-2结构域的蛋白酪氨酸磷酸酶底物,属于免疫球蛋白超家族的跨膜蛋白。SIRPα胞内含有免疫受体酪氨酸抑制基序(Immune-receptor tyrosin-based inhibitory motif,ITIM),当细胞受到生长因子刺激时,SIRPα可以通过ITIM发生磷酸化而抑制相应的生长因子的活性。SIRPα可以广泛表达于巨噬细胞和树突状细胞等髓系细胞表面。CD47又称整合素相关蛋白(Integrin-associated protein,IAP),同属于免疫球蛋白超家族的膜蛋白,主要在各种肿瘤细胞膜上高表达。SIRPα是CD47的一种重要的表面受体,两者结合后产生的CD47-SIRPα信号在免疫系统中存在负性调节作用,在巨噬细胞吞噬过程中具有重要意义。Signal regulatory protein α (SIRPα) is a widely expressed glycoprotein molecule, also known as a protein tyrosine phosphatase substrate containing the SHP-2 domain, and belongs to the transmembrane protein of the immunoglobulin superfamily. SIRPα contains an Immune-receptor tyrosin-based inhibitory motif (ITIM). When cells are stimulated by growth factors, SIRPα can inhibit the growth factor activity by phosphorylation of ITIM. SIRPα can be widely expressed on the surface of myeloid cells such as macrophages and dendritic cells. CD47, also known as Integrin-associated protein (IAP), belongs to the membrane protein of the immunoglobulin superfamily and is highly expressed on various tumor cell membranes. SIRPα is an important surface receptor of CD47. The CD47-SIRPα signal produced by the combination of the two has a negative regulatory effect in the immune system and is of great significance in the process of macrophage phagocytosis.
当SIRPα与CD47结合后,导致受体分子聚集引起酪氨酸的磷酸化和激活并且抑制巨噬细胞突触肌球蛋白的积累,在此过程中带有磷酸化ITIM的SIRPα可以募集并且激活酪氨酸磷酸酶SHP-1和SHP-2,传递抑制信号从而抑制巨噬细胞的吞噬作用,最终导致肿瘤细胞的免疫逃逸。因此阻断SIRPα与CD47结合可以恢复巨噬细胞的相关功能,最终达到治疗肿瘤的效果。When SIRPα binds to CD47, it leads to aggregation of receptor molecules leading to tyrosine phosphorylation and activation and inhibition of macrophage synaptophysin accumulation, in which SIRPα with phosphorylated ITIM can recruit and activate tyrosine The phosphorylases SHP-1 and SHP-2, which transmit an inhibitory signal, thereby inhibiting the phagocytosis of macrophages, ultimately leading to immune escape of tumor cells. Therefore, blocking the binding of SIRPα to CD47 can restore the related functions of macrophages, and finally achieve the effect of treating tumors.
目前已有三种针对于CD47的药物进入临床试验,其中包括两种单克隆抗体(Hu5F9-G4,CC-90002)和一种融合蛋白(TTI-621),临床效果较好。此外,还有1个处于IND申报阶段以及4个处于临床前阶段的抗体类药物。目前尚未有报道靶向于SIRPα的小分子阻断剂,而且大分子的抗体药物具有生产成本高、易产生免疫原性等缺点。因此研究生产成本低、组织渗透性好、不易产生免疫原性,具有更好的稳定性的小分子药物具有良好的应用前景。At present, three drugs targeting CD47 have entered clinical trials, including two monoclonal antibodies (Hu5F9-G4, CC-90002) and one fusion protein (TTI-621), which have good clinical effects. In addition, there is one antibody in the IND reporting phase and four in the preclinical phase. At present, small molecule blockers targeting SIRPα have not been reported, and macromolecular antibody drugs have disadvantages such as high production cost and easy generation of immunogenicity. Therefore, research on small-molecule drugs with low production cost, good tissue permeability, and low immunogenicity, and better stability has a good application prospect.
综上所述,研究开发靶向于SIRPα的小分子阻断剂作为阻断SIRPα与CD47相互作用的候选药物具有重要的临床意义和应用前景。In summary, the research and development of small molecule blockers targeting SIRPα has important clinical significance and application prospects as a candidate for blocking the interaction between SIRPα and CD47.
发明内容Summary of the invention
本发明的目的在于提供一种能够阻断SIRPα与CD47相互作用的小分子阻断剂。It is an object of the present invention to provide a small molecule blocker capable of blocking the interaction of SIRPα with CD47.
在第一方面,本发明提供式I所示化合物或其药学上可接受的盐、前药、溶剂化物在制备阻断SIRPα蛋白与CD47相互作用的药物中的用途,In a first aspect, the present invention provides the use of a compound of Formula I, or a pharmaceutically acceptable salt, prodrug, or solvate thereof, for the manufacture of a medicament for blocking the interaction of a SIRPα protein with CD47,

式中In the middle
X选自CH2、CHOH、C=O、C=S;X is selected from the group consisting of CH2 , CHOH, C=O, C=S;

Y选自N或C,其中当Y为N时,R3不存在;Y is selected from N or C, wherein when Y is N, R3 is absent;
R1选自:氢、任选取代的C1-C10烷基、卤素、任选取代的C1-C10烷氧基、任选取代的C6-C10芳基、任选取代的苄基、硝基、CN;R1 is selected from the group consisting of hydrogen, optionally substituted C1 -C10 alkyl, halogen, optionally substituted C1 -C10 alkoxy, optionally substituted C6 -C10 aryl, optionally substituted Benzyl, nitro, CN;
R2选自:氢、C1-C10烷基或环烷基、卤素、C1-C10烷氧基、任选取代的C6-C10芳基、NRaRb、

或者,R1和R2可以相连形成
R3选自:氢、卤素、任选取代的C1-C10烷氧基、任选取代的烷硫基;R3 is selected from the group consisting of hydrogen, halogen, optionally substituted C1 -C10 alkoxy, optionally substituted alkylthio;
R4选自:氢、任选取代的C1-C10烷基、任选取代的C3-C10环烷基、任选取代的C6-C10芳基、任选取代的苄基;R4 is selected from the group consisting of hydrogen, optionally substituted C1 -C10 alkyl, optionally substituted C3 -C10 cycloalkyl, optionally substituted C6 -C10 aryl, optionally substituted benzyl ;
或者,R3与R4形成任选取代的六元环,或任选取代的含氧或含硫六元杂环;Or, R3 and R4 form an optionally substituted six-membered ring, or an optionally substituted oxygen-containing or sulfur-containing six-membered heterocyclic ring;
R5选自:氢、任选取代的C1-C10烷基、卤素;R5 is selected from the group consisting of: hydrogen, optionally substituted C1 -C10 alkyl, halogen;
R6选自:H、Rc-COOH、Rc-COORd、Rc-CONR8R9、任选取代的羟基C1-C3烷基;R6 is selected from the group consisting of: H, Rc —COOH, Rc —COORd , Rc —CONR8 R9 , optionally substituted hydroxy C1 -C3 alkyl;
其中,Rc不存在或是任选取代的-(CH2)m-,m为选自1-3的整数;Rd是任选取代的C1-C3烷基;R8和R9独立选自:H、任选取代的C1-C10烷基、任选取代的C6-C10芳基;Wherein Rc is absent or optionally substituted -(CH2 )m -, m is an integer selected from 1 to 3; Rd is an optionally substituted C1 -C3 alkyl group; R8 and R9 Independently selected from: H, optionally substituted C1 -C10 alkyl, optionally substituted C6 -C10 aryl;
R7选自:氢、任选取代的C1-C10烷基、卤素、氨基。R7 is selected from the group consisting of hydrogen, optionally substituted C1 -C10 alkyl, halogen, amino.
在具体的实施方式中,所述化合物如式II所示,In a specific embodiment, the compound is as shown in Formula II,

式中In the middle
R1选自:氢、任选取代的C1-C10烷基、卤素、任选取代的C6-C10芳基;R1 is selected from the group consisting of: hydrogen, optionally substituted C1 -C10 alkyl, halogen, optionally substituted C6 -C10 aryl;
R2选自:氢、C1-C3烷基、
R3选自:氢、卤素、任选取代的C1-C10烷氧基;R3 is selected from the group consisting of: hydrogen, halogen, optionally substituted C1 -C10 alkoxy;
R4选自:任选取代的环丙基、任选取代的C1-C6烷基、任选取代的苯基、任选取代的苄基;R4 is selected from the group consisting of: an optionally substituted cyclopropyl, an optionally substituted C1 -C6 alkyl group, an optionally substituted phenyl group, an optionally substituted benzyl group;
或者,R3与R4形成任选取代的六元环;Or, R3 and R4 form an optionally substituted six-membered ring;
R7选自:氢、氨基;R7 is selected from the group consisting of hydrogen and amino groups;
Y选自:C或N,其中当Y为N时,R3不存在。Y is selected from: C or N, wherein when Y is N, R3 is absent.
在具体的实施方式中,所述化合物如式III所示,In a specific embodiment, the compound is as shown in Formula III,

式中In the middle
R1选自:氢、任选取代的C1-C10烷基、卤素;R1 is selected from the group consisting of hydrogen, optionally substituted C1 -C10 alkyl, halogen;
R2选自:氢、
R7选自:氢、氨基;R7 is selected from the group consisting of hydrogen and amino groups;
R10选自:氢、任选取代的C1-C10烷基;R10 is selected from the group consisting of: hydrogen, optionally substituted C1 -C10 alkyl;
Z选自:C、O、S;Z is selected from: C, O, S;
R6如上所述。R6 is as described above.
在优选的实施方式中,R6是Rc-COOH或Rc-COORd;其中,Rc不存在或是任选取代的-(CH2)m-,m为选自1-3的整数,Rd是任选取代的C1-C3烷基;更优选地,R6是-COOH。In a preferred embodiment, R6 is Rc -COOH or Rc -COORd ; wherein Rc is absent or optionally substituted -(CH2 )m -, m is an integer selected from 1 to 3 Rd is an optionally substituted C1 -C3 alkyl group; more preferably, R6 is -COOH.
在具体的实施方式中,所述化合物选自下组:In a specific embodiment, the compound is selected from the group consisting of:



以及as well as


在具体的实施方式中,所述化合物选自下组:In a specific embodiment, the compound is selected from the group consisting of:


在具体的实施方式中,所述药物用于抑制肿瘤、或预防或治疗细菌、病毒或真菌引起的感染或治疗炎性疾病。In a specific embodiment, the medicament is for inhibiting a tumor, or preventing or treating an infection caused by a bacterium, a virus or a fungus, or treating an inflammatory disease.
在具体的实施方式中,所述肿瘤包括但不限于:黑色素瘤、肺癌(优选非小细胞肺癌)、肾癌、卵巢癌、前列腺癌、乳腺癌、结肠癌、骨癌、胰腺癌、皮肤癌、头颈癌、子宫癌、直肠癌、肛门癌、胃癌、睾丸癌、输卵管癌、子宫内膜癌、子宫颈癌、阴道癌、外阴癌、何杰金氏病、非何杰金淋巴瘤、食道癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、急性髓性白血病、慢性髓性白血病、急性淋巴细胞白血病、慢性淋巴细胞白血病、小儿实体瘤、淋巴细胞性淋巴瘤、膀胱癌、肾或输尿管癌、肾盂癌、中枢神经系统(CNS)肿瘤、原发性CNS淋巴瘤、肿瘤血管生成、脊轴瘤、脑干神经胶质瘤、垂体腺瘤、卡波西肉瘤、表皮样癌、鳞状细胞癌、T细胞淋巴瘤;In a specific embodiment, the tumor includes, but is not limited to, melanoma, lung cancer (preferably non-small cell lung cancer), kidney cancer, ovarian cancer, prostate cancer, breast cancer, colon cancer, bone cancer, pancreatic cancer, skin cancer , head and neck cancer, uterine cancer, rectal cancer, anal cancer, stomach cancer, testicular cancer, fallopian tube cancer, endometrial cancer, cervical cancer, vaginal cancer, vulvar cancer, Hodgkin's disease, non-Hodgkin's lymphoma, esophagus Cancer, small intestine cancer, endocrine system cancer, thyroid cancer, parathyroid carcinoma, adrenal cancer, soft tissue sarcoma, urethral cancer, penile cancer, acute myeloid leukemia, chronic myelogenous leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, pediatric Solid tumor, lymphocytic lymphoma, bladder cancer, renal or ureteral cancer, renal pelvic cancer, central nervous system (CNS) tumor, primary CNS lymphoma, tumor angiogenesis, spinal axon, brain stem glioma, Pituitary adenoma, Kaposi's sarcoma, epidermoid carcinoma, squamous cell carcinoma, T-cell lymphoma;
所述病毒包括但不限于:肝炎病毒(甲型、乙型和丙型)、孢疹病毒、流感病毒、腺病毒、冠状病毒、麻疹病毒、登革热病毒、脊髓灰质炎病毒、狂犬病病毒;The viruses include, but are not limited to, hepatitis viruses (types A, B, and C), herpes viruses, influenza viruses, adenoviruses, coronaviruses, measles viruses, dengue viruses, polioviruses, rabies viruses;
所述细菌包括但不限于:衣原体、立克次氏体、分枝杆菌、葡萄球菌、肺炎球菌、霍乱弧菌、破伤风梭菌;The bacteria include, but are not limited to, Chlamydia, Rickettsia, Mycobacterium, Staphylococcus, Pneumococci, Vibrio cholerae, Clostridium tetanus;
所述真菌包括但不限于:假丝酵母、曲霉、皮炎芽酵母;The fungus includes, but is not limited to, Candida, Aspergillus, dermatitis;
所述炎性疾病包括但不限于:强直性脊柱炎、自身免疫性溶血性贫血、关节炎、重症肌无力、系统性红斑狼疮、类风湿性关节炎、恶性贫血、多肌炎。The inflammatory diseases include, but are not limited to, ankylosing spondylitis, autoimmune hemolytic anemia, arthritis, myasthenia gravis, systemic lupus erythematosus, rheumatoid arthritis, pernicious anemia, polymyositis.
在第二方面,本发明提供下式所示化合物,或其药学上可接受的盐、前药、溶剂化物,In a second aspect, the present invention provides a compound of the formula: or a pharmaceutically acceptable salt, prodrug or solvate thereof,

式中In the middle
R1选自:氢、卤素(优选F或Cl)、任选取代的C1-C10烷基(优选C1-C6烷基,更优选C1-C3烷基)、硝基、任选取代的C6-C10芳基(优选苯基)、氰基;R1 is selected from the group consisting of hydrogen, halogen (preferably F or Cl), optionally substituted C1 -C10 alkyl (preferably C1 -C6 alkyl, more preferably C1 -C3 alkyl), nitro, Optionally substituted C6 -C10 aryl (preferably phenyl), cyano;
R2选自:任选取代的取代苯基、丁基、吗啉基、哌啶基、哌嗪基、NRaRb;Ra和Rb独立选自:氢、任选取代的C1-C6烷基;R2 is selected from the group consisting of: an optionally substituted substituted phenyl, butyl, morpholinyl, piperidinyl, piperazinyl, NRa Rb ; Ra and Rb are independently selected from: hydrogen, optionally substituted C1 -C6 alkyl;
R4选自:任选取代的C3-C8环烷基(优选任选取代的环己基)、任选取代的C1-C6烷基 (例如,任选取代的丁基)、-(CH2)o-任选取代的苯基(优选

R6选自:COOH、C(O)-O-C1-C4烷基(优选COOCH3或COOCH2CH3)、CONR8R9、CONHC6H5、CH2OH;R6 is selected from the group consisting of: COOH, C(O)-OC1 -C4 alkyl (preferably COOCH3 or COOCH2 CH3 ), CONR8 R9 , CONHC6 H5 , CH2 OH;
X选自CH2、CH2CH3、CHOH、C=O、C=S;X is selected from the group consisting of CH2 , CH2 CH3 , CHOH, C=O, C=S;
环中
R8和R9独立选自:H、任选取代的C1-C10烷基(优选任选取代的C1-C6烷基)、任选取代的C6-C10芳基(优选任选取代的苯基)。R8 and R9 are independently selected from: H, optionally substituted C1 -C10 alkyl (preferably optionally substituted C1 -C6 alkyl), optionally substituted C6 -C10 aryl (preferably Optionally substituted phenyl).
在第三方面,本发明提供选自下组的化合物:In a third aspect, the invention provides a compound selected from the group consisting of:


在第四方面,本发明提供一种药物组合物,所述药物组合物含有本发明第二或第三方面所述的化合物或其药学上可接受的盐、前药、溶剂化物,以及任选的药学上可接受的载体或赋形剂。In a fourth aspect, the present invention provides a pharmaceutical composition comprising the compound of the second or third aspect of the present invention, or a pharmaceutically acceptable salt, prodrug, solvate thereof, and optionally A pharmaceutically acceptable carrier or excipient.
在优选的实施方式中,所述药物组合物用于抑制肿瘤、或预防或治疗细菌、病毒或真菌引起的感染或治疗炎性疾病。In a preferred embodiment, the pharmaceutical composition is for inhibiting tumors, or preventing or treating infections caused by bacteria, viruses or fungi or treating inflammatory diseases.
在优选的实施方式中,所述肿瘤包括但不限于:黑色素瘤、肺癌(优选非小细胞肺癌)、肾癌、卵巢癌、前列腺癌、乳腺癌、结肠癌、骨癌、胰腺癌、皮肤癌、头颈癌、子宫癌、直肠癌、肛门癌、胃癌、睾丸癌、输卵管癌、子宫内膜癌、子宫颈癌、阴道癌、外阴癌、何杰金氏病、非何杰金淋巴瘤、食道癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、急性髓性白血病、慢性髓性白血病、急性淋巴细胞白血病、慢性淋巴细胞白血病、小儿实体瘤、淋巴细胞性淋巴瘤、膀胱癌、肾或输尿管癌、肾盂癌、中枢神经系统(CNS)肿瘤、原发性CNS淋巴瘤、肿瘤血管生成、脊轴瘤、脑干神经胶质瘤、垂体腺瘤、卡波西肉瘤、表皮样癌、鳞状细胞癌、T细胞淋巴瘤;In a preferred embodiment, the tumor includes, but is not limited to, melanoma, lung cancer (preferably non-small cell lung cancer), kidney cancer, ovarian cancer, prostate cancer, breast cancer, colon cancer, bone cancer, pancreatic cancer, skin cancer , head and neck cancer, uterine cancer, rectal cancer, anal cancer, stomach cancer, testicular cancer, fallopian tube cancer, endometrial cancer, cervical cancer, vaginal cancer, vulvar cancer, Hodgkin's disease, non-Hodgkin's lymphoma, esophagus Cancer, small intestine cancer, endocrine system cancer, thyroid cancer, parathyroid carcinoma, adrenal cancer, soft tissue sarcoma, urethral cancer, penile cancer, acute myeloid leukemia, chronic myelogenous leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, pediatric Solid tumor, lymphocytic lymphoma, bladder cancer, renal or ureteral cancer, renal pelvic cancer, central nervous system (CNS) tumor, primary CNS lymphoma, tumor angiogenesis, spinal axon, brain stem glioma, Pituitary adenoma, Kaposi's sarcoma, epidermoid carcinoma, squamous cell carcinoma, T-cell lymphoma;
所述病毒包括但不限于:肝炎病毒(甲型、乙型和丙型)、孢疹病毒、流感病毒、腺病毒、冠状病毒、麻疹病毒、登革热病毒、脊髓灰质炎病毒、狂犬病病毒;The viruses include, but are not limited to, hepatitis viruses (types A, B, and C), herpes viruses, influenza viruses, adenoviruses, coronaviruses, measles viruses, dengue viruses, polioviruses, rabies viruses;
所述细菌包括但不限于:衣原体、立克次氏体、分枝杆菌、葡萄球菌、肺炎球菌、霍乱弧菌、破伤风梭菌;The bacteria include, but are not limited to, Chlamydia, Rickettsia, Mycobacterium, Staphylococcus, Pneumococci, Vibrio cholerae, Clostridium tetanus;
所述真菌包括但不限于:假丝酵母、曲霉、皮炎芽酵母;The fungus includes, but is not limited to, Candida, Aspergillus, dermatitis;
所述炎性疾病包括但不限于:强直性脊柱炎、自身免疫性溶血性贫血、关节炎、重症肌无力、系统性红斑狼疮、类风湿性关节炎、恶性贫血、多肌炎。The inflammatory diseases include, but are not limited to, ankylosing spondylitis, autoimmune hemolytic anemia, arthritis, myasthenia gravis, systemic lupus erythematosus, rheumatoid arthritis, pernicious anemia, polymyositis.
在第五方面,本发明提供一种阻断SIRPα与CD47结合的方法,包括利用本发明第一、第二或第三方面所述的化合物或第四方面所述的药物组合物阻断SIRPα与CD47结合的步骤。In a fifth aspect, the present invention provides a method of blocking binding of SIRPα to CD47, comprising blocking SIRPα by using the compound of the first, second or third aspect of the invention or the pharmaceutical composition of the fourth aspect The step of CD47 binding.
图1显示了本发明化合物8与人SIRPα蛋白的SPR图;Figure 1 shows a SPR diagram of
图2显示了本发明化合物9与人SIRPα蛋白的SPR图;Figure 2 shows a SPR diagram of
图3显示了本发明化合物11与人SIRPα蛋白的SPR图;Figure 3 shows a SPR diagram of Compound 11 of the present invention and human SIRPα protein;
图4显示了本发明化合物13与人SIRPα蛋白的SPR图;Figure 4 shows a SPR diagram of Compound 13 of the present invention and human SIRPα protein;
图5显示了本发明化合物14与人SIRPα蛋白的SPR图;Figure 5 shows a SPR diagram of
图6显示了本发明化合物15与人SIRPα蛋白的SPR图;Figure 6 shows a SPR diagram of
图7显示了本发明化合物16与人SIRPα蛋白的SPR图;Figure 7 shows a SPR map of Compound 16 of the present invention and human SIRPα protein;
图8显示了本发明化合物17与人SIRPα蛋白的SPR图;Figure 8 shows a SPR diagram of Compound 17 of the present invention and human SIRPα protein;
图9显示了本发明化合物18与人SIRPα蛋白的SPR图;Figure 9 shows a SPR diagram of Compound 18 of the present invention and human SIRPα protein;
图10显示了本发明化合物19与人SIRPα蛋白的SPR图;Figure 10 shows a SPR diagram of Compound 19 of the present invention and human SIRPα protein;
图11显示了本发明化合物20与人SIRPα蛋白的SPR图;Figure 11 shows a SPR diagram of
图12显示了本发明化合物23与人SIRPα蛋白的SPR图;Figure 12 shows a SPR diagram of Compound 23 of the present invention and human SIRPα protein;
图13显示了本发明化合物24与人SIRPα蛋白的SPR图;Figure 13 shows a SPR map of Compound 24 of the present invention and human SIRPα protein;
图14显示了本发明化合物25与人SIRPα蛋白的SPR图;Figure 14 shows a SPR diagram of
图15显示了本发明化合物D2与人SIRPα蛋白的SPR图;Figure 15 shows a SPR diagram of the compound D2 of the present invention and a human SIRPα protein;
图16显示了本发明化合物D3与人SIRPα蛋白的SPR图;Figure 16 shows an SPR diagram of the compound D3 of the present invention and a human SIRPα protein;
图17显示了本发明化合物D4与人SIRPα蛋白的SPR图;Figure 17 shows a SPR diagram of the compound D4 of the present invention and a human SIRPα protein;
图18显示了本发明化合物D7与人SIRPα蛋白的SPR图;Figure 18 shows an SPR diagram of the compound D7 of the present invention and a human SIRPα protein;
图19显示了本发明化合物D8与人SIRPα蛋白的SPR图;Figure 19 shows an SPR diagram of the compound D8 of the present invention and a human SIRPα protein;
图20显示了本发明化合物D9与人SIRPα蛋白的SPR图;Figure 20 shows an SPR diagram of the compound D9 of the present invention and a human SIRPα protein;
图21显示了本发明化合物D11与人SIRPα蛋白的SPR图;Figure 21 shows a SPR diagram of the compound D11 of the present invention and a human SIRPα protein;
图22显示了本发明化合物D15与人SIRPα蛋白的SPR图;Figure 22 shows a SPR diagram of the compound D15 of the present invention and a human SIRPα protein;
图23显示了本发明化合物D17与人SIRPα蛋白的SPR图;Figure 23 shows a SPR diagram of the compound D17 of the present invention and a human SIRPα protein;
图24显示了本发明化合物D21与人SIRPα蛋白的SPR图。Figure 24 shows the SPR map of the compound D21 of the present invention and the human SIRPα protein.
发明人经过广泛而深入的研究,出乎意料的发现了一系列具备沟通结构的化合物能够与人SIRPα蛋白结合,从而成为阻断SIRPα与CD47相互作用的小分子先导药物,进而为抗肿瘤药物的开发提供物质基础。在此基础上完成了本发明。After extensive and in-depth research, the inventors unexpectedly discovered that a series of compounds with a communication structure can bind to human SIRPα protein, and thus become a small molecule lead drug that blocks the interaction between SIRPα and CD47, and thus an antitumor drug. Development provides a material basis. The present invention has been completed on this basis.
除非另有定义,本文中使用的所有技术和科学术语具有与所公开的发明所属领域的技术人员的普遍理解相同的含义。为便于理解本发明,对本发明涉及的相关术语作如下定义,但本发明的范围并不限于这些具体的定义。Unless defined otherwise, all technical and scientific terms used herein have the same meaning meaning meaning For ease of understanding of the present invention, the related terms to which the present invention relates are defined as follows, but the scope of the present invention is not limited to these specific definitions.
术语定义Definition of Terms
本文中,“SIRPα蛋白”是指信号调节蛋白α(Signal regulatory proteinα,SIRPα),其是一类广泛表达的糖蛋白分子。CD47又称整合素相关蛋白,也是属于免疫球蛋白超家族的膜蛋白。SIRPα是CD47的一种重要的表面受体,两者结合后产生的信号在免疫系统中存在负性调节作用。As used herein, "SIRPα protein" refers to Signal Regulatory Protein α (SIRPα), which is a broad class of glycoprotein molecules. CD47, also known as integrin-related protein, is also a membrane protein belonging to the immunoglobulin superfamily. SIRPα is an important surface receptor for CD47, and the signals produced by the combination of the two have a negative regulatory effect in the immune system.
本文中,“烷基”是指碳链长度为1-10个碳原子的饱和的支链或直链烷基,优选的烷基为1-6个碳原子、更优选1-3个碳原子的烷基。烷基的例子包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、庚基、等。类似地,“环烷基”是指碳链长度为3-10个碳原子的饱和的环状烷基,优选的环烷基为3-6个碳原子的环烷基;例如(但不限于)环丙基。在具体的实施方式中,烷基或环烷基可以被取代,例如被卤素取代。在具体的实施方式中,优选卤素取代的C1-C3烷基。As used herein, "alkyl" refers to a saturated branched or straight chain alkyl group having a carbon chain length of from 1 to 10 carbon atoms, preferably an alkyl group of from 1 to 6 carbon atoms, more preferably from 1 to 3 carbon atoms. Alkyl. Examples of alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, heptyl, and the like. Similarly, "cycloalkyl" refers to a saturated cyclic alkyl group having a carbon chain length of from 3 to 10 carbon atoms, preferably a cycloalkyl group having a cycloalkyl group of from 3 to 6 carbon atoms; for example but not limited to ) cyclopropyl. In a specific embodiment, an alkyl or cycloalkyl group can be substituted, for example by a halogen. In a specific embodiment, a halogen-substituted C1 -C3 alkyl group is preferred.
本文中,“烷氧基”指被烷基取代的氧基。优选的烷氧基是1-10个碳原子,优选1-4个碳原子、更优选1-3个碳原子的烷氧基。烷氧基的例子包括但不限于甲氧基、乙氧基、丙氧 基等。在具体的实施方式中,烷氧基可以被取代,例如被卤素取代。在具体的实施方式中,优选卤素取代的C1-C3烷氧基。As used herein, "alkoxy" refers to an oxy group substituted with an alkyl group. Preferred alkoxy groups are alkoxy groups of 1 to 10 carbon atoms, preferably 1 to 4 carbon atoms, more preferably 1 to 3 carbon atoms. Examples of alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, and the like. In a specific embodiment, the alkoxy group can be substituted, for example by a halogen. In a specific embodiment, a halogen-substituted C1 -C3 alkoxy group is preferred.
本文所用“杂环基”包括但不限于含有1-3个选自O、S或N的杂原子的5元或6元杂环基团,包括但不限于呋喃基、噻吩基、吡咯基、吡咯烷基、吡唑基、咪唑基、三唑基、噁唑基、吡喃基、吡啶基、嘧啶基、吡嗪基、哌啶基、吗啉基、异吲哚基等。在具体的实施方式中,杂环基可以被取代,例如被卤素取代。As used herein, "heterocyclyl" includes, but is not limited to, a 5- or 6-membered heterocyclic group containing from 1 to 3 heteroatoms selected from O, S or N, including but not limited to furyl, thienyl, pyrrolyl, Pyrrolidinyl, pyrazolyl, imidazolyl, triazolyl, oxazolyl, pyranyl, pyridyl, pyrimidinyl, pyrazinyl, piperidinyl, morpholinyl, isodecyl and the like. In a specific embodiment, a heterocyclic group can be substituted, for example, by a halogen.
本文中,“氨基”是指结构式为“NRaRb”的基团,其中,Ra和Rb可独立选自H或任选取代的C1-C6烷基,例如C1-C6卤代烷基。在具体的实施方式中,本文所述的“氨基”可以是-NH2。Herein, "amino" refers to a group of the formula "NRa Rb ", wherein Ra and Rb may be independently selected from H or an optionally substituted C1 -C6 alkyl group, for example, C1 -C6 haloalkyl. In specific embodiments herein the "amino" may be -NH2.
本文中,“卤素”指氟、氯、溴和碘。在优选的实施方式中,卤素是氯或氟。As used herein, "halogen" refers to fluoro, chloro, bromo and iodo. In a preferred embodiment, the halogen is chlorine or fluorine.
在本文中,“羧基”或“羧酸基”是指结构式为“Rc-COOH”的基团,其中,Rc是任选取代的C1-C3烷基。在具体的实施方式中,“羧基”或“羧酸基”是指“-COOH”。As used herein, "carboxy" or "carboxylic acid group" refers to a group of the formula "Rc -COOH" wherein Rc is optionally substituted C1 -C3 alkyl. In a specific embodiment, "carboxy" or "carboxylic acid group" means "-COOH".
在本文中,“酯基”或“羧酸酯基”是指结构式为“Rc-COORd”的基团,其中,Rc如上所述;Rd是任选取代的C1-C3烷基。As used herein, "ester group" or "carboxylate group" refers to a group of the formula "Rc -COORd ", wherein Rc is as defined above; Rd is optionally substituted C1 -C3 alkyl.
本文所用的术语“任选取代的”表示在语法上被该术语所修饰的基团可以具有或不具有取代基,而取代基的类型和数量只要符合被取代基团的价键规则即可。在具体的实施方式中,可用来取代其它基团的取代基包括但不限于:羟基、卤素、C1-C6烷基、C3-C6环烷基、C1-C6烷氧基、苯基、氨基、硝基。The term "optionally substituted" as used herein means that a group which is grammatically modified by the term may or may not have a substituent, and the type and number of the substituents may be as long as they conform to the valence bond rule of the substituted group. In a specific embodiment, substituents which may be substituted for other groups include, but are not limited to, hydroxyl, halogen, C1 -C6 alkyl, C3 -C6 cycloalkyl, C1 -C6 alkoxy , phenyl, amino, nitro.
本发明的化合物Compound of the invention
本发明提供了一种具备共同母核的化合物,这些化合物能够结合SIRPα,从而阻断SIRPα与CD47的相互作用。这些化合物的毒性低,安全性好。The present invention provides a compound having a common parent core which is capable of binding SIRPα, thereby blocking the interaction of SIRPα with CD47. These compounds are low in toxicity and safe.
在具体的实施方式中,本发明提供式I所示化合物或其药学上可接受的盐、前药、溶剂化物,In a specific embodiment, the invention provides a compound of Formula I, or a pharmaceutically acceptable salt, prodrug, solvate thereof,

式中In the middle
X选自CH2、CHOH、C=O、C=S;X is selected from the group consisting of CH2 , CHOH, C=O, C=S;

Y选自N或C,其中当Y为N时,R3不存在;Y is selected from N or C, wherein when Y is N, R3 is absent;
R1选自:氢、任选取代的C1-C10烷基、卤素、任选取代的C1-C10烷氧基、任选取代的C6-C10芳基、任选取代的苄基、硝基、CN;R1 is selected from the group consisting of hydrogen, optionally substituted C1 -C10 alkyl, halogen, optionally substituted C1 -C10 alkoxy, optionally substituted C6 -C10 aryl, optionally substituted Benzyl, nitro, CN;
R2选自:氢、C1-C10烷基或环烷基、卤素、C1-C10烷氧基、任选取代的C6-C10芳基、NRaRb、

或者,R1和R2可以相连形成
R3选自:氢、卤素、任选取代的C1-C10烷氧基、任选取代的烷硫基;R3 is selected from the group consisting of hydrogen, halogen, optionally substituted C1 -C10 alkoxy, optionally substituted alkylthio;
R4选自:氢、任选取代的C1-C10烷基、任选取代的C3-C10环烷基、任选取代的C6-C10芳基、任选取代的苄基;R4 is selected from the group consisting of hydrogen, optionally substituted C1 -C10 alkyl, optionally substituted C3 -C10 cycloalkyl, optionally substituted C6 -C10 aryl, optionally substituted benzyl ;
或者,R3与R4形成任选取代的六元环,或任选取代的含氧或含硫六元杂环;Or, R3 and R4 form an optionally substituted six-membered ring, or an optionally substituted oxygen-containing or sulfur-containing six-membered heterocyclic ring;
R5选自:氢、任选取代的C1-C10烷基、卤素;R5 is selected from the group consisting of: hydrogen, optionally substituted C1 -C10 alkyl, halogen;
R6选自:H、Rc-COOH、Rc-COORd、Rc-CONR8R9、任选取代的羟基C1-C3烷基;R6 is selected from the group consisting of: H, Rc —COOH, Rc —COORd , Rc —CONR8 R9 , optionally substituted hydroxy C1 -C3 alkyl;
其中,Rc不存在或是任选取代的-(CH2)m-,m为选自1-3的整数;Rd是任选取代的C1-C3烷基;R8和R9独立选自:H、任选取代的C1-C10烷基、任选取代的C6-C10芳基;Wherein Rc is absent or optionally substituted -(CH2 )m -, m is an integer selected from 1 to 3; Rd is an optionally substituted C1 -C3 alkyl group; R8 and R9 Independently selected from: H, optionally substituted C1 -C10 alkyl, optionally substituted C6 -C10 aryl;
R7选自:氢、任选取代的C1-C10烷基、卤素、氨基。R7 is selected from the group consisting of hydrogen, optionally substituted C1 -C10 alkyl, halogen, amino.
在优选的实施方式中,本发明的化合物如式II所示,In a preferred embodiment, the compound of the invention is as shown in Formula II,

式中In the middle
R1选自:氢、任选取代的C1-C10烷基、卤素、任选取代的C6-C10芳基;R1 is selected from the group consisting of: hydrogen, optionally substituted C1 -C10 alkyl, halogen, optionally substituted C6 -C10 aryl;
R2选自:氢、C1-C3烷基、
R3选自:氢、卤素、任选取代的C1-C10烷氧基;R3 is selected from the group consisting of: hydrogen, halogen, optionally substituted C1 -C10 alkoxy;
R4选自:任选取代的环丙基、任选取代的C1-C6烷基、任选取代的苯基、任选取代的苄基;R4 is selected from the group consisting of: an optionally substituted cyclopropyl, an optionally substituted C1 -C6 alkyl group, an optionally substituted phenyl group, an optionally substituted benzyl group;
或者,R3与R4形成任选取代的六元环;Or, R3 and R4 form an optionally substituted six-membered ring;
R7选自:氢、氨基;R7 is selected from the group consisting of hydrogen and amino groups;
Y选自:C或N,其中当Y为N时,R3不存在。Y is selected from: C or N, wherein when Y is N, R3 is absent.
在具体的实施方式中,本发明的化合物中,R3和R4可以成环,从而形成下式III所示化合物,In a specific embodiment, in the compound of the present invention, R3 and R4 may form a ring to form a compound of the following formula III.

式中In the middle
R1选自:氢、任选取代的C1-C10烷基、卤素;R1 is selected from the group consisting of hydrogen, optionally substituted C1 -C10 alkyl, halogen;
R2选自:氢、
R7选自:氢、氨基;R7 is selected from the group consisting of hydrogen and amino groups;
R10选自:氢、任选取代的C1-C10烷基;R10 is selected from the group consisting of: hydrogen, optionally substituted C1 -C10 alkyl;
Z选自:C、O、S;Z is selected from: C, O, S;
R6如上限定。R6 is as defined above.
或者,本发明的化合物是式IV所示化合物,或其药学上可接受的盐、前药、溶剂化物,Alternatively, the compound of the invention is a compound of formula IV, or a pharmaceutically acceptable salt, prodrug, solvate thereof,

式中In the middle
R1选自:氢、卤素(优选F或Cl)、任选取代的C1-C10烷基(优选C1-C6烷基,更优选C1-C3烷基)、硝基、任选取代的C6-C10芳基(优选苯基)、氰基;R1 is selected from the group consisting of hydrogen, halogen (preferably F or Cl), optionally substituted C1 -C10 alkyl (preferably C1 -C6 alkyl, more preferably C1 -C3 alkyl), nitro, Optionally substituted C6 -C10 aryl (preferably phenyl), cyano;
R2选自:任选取代的取代苯基、丁基、吗啉基、哌啶基、哌嗪基、NRaRb;Ra和Rb独立选自:氢、任选取代的C1-C6烷基;R2 is selected from the group consisting of: an optionally substituted substituted phenyl, butyl, morpholinyl, piperidinyl, piperazinyl, NRa Rb ; Ra and Rb are independently selected from: hydrogen, optionally substituted C1 -C6 alkyl;
R4选自:任选取代的C3-C8环烷基(优选任选取代的环己基)、任选取代的C1-C6烷基(例如,任选取代的丁基)、-(CH2)o-任选取代的苯基(优选

R6选自:COOH、C(O)-O-C1-C4烷基(优选COOCH3或COOCH2CH3)、CONR8R9、CONHC6H5、CH2OH;R6 is selected from the group consisting of: COOH, C(O)-OC1 -C4 alkyl (preferably COOCH3 or COOCH2 CH3 ), CONR8 R9 , CONHC6 H5 , CH2 OH;
X选自CH2、CH2CH3、CHOH、C=O、C=S;X is selected from the group consisting of CH2 , CH2 CH3 , CHOH, C=O, C=S;
环中
R8和R9独立选自:H、任选取代的C1-C10烷基(优选任选取代的C1-C6烷基)、任选取代的C6-C10芳基(优选任选取代的苯基)。R8 and R9 are independently selected from: H, optionally substituted C1 -C10 alkyl (preferably optionally substituted C1 -C6 alkyl), optionally substituted C6 -C10 aryl (preferably Optionally substituted phenyl).
基于本发明的教导以及本领域的公知常识,本领域技术人员可以理解,本发明的通式还包括本发明具体公开的化合物中的具体取代基与通式中其它取代基选择构成的化合物;本领域技术人员也可以理解,这样的化合物能够获得并且能够具备与实施例具体公开的化合物相同或相似的活性。因此,在具体的实施方式中,本发明的通式中的所有取代基可以分别是本发明具体公开的任一化合物中的相应基团。Based on the teachings of the present invention and the common general knowledge in the art, those skilled in the art will appreciate that the formula of the present invention also includes a compound in which a specific substituent in the compound specifically disclosed by the present invention is selected from other substituents in the formula; It will also be understood by those skilled in the art that such compounds are capable of obtaining and capable of having the same or similar activities as the compounds specifically disclosed in the Examples. Thus, in particular embodiments, all of the substituents in the formula of the invention may each be a corresponding group in any of the compounds specifically disclosed herein.
在上述化合物的基础上,本发明进一步提供一种药物组合物,该组合物含有治疗有效量的本发明化合物或其药学上可接受的盐,以及药学上可接受的载体或赋形剂。Based on the above compounds, the present invention further provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of the present invention or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
本发明化合物的药学上可接受的盐的例子包括但不限于无机和有机酸盐,例如盐酸盐、氢溴酸盐、硫酸盐、柠檬酸盐、乳酸盐、酒石酸盐、马来酸盐、富马酸盐、扁桃酸盐和草酸盐;以及与碱例如钠羟基、三(羟基甲基)胺基甲烷(TRIS,氨基丁三醇)和N-甲基葡糖胺形成的无机和有机碱盐。虽然每个人的需求各不相同,本领域技术人员可确定本发明药物组合物中每10种活性成分的佳剂量。一般情况下,本发明的化合物或其药学上可接受的盐,对哺乳动物每天口服给药,药量按照约0.0025到50毫克/公斤体重。但好是每公斤口服给药约0.01到10毫克。例如,单位口服剂量可以包括约0.01到50毫克,好是约0.1到10毫克的本发明化合物。单位剂量可给予一次或多次,每天为一片或多片,每片含有约0.1到50毫克,合宜地约0.25到10毫克的本发明化合物或其溶剂化物。本发明的药物组合物可被配制成适合各种给药途径的制剂形式,包括但不限于被配制成用于肠外,皮下,静脉,肌肉,腹腔内,透皮,口腔,鞘内,颅内,鼻腔或外用途径给药的形式,用于治疗肿瘤和其他疾病。给药量是有效地改善或消除一个或多个病症的药量。对于特定疾病的治疗,有效量是足以改善或以某些方式减轻与疾病有关的症状的药量。这样的药量可作为单一剂量施用,或者可依据有效的治疗方案给药。给药量也许可治愈疾病,但是给药通常是为了改善疾病的症状。一般需要反复给药来实现所需的症状改善。药的剂量将根据病人的年龄,健康与体重,并行治疗的种类,治疗的频率,以及所需治疗效益来决定。本发明的药物制剂可以给予任何哺乳动物,只要他们能获得本发明化合物的治疗效果。在这些哺乳动物中重要的是人类。本发明的化合物或其药物组合物可用于治疗SIRPα与CD47相互作用介导的疾病。本文中,由SIRPα与CD47相互作用介导的疾病为各种癌症。所述癌症包括但不限于:黑色素瘤、肺癌(优选非小细胞肺癌)、肾癌、卵巢癌、前列腺癌、乳腺癌、结肠癌、骨癌、胰腺癌、皮肤癌、头颈癌、子宫癌、直肠癌、肛门癌、胃癌、睾丸癌、输卵管癌、子宫内膜癌、子宫颈癌、阴道癌、外阴癌、何杰金氏病、非何杰金淋巴瘤、食道癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、急性髓性白血病、慢性髓性白血病、急性淋巴细胞白血病、慢性淋巴细胞白血病、小儿实体瘤、淋巴细胞性淋巴瘤、膀胱癌、肾或输尿管癌、肾盂癌、中枢神经系统(CNS)肿瘤、原发性CNS淋巴瘤、肿瘤血管生成、脊轴瘤、脑干神经胶质瘤、垂体腺瘤、卡波西肉瘤、表皮样癌、鳞状细胞癌和T细胞淋巴瘤。Examples of pharmaceutically acceptable salts of the compounds of the invention include, but are not limited to, inorganic and organic acid salts such as the hydrochloride, hydrobromide, sulfate, citrate, lactate, tartrate, maleate salts. , fumarate, mandelate and oxalate; and inorganic and formed with bases such as sodium hydroxy, tris(hydroxymethyl)aminomethane (TRIS, tromethamine) and N-methylglucamine Organic base salt. While each person's needs vary, one skilled in the art can determine the preferred dosage for every 10 active ingredients in the pharmaceutical compositions of the present invention. In general, the compound of the present invention or a pharmaceutically acceptable salt thereof is orally administered to a mammal daily in an amount of from about 0.0025 to 50 mg/kg body weight. But it is good to take about 0.01 to 10 mg per kilogram of oral administration. For example, a unit oral dose can include from about 0.01 to 50 mg, preferably from about 0.1 to 10 mg, of a compound of the invention. The unit dose may be administered one or more times per day in one or more tablets, each tablet containing from about 0.1 to 50 mg, conveniently from about 0.25 to 10 mg of the compound of the invention or a solvate thereof. The pharmaceutical composition of the present invention can be formulated into a form suitable for various administration routes, including but not limited to being formulated for parenteral, subcutaneous, intravenous, intramuscular, intraperitoneal, transdermal, oral, intrathecal, intracranial A form of administration, intranasal or topical, for the treatment of tumors and other diseases. The amount administered is an amount effective to ameliorate or eliminate one or more conditions. For the treatment of a particular disease, an effective amount is an amount sufficient to ameliorate or in some way alleviate the symptoms associated with the disease. Such doses can be administered as a single dose or can be administered according to an effective therapeutic regimen. The amount administered may cure the disease, but administration is usually to improve the symptoms of the disease. Repeated administration is generally required to achieve the desired improvement in symptoms. The dosage of the drug will be determined by the age of the patient, the health and weight, the type of concurrent treatment, the frequency of treatment, and the desired therapeutic benefit. The pharmaceutical preparation of the present invention can be administered to any mammal as long as they can obtain the therapeutic effect of the compound of the present invention. Important among these mammals is humans. The compounds of the invention or pharmaceutical compositions thereof are useful for treating diseases mediated by the interaction of SIRPα with CD47. Herein, the disease mediated by the interaction of SIRPα and CD47 is various cancers. The cancer includes, but is not limited to, melanoma, lung cancer (preferably non-small cell lung cancer), kidney cancer, ovarian cancer, prostate cancer, breast cancer, colon cancer, bone cancer, pancreatic cancer, skin cancer, head and neck cancer, uterine cancer, Rectal cancer, anal cancer, gastric cancer, testicular cancer, fallopian tube cancer, endometrial cancer, cervical cancer, vaginal cancer, vulvar cancer, Hodgkin's disease, non-Hodgkin's lymphoma, esophageal cancer, small intestine cancer, endocrine system Cancer, thyroid cancer, parathyroid carcinoma, adrenal cancer, soft tissue sarcoma, urethral cancer, penile cancer, acute myeloid leukemia, chronic myelogenous leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia, pediatric solid tumor, lymphocytic lymph Tumor, bladder cancer, kidney or ureteral cancer, renal pelvic cancer, central nervous system (CNS) tumor, primary CNS lymphoma, tumor angiogenesis, spinal axon, brain stem glioma, pituitary adenoma, Kaposi Sarcoma, epidermoid carcinoma, squamous cell carcinoma, and T-cell lymphoma.
本发明的药物组合物或药物制剂可用已知的方式制造。例如,由传统的混合,制粒,制锭,溶解,或冷冻干燥过程制造。制造口服制剂时,可结合固体辅料和活性化合物,选择性研磨混合物。如果需要或必要时加入适量助剂后,加工颗粒混合物,获得片剂或锭剂芯。 合适的辅料特别是填料,例如糖类如乳糖或蔗糖,甘露醇或山梨醇;纤维素制剂或钙磷酸盐,例如磷酸三钙或磷酸氢钙;以及粘结剂,例如淀粉糊,包括玉米淀粉,小麦淀粉,大米淀粉,马铃薯淀粉,明胶,黄芪胶,甲基纤维素,羟丙基甲25基纤维素,羧甲基纤维素钠或聚乙烯吡咯烷酮。如果需要,可增加崩解剂,比如上面提到的淀粉,以及羧甲基淀粉,交联聚乙烯吡咯烷酮,琼脂,或褐藻酸或其盐,如海藻酸钠。辅助剂特别是流动调节剂和润滑剂,例如,硅石,滑石,硬脂酸盐类,如镁硬脂酸钙,硬脂酸或聚乙二醇。如果需要,可以給锭剂核芯提供可以抵抗胃液的合适包衣。为此,可以应用浓缩糖类溶液。这个溶液可以含有阿拉伯树胶,滑石,聚乙烯吡咯烷酮,聚乙二醇和/或二氧化钛,漆溶液和合适的有机溶剂或溶剂混合物。为了制备耐胃液的包衣,可使用适当的纤维素溶液,例如醋酸纤维素邻苯二甲酸或羟丙基甲基纤维素邻苯二甲酸。可向药片或锭剂核芯的包衣加入染料或色素。例如,用于识别或为了表征活性成分剂量的组合。本领域技术人员可以基于其专业知识以及实际需求,将本发明的药物组合物制备成任何剂型。例如,在具体的实施方式中,本发明的药物组合物是适于口服的5剂型,包括但不限于片剂、溶液剂、混悬液、胶囊剂、颗粒剂、粉剂。The pharmaceutical or pharmaceutical preparation of the invention can be produced in a known manner. For example, it is manufactured by a conventional mixing, granulating, tableting, dissolving, or freeze drying process. In the manufacture of oral formulations, the mixture can be selectively milled by combining the solid adjuvant with the active compound. If necessary or necessary, after adding an appropriate amount of auxiliary agent, the mixture of particles is processed to obtain a tablet or tablet core. Suitable excipients are, in particular, fillers, such as sugars such as lactose or sucrose, mannitol or sorbitol; cellulose preparations or calcium phosphates, such as tricalcium phosphate or calcium hydrogen phosphate; and binders, such as starch pastes, including corn starch. , wheat starch, rice starch, potato starch, gelatin, tragacanth, methyl cellulose, hydroxypropylmethyl 25-cell cellulose, sodium carboxymethyl cellulose or polyvinylpyrrolidone. If necessary, a disintegrating agent such as the above-mentioned starch, and carboxymethyl starch, crosslinked polyvinylpyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate may be added. Adjuvants are especially flow regulators and lubricants, for example, silica, talc, stearates such as calcium magnesium stearate, stearic acid or polyethylene glycol. If desired, the tablet core can be provided with a suitable coating that is resistant to gastric juice. For this purpose, a concentrated sugar solution can be applied. This solution may contain gum arabic, talc, polyvinylpyrrolidone, polyethylene glycol and/or titanium dioxide, a lacquer solution and a suitable organic solvent or solvent mixture. For the preparation of a gastric juice resistant coating, a suitable cellulose solution such as cellulose acetate phthalic acid or hydroxypropyl methylcellulose phthalic acid can be used. A dye or pigment can be added to the coating of the tablet or tablet core. For example, a combination for identifying or for characterizing the dosage of an active ingredient. One skilled in the art can prepare the pharmaceutical compositions of the present invention into any dosage form based on their expertise and actual needs. For example, in a specific embodiment, the pharmaceutical compositions of the present invention are in a dosage form suitable for oral administration, including, but not limited to, tablets, solutions, suspensions, capsules, granules, powders.
基于上述化合物和药物组合物,本发明进一步提供一种治疗SIRPα与CD47相互作用介导的疾病的方法,该方法包括给予需要的对象以本发明的化合物或药物组合物。给药方法包括但不限于本领域周知的各种给药方法,可根据患者的实际情况加以确定。这些方法包括但不限于肠外、皮下、静脉、肌肉、腹腔内、透皮、口腔、鞘内、颅内、鼻腔或外用途径给药。本发明也包括本发明化合物在制备预防或治疗SIRPα与CD47相互作用介导的疾病的药物中的用途。Based on the above compounds and pharmaceutical compositions, the invention further provides a method of treating a disease mediated by the interaction of SIRPα with CD47, the method comprising administering to a subject in need thereof a compound or pharmaceutical composition of the invention. Methods of administration include, but are not limited to, various methods of administration well known in the art, which can be determined based on the actual circumstances of the patient. These methods include, but are not limited to, parenteral, subcutaneous, intravenous, intramuscular, intraperitoneal, transdermal, buccal, intrathecal, intracranial, nasal or topical routes of administration. The invention also encompasses the use of a compound of the invention in the manufacture of a medicament for the prophylaxis or treatment of a disease mediated by the interaction of SIRPα with CD47.
本发明的优点:Advantages of the invention:
1.本发明首次发现了一系列化合物能够结合SIRPα,从而阻断SIRPα与CD47的相互作用;1. The present inventors have for the first time discovered that a series of compounds can bind to SIRPα, thereby blocking the interaction of SIRPα with CD47;
2.本发明的化合物是高效、低毒的SIRPα阻断剂,从而具备很重要的学术价值与现实意义。2. The compound of the present invention is a highly effective and low-toxic SIRPα blocker, and thus has important academic value and practical significance.
以下结合具体实施案例对本发明的技术方案进一步描述,但以下实施案例不构成对本发明的限制,所有依据本发明的原理和技术手段采用的各种施用方法,均属于本发明范围。下列实施案例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。The technical solutions of the present invention are further described below in conjunction with the specific embodiments, but the following embodiments are not intended to limit the present invention, and all the application methods according to the principles and technical means of the present invention are within the scope of the present invention. The experimental methods in the following examples that do not specify the specific conditions are usually in accordance with conventional conditions or according to the conditions recommended by the manufacturer. Percentages and parts are by weight unless otherwise stated.
实施例1. 1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(1-哌嗪基)-3-喹啉乙酸甲酯(D2)的合成Example 1. Synthesis of 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinolineacetic acid methyl ester (D2)

将沙拉沙星原料药(500mg,1.2mmol)放于50mL反应瓶中,加甲醇10mL和浓硫酸 1.5mL,加热回流,5小时后反应完成。在冰浴下,加NaOH中和至pH=8~9,加DCM萃取,有机相旋干,得到白色固体360mg,产率为76.1%。1H NMR(400MHz,DMSO-d6)δ8.39(s,1H),7.80(d,J=13.6Hz,1H),7.76(dd,J1=4.8Hz,J2=4Hz,2H),7.52(t,J=8.8Hz,2H),6.26(d,J=7.2Hz,1H),3.72(s,3H),2.88-2.86(m,4H),2.78-2.77(m,4H)。The sarafloxacin bulk drug (500 mg, 1.2 mmol) was placed in a 50 mL reaction flask, and 10 mL of methanol and 1.5 mL of concentrated sulfuric acid were added, and the mixture was heated to reflux, and the reaction was completed after 5 hours. In an ice bath, NaOH was added to neutralize to pH = 8 to 9, extracted with DCM, and then evaporated to dryness.1 H NMR (400MHz, DMSO- d 6) δ8.39 (s, 1H), 7.80 (d, J = 13.6Hz, 1H), 7.76 (dd, J1 = 4.8Hz, J2 = 4Hz, 2H), 7.52 ( t, J = 8.8 Hz, 2H), 6.26 (d, J = 7.2 Hz, 1H), 3.72 (s, 3H), 2.88-2.86 (m, 4H), 2.78-2.77 (m, 4H).
实施例2. 1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(1-哌嗪基)-3-喹啉乙酸乙酯(D3)的合成Example 2. Synthesis of ethyl p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinoline (D3)

1)2,4-二氯-5-氟苯甲酰乙酸乙酯的合成1) Synthesis of 2,4-dichloro-5-fluorobenzoylacetic acid ethyl acetate

先将NaH(466mg,24.3mmol)放入100mL干燥的三口瓶中,加无水THF和碳酸二乙酯溶解,氩气保护,在45℃下搅拌30分钟,缓慢滴加含2,4-二氯-5-氟苯乙酮(1g,4.9mmol)的碳酸二乙酯溶液,保持内温在50-55℃,滴毕,搅拌5小时,反应完成,将反应液倒入含醋酸的冰水中,乙酸乙酯萃取,过硅胶柱(PE:DCM=15:1),得到黄色油状物0.65g,产率为48%。投到量得到产物4.1g,产率为61%。LC-MS:m/z:279.05(M+H)+。First, NaH (466 mg, 24.3 mmol) was placed in a 100 mL dry three-necked flask, dissolved in anhydrous THF and diethyl carbonate, argon-protected, stirred at 45 ° C for 30 minutes, and slowly added with 2,4-di Chloro-5-fluoroacetophenone (1g, 4.9mmol) in diethyl carbonate solution, keep the internal temperature at 50-55 ° C, drip, stir for 5 hours, the reaction is completed, the reaction solution is poured into ice water containing acetic acid The mixture was extracted with ethyl acetate. EtOAc (EtOAc:EtOAc) The amount obtained was 4.1 g, and the yield was 61%. LC-MS: m / z: 279.05 (M + H) +.
2)2-(2,4-二氯-5-氟苯甲酰)-3-对氟苯胺基丙烯酸乙酯的合成2) Synthesis of 2-(2,4-dichloro-5-fluorobenzoyl)-3-p-fluoroanilinoethyl acrylate

向25mL反应瓶中加入上一步产物(0.3g,1.08mmol),原甲酸三乙酯(269μL,1.62mmol)及乙酸酐10mL,搅拌下缓缓升温至125℃反应3小时,减压除去生成的乙醇,向得到的固体物中加入无水乙醇3mL,在0℃左右滴加对氟苯胺(120mg,1.08mmol),加毕室温反应1.5小时,抽滤,烘干,得到白色固体0.278g,收率64.6%。1H NMR(400MHz,DMSO-d6)δ12.41(d,J=11.2Hz,1H),8.54(d,J=12.4Hz,1H),7.83(d,J=6.8Hz,1H),7.62(s,2H),7.48(d, J=9.2Hz,1H),7.30(t,J=8.8Hz,2H),3.96(q,J=7.2Hz,2H),0.98(t,J=6Hz,3H)。LC-MS:m/z:400.10(M+H)+。The product of the previous step (0.3 g, 1.08 mmol), triethyl orthoformate (269 μL, 1.62 mmol) and 10 mL of acetic anhydride were added to a 25 mL reaction flask, and the mixture was slowly heated to 125 ° C for 3 hours while stirring, and the resulting product was removed under reduced pressure. Ethanol, 3 mL of absolute ethanol was added to the obtained solid, p-fluoroaniline (120 mg, 1.08 mmol) was added dropwise at about 0 ° C, and the mixture was reacted at room temperature for 1.5 hours, filtered with suction, and dried to give a white solid. The rate is 64.6%.1 H NMR (400MHz, DMSO- d 6) δ12.41 (d, J = 11.2Hz, 1H), 8.54 (d, J = 12.4Hz, 1H), 7.83 (d, J = 6.8Hz, 1H), 7.62 (s, 2H), 7.48 (d, J = 9.2 Hz, 1H), 7.30 (t, J = 8.8 Hz, 2H), 3.96 (q, J = 7.2 Hz, 2H), 0.98 (t, J = 6 Hz, 3H). LC-MS: m/z: 400.10 (M+H)+ .
3)7-氯-6-氟-1-对氟苯基-1,4-二氢-4-氧代喹啉-3-乙酸乙酯的合成3) Synthesis of 7-chloro-6-fluoro-1-p-fluorophenyl-1,4-dihydro-4-oxoquinoline-3-acetic acid ethyl acetate

将上述产物(150mg,0.376mmol)、无水K2CO3(78mg,0.564mmol)和DMF 10mL投入25mL反应瓶中,在125℃反应2小时,减压旋干DMF,降温至室温,向固体中加水15mL,常温搅拌30分钟,抽滤,用少量水洗涤,烘干。得到浅黄色固体120mg,产率为88%。1H NMR(400MHz,DMSO-d6)δ8.47(s,1H),8.07(d,J=9.2Hz,1H),7.78(dd,J1=4.8Hz,J2=4Hz,2H),7.53(t,J=8.8Hz,2H),7.12(d,J=6Hz,1H),4.21(q,J=6.8Hz,2H),1.26(t,J=6.8Hz,3H)。LC-MS:m/z:364.10(M+H)+。The above product (150 mg, 0.376 mmol), anhydrous K2 CO3 (78 mg, 0.564 mmol) and
4)1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(1-哌嗪基)-3-喹啉乙酸乙酯4) ethyl p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinoline

将上一步产物(65mg,0.18mmol),无水哌嗪(62mg,0.72mmol)加入25mL反应瓶中,加入吡啶溶解,搅拌下缓缓升温回流,反应8小时后,减压蒸除吡啶,得到的粗产品加1mL乙醇加热回流,30分钟后冷却至室温,析出固体,抽滤,少量乙醇洗涤,烘干。得到淡黄色固体20mg,产率为27%。产率较低,之后用购买的原料药酯化也可获得产物,产率66.3%。1H NMR(400MHz,DMSO-d6)δ8.37(s,1H),7.81-7.75(m,3H),7.53(t,J=8.8Hz,2H),6.26(d,J=7.2Hz,1H),4.19(q,J=7.2Hz,2H),3.64-3.57(m,4H),3.02-2.86(m,4H),1.25(t,J=7.2Hz,3H)。LC-MS:m/z:364.10(M+H)+。HRMS(ESI)(m/z):[M+H]+calcd for C22H22F2N3O3,414.1629;found,414.1633.The product of the previous step (65 mg, 0.18 mmol) and anhydrous piperazine (62 mg, 0.72 mmol) were added to a 25 mL reaction flask, dissolved in pyridine, and slowly refluxed under stirring. After reacting for 8 hours, the pyridine was distilled off under reduced pressure to obtain The crude product was heated to reflux with 1 mL of ethanol, and after 30 minutes, it was cooled to room temperature, and the solid was precipitated, suction filtered, washed with a small amount of ethanol, and dried. 20 mg of a pale yellow solid was obtained in a yield of 27%. The yield was low, and the product was also obtained by esterification with the purchased bulk drug, with a yield of 66.3%.1 H NMR (400 MHz, DMSO-d6 ) δ 8.37 (s, 1H), 7.81 - 7.75 (m, 3H), 7.53 (t, J = 8.8 Hz, 2H), 6.26 (d, J = 7.2 Hz, 1H), 4.19 (q, J = 7.2 Hz, 2H), 3.64 - 3.57 (m, 4H), 3.02 - 2.86 (m, 4H), 1.25 (t, J = 7.2 Hz, 3H). LC-MS: m/z: 364.10 (M+H)+. HRMS (ESI) (m/z): [M+H]+ calcd for C22 H22 F2 N3 O3 , 414.1629; found, 414.1633.
实施例3. 1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(1-哌嗪基)-3-喹啉乙酰丁胺(D4)的合成Example 3. Synthesis of 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinoline acetobutylamine (D4)

将甲酯产物(50mg,0.13mmol)放于25mL反应瓶中,加正丁胺溶解,66℃下搅拌,6小时反应完成,旋干溶剂,得到黄色产物40mg,产率为72.7%。1H NMR(400MHz,CD3OD)δ8.63(s,1H),7.95(d,J=13.6Hz,1H),7.64(dd,J1=4.8Hz,J2=4.4Hz,2H),7.45(t,J=8.4Hz,2H),6.42(d,J=7.2Hz,1H),3.43(t,J=6.8Hz,2H),3.09-3.07(m,4H),3.01-2.98(m,4H), 1.65-1.58(m,2H),1.50-1.41(m,2H),0.98(t,J=7.6Hz,3H)。HRMS(ESI)(m/z):[M+H]+calcd for C24H27F2N4O2,441.2102;found,441.21The methyl ester product (50 mg, 0.13 mmol) was placed in a 25 mL reaction flask, dissolved in n-butylamine, stirred at 66 ° C, and the reaction was completed in 6 hours, and the solvent was evaporated to give a yellow product (yield: 72.7%).1 H NMR (400 MHz, CD3 OD) δ 8.63 (s, 1H), 7.95 (d, J = 13.6 Hz, 1H), 7.64 (dd, J1 = 4.8 Hz, J2 = 4.4 Hz, 2H), 7.45 ( t, J = 8.4 Hz, 2H), 6.42 (d, J = 7.2 Hz, 1H), 3.43 (t, J = 6.8 Hz, 2H), 3.09 - 3.07 (m, 4H), 3.01-2.98 (m, 4H) ), 1.65-1.58 (m, 2H), 1.50-1.41 (m, 2H), 0.98 (t, J = 7.6 Hz, 3H). HRMS (ESI) (m/z): [M+H]+calcd for C24 H27 F2 N4 O2 , 441.2102; found,441.21.
实施例4. 1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(哌嗪-1-基)-3-喹啉乙酰苯胺(D5)的合成Example 4. Synthesis of 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(piperazin-1-yl)-3-quinolineacetanilide (D5)
1)1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(4-(叔丁氧基羰基)哌嗪-1-基)-3-喹啉乙酸的合成1) 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(4-(tert-butoxycarbonyl)piperazin-1-yl)-3-quinolineacetic acid Synthesis

将上一步产物(200mg,0.6mmol),Bo c-哌嗪(440mg,2.4mmol)加入25mL反应瓶中,加入5mLNMP溶解,N2保护下115℃反应24小时后,减压蒸除溶剂,得到的粗产品加8mL甲醇加热回流,10分钟后冷却至室温,析出固体,抽滤,少量甲醇洗涤,烘干。得到白色固体190mg,产率为65.68%。产品未经纯化直接投下步反应。The product of the previous step (200 mg, 0.6 mmol), Bo c-piperazine (440 mg, 2.4 mmol) was added to a 25 mL reaction flask, dissolved in5 mL of NMP, and reacted at 115 ° C for 24 hours under N2 protection. The crude product was heated to reflux with 8 mL of methanol, and after 10 minutes, it was cooled to room temperature, and the solid was precipitated, suction filtered, washed with a small amount of methanol, and dried. 190 mg of a white solid were obtained in a yield of 65.68%. The product was directly subjected to a step reaction without purification.
2)1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(4-(叔丁氧基羰基)哌嗪-1-基)-3-喹啉乙酰苯胺的合成2) 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(4-(tert-butoxycarbonyl)piperazin-1-yl)-3-quinoline acetyl Synthesis of aniline

将上一步产物(190mg,0.4mmol)放于25mL反应瓶中,加苯胺(44mg,0.48mmol)、HATU(0.22mg,0.6mmol)、DIPEA(152mg、12mmol),加4mLDMF溶解,室温搅拌过夜,抽滤,得到白色产物125mg,产率为56.97%。产品未经纯化直接投下步反应。The product of the previous step (190 mg, 0.4 mmol) was placed in a 25 mL reaction flask, aniline (44 mg, 0.48 mmol), HATU (0.22 mg, 0.6 mmol), DIPEA (152 mg, 12 mmol), dissolved in 4 mL DMF and stirred at room temperature overnight. Filtration with suction gave a white product (yield: mp. The product was directly subjected to a step reaction without purification.
3)1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(哌嗪-1-基)-3-喹啉乙酰苯胺的合成3) Synthesis of 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(piperazin-1-yl)-3-quinolineacetanilide

将上一步产物(125mg,0.22mmol)加入25mL反应瓶中,加入5mLDCM溶解,缓慢滴入TFA 0.8mL,室温搅拌过夜。加入饱和碳酸氢钠溶液中和至碱性,DCM萃取,收集有机层,减压蒸除溶剂。得到无色固体67.7mg,产率为65.94%。1H NMR(400MHz,DMSO)δ12.33(s,1H),8.61(s,1H),7.95(d,J=13.6Hz,1H),7.83-7.79(m,2H),7.70(d,J=8Hz,2H),7.55(t,J=8Hz,2H),7.36(t,J=8Hz,2H),7.10(t,J=7.2Hz,1H),6.33(d,J=7.2Hz,1H),2.92(s,4H),2.78(s, 4H),7.70(d,J=8Hz,2H),1.23(s,1H)。LC-MS:m/z:460.38(M+H)+。The product of the previous step (125 mg, 0.22 mmol) was added to a 25 mL reaction flask, dissolved in 5 m LDCM, and slowly added dropwise to TFA 0.8 mL. The mixture was neutralized with a saturated aqueous solution of sodium hydrogencarbonate and evaporated to dryness. 67.7 mg of a colorless solid were obtained with a yield of 65.94%.1 H NMR (400MHz, DMSO) δ12.33 (s, 1H), 8.61 (d, J = 13.6Hz, 1H) (s, 1H), 7.95, 7.83-7.79 (m, 2H), 7.70 (d, J =8 Hz, 2H), 7.55 (t, J = 8 Hz, 2H), 7.36 (t, J = 8 Hz, 2H), 7.10 (t, J = 7.2 Hz, 1H), 6.33 (d, J = 7.2 Hz, 1H) ), 2.92 (s, 4H), 2.78 (s, 4H), 7.70 (d, J = 8 Hz, 2H), 1.23 (s, 1H). LC-MS: m/z: 460.38 (M+H)+ .
实施例5. 1-对氟苄基-6-氟-1,4-二氢-4-氧代-7-(1-哌嗪基)-3-喹啉乙酸(D9)的合成Example 5. Synthesis of 1-p-fluorobenzyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinolinic acid (D9)

1)2-(2,4-二氯-5-氟苯甲酰)-3-对氟苄胺基丙烯酸乙酯的合成1) Synthesis of 2-(2,4-dichloro-5-fluorobenzoyl)-3-p-fluorobenzylamino acrylate

得到白色固体420mg,收率56.5%。1H NMR(400MHz,DMSO-d6)δ11.13-11.08(m,1H),8.35(d,J=14.4Hz,1H),7.76(d,J=6.8Hz,1H),7.47-7.44(m,2H),7.38(d,J=9.2Hz,1H),7.24(t,J=9.2Hz,2H),4.71(d,J=6.4Hz,2H),3.88(q,J=6.8Hz,2H),0.92(t,J=7.2Hz,3H)。LC-MS:m/z:414.10(M+H)+。420 mg of a white solid were obtained in a yield of 56.5%.1 H NMR (400 MHz, DMSO-d6 ) δ 11.13-11.08 (m, 1H), 8.35 (d, J = 14.4 Hz, 1H), 7.76 (d, J = 6.8 Hz, 1H), 7.47-7.44 ( m, 2H), 7.38 (d, J = 9.2 Hz, 1H), 7.24 (t, J = 9.2 Hz, 2H), 4.71 (d, J = 6.4 Hz, 2H), 3.88 (q, J = 6.8 Hz, 2H), 0.92 (t, J = 7.2 Hz, 3H). LC-MS: m / z: 414.10 (M + H) +.
2)7-氯-6-氟-1-对氟苯苄基-1,4-二氢-4-氧代喹啉-3-乙酸乙酯的合成2) Synthesis of 7-chloro-6-fluoro-1-p-fluorophenylbenzyl-1,4-dihydro-4-oxoquinoline-3-acetic acid ethyl acetate

得到白色固体0.34g,收率88.8%。1H NMR(400MHz,DMSO-d6)δ8.91(s,1H),8.05(d,J=9.6Hz,1H),8.00(d,J=6Hz,1H),7.36-7.33(m,2H),7.22(t,J=8.8Hz,2H),5.70(s,2H),4.29(q,J=7.2Hz,2H),1.29(t,J=7.2Hz,3H)。LC-MS:m/z:378.10(M+H)+。0.34 g of a white solid was obtained in a yield of 88.8%.1 H NMR (400 MHz, DMSO-d6 ) δ 8.91 (s, 1H), 8.05 (d, J = 9.6 Hz, 1H), 8.00 (d, J = 6 Hz, 1H), 7.36-7.33 (m, 2H) ), 7.22 (t, J = 8.8 Hz, 2H), 5.70 (s, 2H), 4.29 (q, J = 7.2 Hz, 2H), 1.29 (t, J = 7.2 Hz, 3H). LC-MS: m / z: 378.10 (M + H) +.
3)7-氯-6-氟-1-对氟苯苄基-1,4-二氢-4-氧代喹啉-3-乙酸的合成3) Synthesis of 7-chloro-6-fluoro-1-p-fluorobenzylbenzyl-1,4-dihydro-4-oxoquinoline-3-acetic acid

将乙酯产物(100mg,0.26mmol)放于25mL反应瓶中,加5%NaOH溶液,100℃下回流,4小时后反应完成,调pH=2~3,析出固体,抽滤,水洗,烘干,得到淡黄色固体55mg,产率为59.8%。1H NMR(400MHz,DMSO-d6)δ14.67(s,1H),9.26(s,1H),8.25(d,J=6Hz,1H),8.22(d,J=9.2Hz,1H),7.41-7.37(m,2H),7.22(t,J=8.8Hz,2H),5.88(s,2H)。LC-MS:m/z: 350.10(M+H)+。The ethyl ester product (100 mg, 0.26 mmol) was placed in a 25 mL reaction flask, 5% NaOH solution was added, and refluxed at 100 ° C. After 4 hours, the reaction was completed, pH was adjusted to 2 to 3, solids were precipitated, suction filtered, washed with water, and dried. Dry to give a pale yellow solid (yield: 55 mg).1 H NMR (400MHz, DMSO- d 6) δ14.67 (s, 1H), 9.26 (s, 1H), 8.25 (d, J = 6Hz, 1H), 8.22 (d, J = 9.2Hz, 1H), 7.41-7.37 (m, 2H), 7.22 (t, J = 8.8 Hz, 2H), 5.88 (s, 2H). LC-MS: m/z: 350.10 (M+H)+ .
4)1-对氟苄基-6-氟-1,4-二氢-4-氧代-7-(1-哌嗪基)-3-喹啉乙酸4) 1-p-fluorobenzyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)-3-quinolineacetic acid

1H NMR(400MHz,DMSO-d6)δ9.20(s,1H),7.90(d,J=13.2Hz,1H),7.42-7.37(m,2H),7.22(t,J=9.2Hz,2H),7.06(d,J=7.2Hz,1H),5.84(s,2H),3.15-3.13(m,4H),2.92-2.89(m,4H)。HRMS(ESI)(m/z):[M+H]+calcd for C21H20F2N3O3,400.1472;found,400.1468。1 H NMR (400MHz, DMSO- d 6) δ9.20 (s, 1H), 7.90 (d, J = 13.2Hz, 1H), 7.42-7.37 (m, 2H), 7.22 (t, J = 9.2Hz, 2H), 7.06 (d, J = 7.2 Hz, 1H), 5.84 (s, 2H), 3.15 - 3.13 (m, 4H), 2.92 - 2.89 (m, 4H). HRMS (ESI) (m/z): [M+H] + calcd for C21 H20 F2 N3 O3 , 400.1472; found, 400.1468.
实施例6. 1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-苯基-3-喹啉乙酸(D11)的合成Example 6. Synthesis of 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-phenyl-3-quinolineacetic acid (D11)

将乙酯产物(0.315mg,0.87mmol),氟化铯(0.143mg,0.87mmol),苯硼酸(0.184mg,1.13mmol)放于50mL三口瓶中,加甲苯溶解,除氧15分钟后加入催化量的钯催化剂,再除氧15分钟,在氩气保护下,加热回流,24小时后,大部分原料反应完成,抽滤,滤液旋干,过柱,得到粗产物100mg,HRMS(ESI)(m/z):[M+H]+calcd for C24H18F2NO3,406.1255;found,406.1248。向粗品中加入5%NaOH溶液,加热回流,反应完成后用盐酸溶液调节pH=2~3,析出固体,抽滤,得到白色固体40mg,产率12.2%。1H NMR(400MHz,DMSO-d6)δ14.77(s,1H),8.80(s,1H),8.24(d,J=10Hz,1H),7.85(d,J=0.8Hz,2H),7.56-7.50(m,7H),7.15(d,J=5.6Hz,1H)。The ethyl ester product (0.315 mg, 0.87 mmol), cesium fluoride (0.143 mg, 0.87 mmol), phenylboronic acid (0.184 mg, 1.13 mmol) was placed in a 50 mL three-necked flask, dissolved in toluene, and deoxygenated for 15 minutes. The amount of palladium catalyst was deoxygenated for 15 minutes, and heated under reflux with argon. After 24 hours, most of the starting material was reacted, suction filtered, and the filtrate was evaporated to dryness to give the
实施例7. 1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(1-吗啉基)-3-喹啉乙酸(D13)的合成Example 7. Synthesis of 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-morpholinyl)-3-quinolineacetic acid (D13)
1)7-氯-6-氟-1-对氟苯基-1,4-二氢-4-氧代喹啉-3-乙酸的合成1) Synthesis of 7-chloro-6-fluoro-1-p-fluorophenyl-1,4-dihydro-4-oxoquinoline-3-acetic acid

将乙酯产物(500mg,1.37mmol)放于100mL反应瓶中,加5%NaOH溶液,100℃下回流,4小时后反应完成,调pH=2~3,析出固体,抽滤,水洗,烘干,得到白色固体296mg,产率为64.14%。1H NMR(400MHz,DMSO-d6)δ14.67(s,1H),9.26(s,1H),8.25(d,J=6Hz,1H),8.22(d,J=9.2Hz,1H),7.41-7.37(m,2H),7.22(t,J=8.8Hz,2H)。LC-MS:m/z:335.69。The ethyl ester product (500 mg, 1.37 mmol) was placed in a 100 mL reaction flask, and a 5% NaOH solution was added thereto, and refluxed at 100 ° C. After 4 hours, the reaction was completed, pH was adjusted to 2 to 3, solids were precipitated, suction filtered, washed with water, and dried. Dry to give 296 mg of a white solid (yield: 64.14%).1 H NMR (400MHz, DMSO- d6) δ14.67 (s, 1H), 9.26 (s, 1H), 8.25 (d, J = 6Hz, 1H), 8.22 (d, J = 9.2Hz, 1H), 7.41 -7.37 (m, 2H), 7.22 (t, J = 8.8 Hz, 2H). LC-MS: m/z: 335.69.
2)1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(1-吗啉基)-3-喹啉乙酸2) 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-morpholinyl)-3-quinolineacetic acid

将上一步产物(500mg,1.49mmol),吗啉(520mg,5.97mmol)加入100mL反应瓶中,加入20mLNMP溶解,搅拌下缓缓升温至115℃,反应24小时后,减压蒸除溶剂,得到的粗产品加20mL甲醇加热回流,30分钟后冷却至室温,析出固体,抽滤,少量乙醇洗涤,烘干。得到白色固体363mg,产率为63.08%。1H NMR(400MHz,CDCl3)δ14.79(s,1H),8.52(d,1H),7.38-7.27(m,5H),6.24(d,J=7.2Hz,1H),3.74(t,J=4.4Hz,4H),3.01(t,J=4.4Hz,4H)。LC-MS:m/z:387.15(M+H)+。The product of the previous step (500 mg, 1.49 mmol), morpholine (520 mg, 5.97 mmol) was added to a 100 mL reaction flask, dissolved in 20 mL of NMP, and slowly heated to 115 ° C under stirring. After reacting for 24 hours, the solvent was evaporated under reduced pressure. The crude product was heated and refluxed with 20 mL of methanol, and after 30 minutes, it was cooled to room temperature, and the solid was precipitated, suction filtered, washed with a small amount of ethanol, and dried. 363 mg of a white solid were obtained in a yield of 63.08%.1 H NMR (400MHz, CDCl 3 ) δ14.79 (s, 1H), 8.52 (d, 1H), 7.38-7.27 (m, 5H), 6.24 (d, J = 7.2Hz, 1H), 3.74 (t, J = 4.4 Hz, 4H), 3.01 (t, J = 4.4 Hz, 4H). LC-MS: m/z: 387.15 (M+H)+ .
实施例8. 1-对氟苯基-6-氟-1,4-二氢-4-氧代-7-(1-哌啶基)-3-喹啉乙酸(D14)的合成Example 8. Synthesis of 1-p-fluorophenyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperidinyl)-3-quinolineacetic acid (D14)

将上一步产物(100mg,0.3mmol),哌啶(100mg,1.2mmol)加入25mL反应瓶中,加入3mLDMF溶解,再加10d三乙胺,搅拌下缓缓升温至90℃,反应9小时后,减压蒸除溶剂,得到的粗产品加2mL乙醇加热回流,30分钟后冷却至室温,析出固体,抽滤,少量乙醇洗涤,烘干。得到白色固体56.2mg,产率为49.08%。1H NMR(400MHz,CDCl3)δ14.93(s,1H),8.54(s,1H),7.93(d,J=12.8,1H),7.35-7.17(m,4H),6.21(d,J=7.2Hz,1H),2.96(t,J=4.8Hz,4H),1.57-1.58(m,4H),1.50(t,J=7.2Hz,2H)。LC-MS:m/z:385.38(M+H)+。The product of the previous step (100 mg, 0.3 mmol), piperidine (100 mg, 1.2 mmol) was added to a 25 mL reaction flask, dissolved in 3 mL of DMF, added with 10 d of triethylamine, and slowly warmed to 90 ° C with stirring. After 9 hours of reaction, The solvent was evaporated under reduced pressure, and the obtained crude product was heated and refluxed with 2 mL of ethanol. After 30 minutes, the mixture was cooled to room temperature to precipitate a solid, which was filtered, washed with a small amount of ethanol, and dried. 56.2 mg of a white solid were obtained in a yield of 49.08%.1 H NMR (400MHz, CDCl 3 ) δ14.93 (s, 1H), 8.54 (s, 1H), 7.93 (d, J = 12.8,1H), 7.35-7.17 (m, 4H), 6.21 (d, J = 7.2 Hz, 1H), 2.96 (t, J = 4.8 Hz, 4H), 1.57-1.58 (m, 4H), 1.50 (t, J = 7.2 Hz, 2H). LC-MS: m / z: 385.38 (M + H) +.
实施例9. 6-氟-1-(4-对氟苯基)-7-(哌嗪基)-4-氧代-2,3-二氢喹啉(D21)的合成Example 9. Synthesis of 6-fluoro-1-(4-p-fluorophenyl)-7-(piperazinyl)-4-oxo-2,3-dihydroquinoline (D21)

将上述原料药的乙酸盐(80mg,0.19mmol)溶于甲醇中,滴加含有NaBH4(29mg,0.76mmol)的甲醇溶液,搅拌1小时,加入催化量的对甲苯磺酸,加热至回流,2小时后反应完成,降温,DCM萃取,过柱(DCM:MeOH=50:1),得到黄色固体40mg,产率为61.5%。1H NMR(400MHz,DMSO-d6)δ7.42-7.39(m,2H),7.35(d,J=11.2Hz,1H),7.33-7.29(m,2H),5.89(d,J=7.6Hz,1H),3.81(t,J=7.2Hz,2H),2.85-2.82(m,4H),2.75-2.73(m,4H),2.68(d,J=6.8Hz,2H)。LC-MS:m/z:342.20(M+H)+。The acetate (80 mg, 0.19 mmol) of the above-mentioned drug substance was dissolved in methanol, and a methanol solution containing NaBH4 (29 mg, 0.76 mmol) was added dropwise thereto, and the mixture was stirred for 1 hour, and a catalytic amount of p-toluenesulfonic acid was added thereto, followed by heating to reflux. After 2 hours, the reaction was completed, cooled, DCM was applied,jjjjjjjjjj1 H NMR (400 MHz, DMSO-d6 ) δ 7.42 - 7.39 (m, 2H), 7.35 (d, J = 11.2 Hz, 1H), 7.33 - 7.29 (m, 2H), 5.89 (d, J = 7.6 Hz, 1H), 3.81 (t, J = 7.2 Hz, 2H), 2.85-2.82 (m, 4H), 2.75-2.73 (m, 4H), 2.68 (d, J = 6.8 Hz, 2H). LC-MS: m / z: 342.20 (M + H) +.
实施例10.本发明化合物与SIRPα的结合常数的测定Example 10. Determination of Binding Constants of Compounds of the Invention and SIRPα
在本实施例中,采用表面等离子体共振测定本发明化合物与SIRPα的结合常数。In the present example, the binding constant of the compound of the present invention to SIRPα was determined by surface plasmon resonance.
本实施例中所用的以下化合物1-24购买于Aladdin公司。利用Biacore T200进行表面等离子体共振(SPR)实验,测定本发明化合物与人SIRPα蛋白之间的结合常数。具体实验步骤如下:首先将购买的SIRPα蛋白(北京义翘神州生物技术有限公司)用pH 4.5的醋酸钠稀释至50μg/ml,使用氨基偶联试剂盒将蛋白偶联在CM7芯片上,以10μl/min的流速结合600s,最终偶联量约为15000RU。偶联结束后,将CM7芯片用缓冲液(1.05×PBS,0.05%P20)平衡至稳定状态。随后用运行缓冲液(1.05×PBS,0.05%P20,1%DMSO)将化合物稀释至一系列不同浓度,随运行缓冲液流经芯片表面,流速30μL/min,结合时间90s,解离时间120s。最后数据用BIAevaluation2.0软件分析,利用稳态拟合得到KD值。下表1中列出了所测化合物的KD值。The following compounds 1-24 used in this example were purchased from Aladdin Corporation. The surface plasmon resonance (SPR) experiment was performed using a Biacore T200 to determine the binding constant between the compound of the present invention and the human SIRPα protein. The specific experimental steps are as follows: First, the purchased SIRPα protein (Beijing Yiqiao Shenzhou Biotechnology Co., Ltd.) was diluted to 50 μg/ml with sodium acetate pH 4.5, and the protein was coupled to the CM7 chip using an amino coupling kit to 10 μl. The flow rate of /min is combined for 600 s and the final coupling amount is approximately 15000 RU. After the end of the coupling, the CM7 chip was equilibrated to a steady state with a buffer (1.05 x PBS, 0.05% P20). The compound was then diluted with running buffer (1.05 x PBS, 0.05% P20, 1% DMSO) to a range of different concentrations, with running buffer flowing through the surface of the chip at a flow rate of 30 μL/min, a binding time of 90 s, and a dissociation time of 120 s. Finally BIAevaluation2.0 data analysis software, using the KD values obtained by fitting the steady state. The KD values of the tested compounds are listed in Table 1 below.
表1.本发明的小分子化合物与重组人SIRPα蛋白的结合亲和力Table 1. Binding affinities of the small molecule compounds of the invention to recombinant human SIRPα protein


实施例11.本发明的合成化合物与SIRPα的结合测定Example 11. Binding assay of synthetic compounds of the invention with SIRPα
本实施例研究了本发明的13个化合物与SIRPα蛋白的结合亲和力。This example investigates the binding affinities of the 13 compounds of the invention to the SIRPα protein.
本实施例采用的仪器及试剂为:The instruments and reagents used in this embodiment are:
Biacore T200、CM5传感芯片、维护芯片、Biacore维护试剂盒2型、二甲基亚砜(DMSO)、滤头(0.22μM)、10X HBS、氨基偶联试剂盒。Biacore T200, CM5 sensor chip, maintenance chip, Biacore
本实施例的实验原理为:The experimental principle of this embodiment is:
将蛋白偶联在CM5芯片表面上,当分析物随溶液流经该表面时,与偶联上的蛋白结合,则产生一定的响应信号,响应信号与结合在传感芯片上的分析物的量成正比。通过稳态拟合得到KD值。The protein is coupled to the surface of the CM5 chip, and when the analyte flows through the surface with the solution, binding to the coupled protein produces a response signal, the response signal and the amount of analyte bound to the sensor chip. In direct proportion. The KD value was obtained by steady state fitting.
本实施例的实验步骤为:The experimental steps of this embodiment are:
1.通过氨基偶联的方法将SIRPα蛋白偶联到CM5芯片表面;1. Coupling the SIRPα protein to the surface of the CM5 chip by amino coupling;
2.将不同浓度的化合物或蛋白随运行buffer(10mM HEPES,140mM NaCl和0.05%表面活性剂P20,pH 7.4)流经芯片表面,流速30μL/min,结合时间90s,解离时间120s。2. Different concentrations of compound or protein were flowed through the chip surface with a running buffer (10 mM HEPES, 140 mM NaCl and 0.05% surfactant P20, pH 7.4) at a flow rate of 30 μL/min, a binding time of 90 s, and a dissociation time of 120 s.
3.用Biacore T200Evaluation2.0软件分析实验结果。(所用蛋白:sirpα;偶联量:8276.1RU)3. Analyze the experimental results using Biacore T200Evaluation 2.0 software. (Used protein: sirpα; coupling amount: 8276.1RU)

在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。All documents mentioned in the present application are hereby incorporated by reference in their entirety in their entireties in the the the the the the the the the In addition, it should be understood that various modifications and changes may be made by those skilled in the art in the form of the present invention.
Claims (11)
Translated fromChinese






















Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710785796.X | 2017-09-04 | ||
| CN201710785796.XACN109422726B (en) | 2017-09-04 | 2017-09-04 | Blockers of CD47/SIRPα and their applications |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019042470A1true WO2019042470A1 (en) | 2019-03-07 |
Family
ID=65513313
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2018/103974CeasedWO2019042470A1 (en) | 2017-09-04 | 2018-09-04 | BLOCKER OF CD47/SIRPα AND APPLICATION THEREOF |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN109422726B (en) |
| WO (1) | WO2019042470A1 (en) |
Cited By (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111297856A (en)* | 2020-03-13 | 2020-06-19 | 黑龙江中医药大学 | Medicinal composition for resisting fungal infection and application thereof |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021011544A1 (en) | 2019-07-16 | 2021-01-21 | Gilead Sciences, Inc. | Hiv vaccines and methods of making and using |
| WO2021042046A1 (en)* | 2019-08-30 | 2021-03-04 | Trustees Of Dartmouth College | Membrane-active anti-bacterial compounds and uses thereof |
| WO2021076908A1 (en) | 2019-10-18 | 2021-04-22 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021087064A1 (en) | 2019-10-31 | 2021-05-06 | Forty Seven, Inc. | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021163064A2 (en) | 2020-02-14 | 2021-08-19 | Jounce Therapeutics, Inc. | Antibodies and fusion proteins that bind to ccr8 and uses thereof |
| CN115043907A (en)* | 2022-06-02 | 2022-09-13 | 中山大学 | Parent peptide or derived peptide with CD47/SIRP alpha blocking effect and application thereof |
| WO2022190058A1 (en) | 2021-03-12 | 2022-09-15 | Dcprime B.V. | Methods of vaccination and use of cd47 blockade |
| WO2022221304A1 (en) | 2021-04-14 | 2022-10-20 | Gilead Sciences, Inc. | CO-INHIBITION OF CD47/SIRPα BINDING AND NEDD8-ACTIVATING ENZYME E1 REGULATORY SUBUNIT FOR THE TREATMENT OF CANCER |
| WO2022245671A1 (en) | 2021-05-18 | 2022-11-24 | Gilead Sciences, Inc. | Methods of using flt3l-fc fusion proteins |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023278377A1 (en) | 2021-06-29 | 2023-01-05 | Seagen Inc. | Methods of treating cancer with a combination of a nonfucosylated anti-cd70 antibody and a cd47 antagonist |
| WO2023039089A1 (en)* | 2021-09-08 | 2023-03-16 | Twentyeight-Seven, Inc. | Papd5 and/or papd7 inhibiting 4-oxo-1,4-dihydroquinoline-3-carboxylic acid derivatives |
| CN115998863A (en)* | 2022-11-17 | 2023-04-25 | 重庆医科大学附属第二医院 | CD47 signal-mediated targeting nanoparticle with photothermal and chemical kinetics treatment effects as well as preparation method and application thereof |
| WO2023077030A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| US20230192680A1 (en)* | 2019-03-11 | 2023-06-22 | Er Stress Research Institute, Inc. | NEW AGENT FOR PROMOTING PHOSPHORYLATION OF eIF2a |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023183817A1 (en) | 2022-03-24 | 2023-09-28 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023196784A1 (en) | 2022-04-05 | 2023-10-12 | Gilead Sciences, Inc. | Combinations of antibody therapies for treating colorectal cancer |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024015741A1 (en) | 2022-07-12 | 2024-01-18 | Gilead Sciences, Inc. | Hiv immunogenic polypeptides and vaccines and uses thereof |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
| WO2024137852A1 (en) | 2022-12-22 | 2024-06-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| US12091681B2 (en) | 2020-03-27 | 2024-09-17 | Mendus B.V. | Ex vivo use of modified cells of leukemic origin for enhancing the efficacy of adoptive cell therapy |
| WO2024215754A1 (en) | 2023-04-11 | 2024-10-17 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2024220917A1 (en) | 2023-04-21 | 2024-10-24 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2025006720A1 (en) | 2023-06-30 | 2025-01-02 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2025024663A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025024811A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025054347A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2025054530A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Pyrimidine-containing polycyclic derivatives as kras g12d modulating compounds |
| WO2025096589A1 (en) | 2023-11-03 | 2025-05-08 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2025137640A1 (en) | 2023-12-22 | 2025-06-26 | Gilead Sciences, Inc. | Azaspiro wrn inhibitors |
| US12364758B2 (en) | 2020-06-30 | 2025-07-22 | Mendus B.V. | Use of leukemia-derived cells in ovarian cancer vaccines |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111771895B (en)* | 2020-08-03 | 2025-04-25 | 广西田园生化股份有限公司 | New application of quinolone compounds in preventing and controlling plant bacterial disease citrus canker |
| CN112076193B (en)* | 2020-10-13 | 2021-10-26 | 苏州大学 | Application of mequindox in preparation of medicine for treating and/or preventing diseases taking T-type calcium channel as treatment target |
| AU2022492644A1 (en)* | 2022-12-26 | 2025-08-07 | Alphamol Science Ltd (Shanghai) | Novel phenylquinolone compound having antibacterial and anticancer functions, and preparation thereof |
| CN118255716A (en) | 2022-12-26 | 2024-06-28 | 深圳阿尔法分子科技有限责任公司 | A novel phenylquinolone compound with antibacterial and anticancer functions and its preparation |
| CN119909071A (en)* | 2025-01-22 | 2025-05-02 | 中国科学院遗传与发育生物学研究所 | Ivacaftor targets NINJ1 to inhibit plasma membrane disruption and limit tissue damage in inflammatory diseases |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN86103954A (en)* | 1985-06-07 | 1987-01-28 | 拜尔公司 | Process for preparing quinolone carboxylates with antibacterial activity |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4959363A (en)* | 1989-06-23 | 1990-09-25 | Sterling Drug Inc. | Quinolonecarboxamide compounds, their preparation and use as antivirals. |
| EP1603569A1 (en)* | 2003-03-07 | 2005-12-14 | Bayer Pharmaceuticals Corporation | Method of treating cancer with quinolone carboxylic acid derivatives |
| WO2014160183A1 (en)* | 2013-03-13 | 2014-10-02 | The United States Of America,As Represented By The Secretary,Department Of Health And Human Services | Methods for modulating chemotherapeutic cytotoxicity |
| CN106467541B (en)* | 2015-08-18 | 2019-04-05 | 暨南大学 | Substituted quinolone analog derivative or its pharmaceutically acceptable salt or stereoisomer and its Pharmaceutical composition and application |
- 2017
- 2017-09-04CNCN201710785796.XApatent/CN109422726B/ennot_activeExpired - Fee Related
- 2018
- 2018-09-04WOPCT/CN2018/103974patent/WO2019042470A1/ennot_activeCeased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN86103954A (en)* | 1985-06-07 | 1987-01-28 | 拜尔公司 | Process for preparing quinolone carboxylates with antibacterial activity |
Non-Patent Citations (3)
| Title |
|---|
| DANIEL: "Synthesis and Structure-Activity Relationships of Novel Arylfluoroquinolone Antibacterial Agents", JOURNAL OF MEDICINAL CHEMISTRY, vol. 28, no. 11, 1 November 1985 (1985-11-01), pages 1558 - 1564, XP001061840, ISSN: 0022-2623, DOI: doi:10.1021/jm00149a003* |
| F. J. MORALES-GUTIERREZ: "High-resolution Mass Spectrometry Applied to the Identification of Transformation Products of Quinolones from Stability Stu- dies and New Metabolites of Enrofloxacin in Chicken Muscle Tissues", JOURNAL OF PHARMACEUTICAL AND BIOMEDICAL ANALYSIS, vol. 92, 24 January 2014 (2014-01-24), pages 165 - 176, XP028620672, ISSN: 0731-7085* |
| SHEU, JIAYUH ET AL: "Synthesis and Antibacterial Activity of 1-(Substituted-ben- zyl)-6-fluoro-1, 4-dihydro-4-oxoquinoline-3-carboxylic Acids and their 6, 8- Difluoro Analogs", JOURNAL OF HETEROCYCLIC CHEMISTRY, vol. 35, no. 4, 31 August 1998 (1998-08-31), pages 955 - 964, XP055579097, ISSN: 0022-152X* |
Cited By (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20230192680A1 (en)* | 2019-03-11 | 2023-06-22 | Er Stress Research Institute, Inc. | NEW AGENT FOR PROMOTING PHOSPHORYLATION OF eIF2a |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021011544A1 (en) | 2019-07-16 | 2021-01-21 | Gilead Sciences, Inc. | Hiv vaccines and methods of making and using |
| WO2021042046A1 (en)* | 2019-08-30 | 2021-03-04 | Trustees Of Dartmouth College | Membrane-active anti-bacterial compounds and uses thereof |
| EP4349413A2 (en) | 2019-10-18 | 2024-04-10 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021076908A1 (en) | 2019-10-18 | 2021-04-22 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021087064A1 (en) | 2019-10-31 | 2021-05-06 | Forty Seven, Inc. | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| EP4445902A2 (en) | 2019-12-24 | 2024-10-16 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021163064A2 (en) | 2020-02-14 | 2021-08-19 | Jounce Therapeutics, Inc. | Antibodies and fusion proteins that bind to ccr8 and uses thereof |
| US12297282B2 (en) | 2020-02-14 | 2025-05-13 | Gilead Sciences, Inc. | Nucleic acids encoding, and methods of producing, antibodies that bind human chemokine (C—C motif) receptor 8 (CCR8) |
| US11692038B2 (en) | 2020-02-14 | 2023-07-04 | Gilead Sciences, Inc. | Antibodies that bind chemokine (C-C motif) receptor 8 (CCR8) |
| CN111297856A (en)* | 2020-03-13 | 2020-06-19 | 黑龙江中医药大学 | Medicinal composition for resisting fungal infection and application thereof |
| CN111297856B (en)* | 2020-03-13 | 2020-12-18 | 黑龙江中医药大学 | A kind of medicinal composition of antifungal infection and use thereof |
| US12091681B2 (en) | 2020-03-27 | 2024-09-17 | Mendus B.V. | Ex vivo use of modified cells of leukemic origin for enhancing the efficacy of adoptive cell therapy |
| US12364758B2 (en) | 2020-06-30 | 2025-07-22 | Mendus B.V. | Use of leukemia-derived cells in ovarian cancer vaccines |
| WO2022190058A1 (en) | 2021-03-12 | 2022-09-15 | Dcprime B.V. | Methods of vaccination and use of cd47 blockade |
| WO2022221304A1 (en) | 2021-04-14 | 2022-10-20 | Gilead Sciences, Inc. | CO-INHIBITION OF CD47/SIRPα BINDING AND NEDD8-ACTIVATING ENZYME E1 REGULATORY SUBUNIT FOR THE TREATMENT OF CANCER |
| WO2022245671A1 (en) | 2021-05-18 | 2022-11-24 | Gilead Sciences, Inc. | Methods of using flt3l-fc fusion proteins |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023278377A1 (en) | 2021-06-29 | 2023-01-05 | Seagen Inc. | Methods of treating cancer with a combination of a nonfucosylated anti-cd70 antibody and a cd47 antagonist |
| WO2023039089A1 (en)* | 2021-09-08 | 2023-03-16 | Twentyeight-Seven, Inc. | Papd5 and/or papd7 inhibiting 4-oxo-1,4-dihydroquinoline-3-carboxylic acid derivatives |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023077030A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| EP4464703A2 (en) | 2022-03-17 | 2024-11-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023178181A1 (en) | 2022-03-17 | 2023-09-21 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023183817A1 (en) | 2022-03-24 | 2023-09-28 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023196784A1 (en) | 2022-04-05 | 2023-10-12 | Gilead Sciences, Inc. | Combinations of antibody therapies for treating colorectal cancer |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| CN115043907A (en)* | 2022-06-02 | 2022-09-13 | 中山大学 | Parent peptide or derived peptide with CD47/SIRP alpha blocking effect and application thereof |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024015741A1 (en) | 2022-07-12 | 2024-01-18 | Gilead Sciences, Inc. | Hiv immunogenic polypeptides and vaccines and uses thereof |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
| CN115998863A (en)* | 2022-11-17 | 2023-04-25 | 重庆医科大学附属第二医院 | CD47 signal-mediated targeting nanoparticle with photothermal and chemical kinetics treatment effects as well as preparation method and application thereof |
| WO2024137852A1 (en) | 2022-12-22 | 2024-06-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2024215754A1 (en) | 2023-04-11 | 2024-10-17 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2024220917A1 (en) | 2023-04-21 | 2024-10-24 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2025006720A1 (en) | 2023-06-30 | 2025-01-02 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2025024663A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025024811A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025054347A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2025054530A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Pyrimidine-containing polycyclic derivatives as kras g12d modulating compounds |
| WO2025096589A1 (en) | 2023-11-03 | 2025-05-08 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2025137640A1 (en) | 2023-12-22 | 2025-06-26 | Gilead Sciences, Inc. | Azaspiro wrn inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| CN109422726B (en) | 2022-10-28 |
| CN109422726A (en) | 2019-03-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019042470A1 (en) | BLOCKER OF CD47/SIRPα AND APPLICATION THEREOF | |
| JP5856673B2 (en) | 7- (3,5-Dimethyl-4-isoxazolyl) -8- (methyloxy) -1H-imidazo [4,5-c] quinoline derivative | |
| US9315487B2 (en) | Tetrahydroquinoline derivatives useful as bromodomain inhibitors | |
| CN115322158B (en) | As KRASG12CSubstituted quinazoline compounds of protein inhibitor | |
| JP2021119151A (en) | Diaryl macrocycle | |
| JP6242390B2 (en) | 2-Aminopyrazine derivatives as CSF-1R kinase inhibitors | |
| CN113773308A (en) | RHO kinase inhibitors | |
| CA2832763A1 (en) | Tetrahydroquinoline derivatives useful as bromodomain inhibitors | |
| CN103930425A (en) | Pteridinone derivatives and their application as EGFR, BLK, FLT3 inhibitors | |
| WO2017028816A1 (en) | Indole derivative, preparation method thereof, and use thereof in pharmaceutical drug | |
| CN110194762B (en) | Phthalazinone derivatives, preparation method and application thereof | |
| WO2022217821A1 (en) | Alkynylphenylbenzamide compound and use thereof | |
| JP4363530B2 (en) | Protein kinase inhibitor | |
| CN116997339A (en) | Inhibitors of cGAS activity as therapeutic agents | |
| CN116600808A (en) | Tetrahydronaphthyridine derivative serving as KRAS mutant G12C inhibitor, and preparation method and application thereof | |
| CN118221698A (en) | KRAS G12D inhibitors | |
| CN110407839B (en) | Preparation and application of triazole heterocyclic compound containing heteroaryl amide structure | |
| KR102046196B1 (en) | Therapeutic use of imidazopyridine derivatives | |
| CN110407854A (en) | Novel tetracyclic compounds | |
| US20170305915A1 (en) | Indazole and indole derivatives as inhibitors of retinoic acid relates orphan receptor gamma (ror gamma) for the treatment of immune-related diseases | |
| CN111108083A (en) | Use of aminomethylene cyclohexane 1, 3-dione compounds | |
| CN117903128A (en) | A class of ASK1/PDK1 dual-targeting inhibitors and preparation methods and applications thereof | |
| JP7110335B2 (en) | Pyridoquinazoline derivatives useful as protein kinase inhibitors | |
| CN115003383A (en) | Bicyclic agonists of interferon gene stimulator STING | |
| CN113493414A (en) | Deuterated substituted butene amide and preparation method and application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | Ref document number:18851940 Country of ref document:EP Kind code of ref document:A1 | |
| NENP | Non-entry into the national phase | Ref country code:DE | |
| 122 | Ep: pct application non-entry in european phase | Ref document number:18851940 Country of ref document:EP Kind code of ref document:A1 |