KR20170040798A - Stable and soluble formulations of receptor tyrosine kinase inhibitors, and methods of preparation thereof - Google Patents
Stable and soluble formulations of receptor tyrosine kinase inhibitors, and methods of preparation thereofDownload PDFInfo
- Publication number
- KR20170040798A KR20170040798AKR1020177006057AKR20177006057AKR20170040798AKR 20170040798 AKR20170040798 AKR 20170040798AKR 1020177006057 AKR1020177006057 AKR 1020177006057AKR 20177006057 AKR20177006057 AKR 20177006057AKR 20170040798 AKR20170040798 AKR 20170040798A
- Authority
- KR
- South Korea
- Prior art keywords
- cyclodextrin
- beta
- formulation
- salt
- agent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 238000000034methodMethods0.000titleclaimsabstractdescription180
- 238000002360preparation methodMethods0.000titleclaimsabstractdescription76
- 239000000203mixtureSubstances0.000titleclaimsdescription353
- 238000009472formulationMethods0.000titleclaimsdescription318
- 229940127361Receptor Tyrosine Kinase InhibitorsDrugs0.000titleabstractdescription4
- 239000003795chemical substances by applicationSubstances0.000claimsabstractdescription68
- 239000003798L01XE11 - PazopanibSubstances0.000claimsabstractdescription59
- CUIHSIWYWATEQL-UHFFFAOYSA-NpazopanibChemical compoundC1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1CUIHSIWYWATEQL-UHFFFAOYSA-N0.000claimsabstractdescription59
- 229960000639pazopanibDrugs0.000claimsabstractdescription59
- 239000003814drugSubstances0.000claimsdescription256
- 150000003839saltsChemical class0.000claimsdescription235
- 229940124597therapeutic agentDrugs0.000claimsdescription217
- 229920000858CyclodextrinPolymers0.000claimsdescription199
- IAZDPXIOMUYVGZ-UHFFFAOYSA-NDimethylsulphoxideChemical compoundCS(C)=OIAZDPXIOMUYVGZ-UHFFFAOYSA-N0.000claimsdescription133
- 229960004853betadexDrugs0.000claimsdescription93
- 230000001225therapeutic effectEffects0.000claimsdescription80
- 239000001116FEMA 4028Substances0.000claimsdescription78
- 239000002904solventSubstances0.000claimsdescription77
- 239000008139complexing agentSubstances0.000claimsdescription76
- XLYOFNOQVPJJNP-UHFFFAOYSA-NwaterSubstancesOXLYOFNOQVPJJNP-UHFFFAOYSA-N0.000claimsdescription69
- RHQDFWAXVIIEBN-UHFFFAOYSA-NTrifluoroethanolChemical compoundOCC(F)(F)FRHQDFWAXVIIEBN-UHFFFAOYSA-N0.000claimsdescription53
- WHNWPMSKXPGLAX-UHFFFAOYSA-NN-Vinyl-2-pyrrolidoneChemical compoundC=CN1CCCC1=OWHNWPMSKXPGLAX-UHFFFAOYSA-N0.000claimsdescription51
- 239000000872bufferSubstances0.000claimsdescription50
- -1halide saltChemical class0.000claimsdescription50
- 239000000463materialSubstances0.000claimsdescription48
- 238000004108freeze dryingMethods0.000claimsdescription47
- 239000008194pharmaceutical compositionSubstances0.000claimsdescription44
- QZNNVYOVQUKYSC-JEDNCBNOSA-N(2s)-2-amino-3-(1h-imidazol-5-yl)propanoic acid;hydron;chlorideChemical groupCl.OC(=O)[C@@H](N)CC1=CN=CN1QZNNVYOVQUKYSC-JEDNCBNOSA-N0.000claimsdescription43
- 235000011175beta-cyclodextrineNutrition0.000claimsdescription40
- 208000037265diseases, disorders, signs and symptomsDiseases0.000claimsdescription38
- HFHDHCJBZVLPGP-UHFFFAOYSA-Nschardinger α-dextrinChemical compoundO1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2COHFHDHCJBZVLPGP-UHFFFAOYSA-N0.000claimsdescription30
- 238000000634powder X-ray diffractionMethods0.000claimsdescription28
- 239000006172buffering agentSubstances0.000claimsdescription25
- 206010029113NeovascularisationDiseases0.000claimsdescription24
- 201000010099diseaseDiseases0.000claimsdescription23
- 229940080345gamma-cyclodextrinDrugs0.000claimsdescription22
- 238000004519manufacturing processMethods0.000claimsdescription22
- 150000003841chloride saltsChemical group0.000claimsdescription19
- WHGYBXFWUBPSRW-FOUAGVGXSA-Nbeta-cyclodextrinChemical classOC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COWHGYBXFWUBPSRW-FOUAGVGXSA-N0.000claimsdescription18
- 239000003960organic solventSubstances0.000claimsdescription18
- 238000003860storageMethods0.000claimsdescription17
- YZOUYRAONFXZSI-SBHWVFSVSA-N(1S,3R,5R,6R,8R,10R,11R,13R,15R,16R,18R,20R,21R,23R,25R,26R,28R,30R,31S,33R,35R,36R,37S,38R,39S,40R,41S,42R,43S,44R,45S,46R,47S,48R,49S)-5,10,15,20,25,30,35-heptakis(hydroxymethyl)-37,39,40,41,42,43,44,45,46,47,48,49-dodecamethoxy-2,4,7,9,12,14,17,19,22,24,27,29,32,34-tetradecaoxaoctacyclo[31.2.2.23,6.28,11.213,16.218,21.223,26.228,31]nonatetracontane-36,38-diolChemical compoundO([C@@H]([C@H]([C@@H]1OC)OC)O[C@H]2[C@@H](O)[C@@H]([C@@H](O[C@@H]3[C@@H](CO)O[C@@H]([C@H]([C@@H]3O)OC)O[C@@H]3[C@@H](CO)O[C@@H]([C@H]([C@@H]3OC)OC)O[C@@H]3[C@@H](CO)O[C@@H]([C@H]([C@@H]3OC)OC)O[C@@H]3[C@@H](CO)O[C@@H]([C@H]([C@@H]3OC)OC)O3)O[C@@H]2CO)OC)[C@H](CO)[C@H]1O[C@@H]1[C@@H](OC)[C@H](OC)[C@H]3[C@@H](CO)O1YZOUYRAONFXZSI-SBHWVFSVSA-N0.000claimsdescription15
- 208000035475disorderDiseases0.000claimsdescription15
- 238000002156mixingMethods0.000claimsdescription15
- 230000008569processEffects0.000claimsdescription15
- ODLHGICHYURWBS-LKONHMLTSA-Ntrappsol cycloChemical compoundCC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)OODLHGICHYURWBS-LKONHMLTSA-N0.000claimsdescription15
- PCWPQSDFNIFUPO-VDQKLNDWSA-N(1S,3R,5R,6S,8R,10R,11S,13R,15R,16S,18R,20R,21S,23R,25R,26S,28R,30R,31S,33R,35R,36R,37S,38R,39S,40R,41S,42R,43S,44R,45S,46R,47S,48R,49S)-37,39,41,43,45,47,49-heptakis(2-hydroxyethoxy)-5,10,15,20,25,30,35-heptakis(hydroxymethyl)-2,4,7,9,12,14,17,19,22,24,27,29,32,34-tetradecaoxaoctacyclo[31.2.2.23,6.28,11.213,16.218,21.223,26.228,31]nonatetracontane-36,38,40,42,44,46,48-heptolChemical compoundOCCO[C@H]1[C@H](O)[C@@H]2O[C@H]3O[C@H](CO)[C@@H](O[C@H]4O[C@H](CO)[C@@H](O[C@H]5O[C@H](CO)[C@@H](O[C@H]6O[C@H](CO)[C@@H](O[C@H]7O[C@H](CO)[C@@H](O[C@H]8O[C@H](CO)[C@@H](O[C@H]1O[C@@H]2CO)[C@@H](O)[C@@H]8OCCO)[C@@H](O)[C@@H]7OCCO)[C@@H](O)[C@@H]6OCCO)[C@@H](O)[C@@H]5OCCO)[C@@H](O)[C@@H]4OCCO)[C@@H](O)[C@@H]3OCCOPCWPQSDFNIFUPO-VDQKLNDWSA-N0.000claimsdescription13
- CUJVBAPGYBSBHJ-YWBSARSQSA-N2-[[(1R,3R,5R,6S,8R,10R,11S,13R,15R,16S,18R,20R,21R,23R,25R,26R,28R,30R,31R,33R,35R,36R,37R,38R,39R,40R,41R,42R,43R,44R,45R,46R,47R,48R,49R)-36,38,40,42-tetrakis(carboxymethoxy)-10,15-bis(carboxymethoxymethyl)-37,39,41,43,44,45,46,47,48,49-decahydroxy-20,25,30,35-tetrakis(hydroxymethyl)-2,4,7,9,12,14,17,19,22,24,27,29,32,34-tetradecaoxaoctacyclo[31.2.2.23,6.28,11.213,16.218,21.223,26.228,31]nonatetracontan-5-yl]methoxy]acetic acidChemical compoundOC[C@H]1O[C@@H]2O[C@H]3[C@H](O)[C@@H](O)[C@H](O[C@@H]3COCC(O)=O)O[C@H]3[C@H](O)[C@@H](O)[C@H](O[C@@H]3COCC(O)=O)O[C@H]3[C@H](O)[C@@H](O)[C@H](O[C@@H]3COCC(O)=O)O[C@@H]3[C@@H](CO)O[C@H](O[C@@H]4[C@@H](CO)O[C@H](O[C@@H]5[C@@H](CO)O[C@H](O[C@H]1[C@H](OCC(O)=O)[C@H]2O)[C@H](O)[C@H]5OCC(O)=O)[C@H](O)[C@H]4OCC(O)=O)[C@H](O)[C@H]3OCC(O)=OCUJVBAPGYBSBHJ-YWBSARSQSA-N0.000claimsdescription13
- 238000013270controlled releaseMethods0.000claimsdescription12
- 206010064930age-related macular degenerationDiseases0.000claimsdescription11
- 208000002780macular degenerationDiseases0.000claimsdescription11
- NOPKOJDDVCBPTP-DJSZNTTKSA-N23739-88-0Chemical compoundCC(=O)OC[C@H]([C@H]([C@H]([C@@H]1OC(C)=O)OC(C)=O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COC(C)=O)[C@H]([C@H]([C@@H]3OC(C)=O)OC(C)=O)O[C@H]3O[C@H](COC(C)=O)[C@H]([C@H]([C@@H]3OC(C)=O)OC(C)=O)O[C@H]3O[C@H](COC(C)=O)[C@H]([C@H]([C@@H]3OC(C)=O)OC(C)=O)O[C@H]3O[C@H](COC(C)=O)[C@H]([C@H]([C@@H]3OC(C)=O)OC(C)=O)O3)[C@@H](OC(C)=O)[C@@H]2OC(C)=O)COC(=O)C)O[C@@H]1O[C@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H]3O[C@@H]1COC(C)=ONOPKOJDDVCBPTP-DJSZNTTKSA-N0.000claimsdescription9
- 229940097346sulfobutylether-beta-cyclodextrinDrugs0.000claimsdescription9
- 208000005590Choroidal NeovascularizationDiseases0.000claimsdescription7
- 206010060823Choroidal neovascularisationDiseases0.000claimsdescription7
- 206010012689Diabetic retinopathyDiseases0.000claimsdescription7
- JVFGXECLSQXABC-UHFFFAOYSA-Nac1l3obqChemical compoundO1C(C(C2O)O)C(COCC(C)O)OC2OC(C(C2O)O)C(COCC(C)O)OC2OC(C(C2O)O)C(COCC(C)O)OC2OC(C(C2O)O)C(COCC(C)O)OC2OC(C(C2O)O)C(COCC(C)O)OC2OC(C(O)C2O)C(COCC(O)C)OC2OC(C(C2O)O)C(COCC(C)O)OC2OC2C(O)C(O)C1OC2COCC(C)OJVFGXECLSQXABC-UHFFFAOYSA-N0.000claimsdescription7
- 230000001575pathological effectEffects0.000claimsdescription7
- 150000004820halidesChemical group0.000claimsdescription6
- 208000010412GlaucomaDiseases0.000claimsdescription5
- 208000007135Retinal NeovascularizationDiseases0.000claimsdescription5
- 201000003142neovascular glaucomaDiseases0.000claimsdescription5
- 208000028867ischemiaDiseases0.000claimsdescription4
- 238000001356surgical procedureMethods0.000claimsdescription4
- 208000001344Macular EdemaDiseases0.000claimsdescription3
- 201000010183PapilledemaDiseases0.000claimsdescription3
- 206010038886Retinal oedemaDiseases0.000claimsdescription3
- 206010038933Retinopathy of prematurityDiseases0.000claimsdescription3
- 206010038935Retinopathy sickle cellDiseases0.000claimsdescription3
- 206010046851UveitisDiseases0.000claimsdescription3
- 150000001805chlorine compoundsChemical group0.000claimsdescription3
- 201000011195retinal edemaDiseases0.000claimsdescription3
- 208000004644retinal vein occlusionDiseases0.000claimsdescription3
- 206010058202Cystoid macular oedemaDiseases0.000claimsdescription2
- 201000010206cystoid macular edemaDiseases0.000claimsdescription2
- 239000013543active substanceSubstances0.000abstractdescription35
- 229940121358tyrosine kinase inhibitorDrugs0.000abstractdescription9
- 239000005483tyrosine kinase inhibitorSubstances0.000abstractdescription9
- 239000003381stabilizerSubstances0.000abstractdescription3
- 230000002459sustained effectEffects0.000abstractdescription3
- 239000000243solutionSubstances0.000description173
- VEXZGXHMUGYJMC-UHFFFAOYSA-NHydrochloric acidChemical compoundClVEXZGXHMUGYJMC-UHFFFAOYSA-N0.000description112
- HEMHJVSKTPXQMS-UHFFFAOYSA-MSodium hydroxideChemical compound[OH-].[Na+]HEMHJVSKTPXQMS-UHFFFAOYSA-M0.000description51
- 238000011282treatmentMethods0.000description44
- 229940079593drugDrugs0.000description36
- 150000001875compoundsChemical class0.000description23
- PEDCQBHIVMGVHV-UHFFFAOYSA-NGlycerineChemical compoundOCC(O)COPEDCQBHIVMGVHV-UHFFFAOYSA-N0.000description21
- 238000010790dilutionMethods0.000description18
- 239000012895dilutionSubstances0.000description18
- 208000024891symptomDiseases0.000description17
- 239000013078crystalSubstances0.000description16
- 238000009792diffusion processMethods0.000description16
- 238000012377drug deliveryMethods0.000description16
- 239000012530fluidSubstances0.000description15
- 210000003786scleraAnatomy0.000description15
- ZAHRKKWIAAJSAO-UHFFFAOYSA-NrapamycinNatural productsCOCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)CZAHRKKWIAAJSAO-UHFFFAOYSA-N0.000description14
- QFJCIRLUMZQUOT-HPLJOQBZSA-NsirolimusChemical compoundC1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1QFJCIRLUMZQUOT-HPLJOQBZSA-N0.000description14
- 229960002930sirolimusDrugs0.000description14
- URAYPUMNDPQOKB-UHFFFAOYSA-NtriacetinChemical compoundCC(=O)OCC(OC(C)=O)COC(C)=OURAYPUMNDPQOKB-UHFFFAOYSA-N0.000description14
- 239000007943implantSubstances0.000description12
- 238000005063solubilizationMethods0.000description12
- 230000007928solubilizationEffects0.000description12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-NEthanolChemical compoundCCOLFQSCWFLJHTTHZ-UHFFFAOYSA-N0.000description11
- 239000002253acidSubstances0.000description11
- 230000001965increasing effectEffects0.000description11
- 239000007788liquidSubstances0.000description11
- 238000012360testing methodMethods0.000description11
- 210000004127vitreous bodyAnatomy0.000description11
- ZZUYEGMKIIREEE-UHFFFAOYSA-N2,2,2-trifluoroethanol;hydrateChemical compoundO.OCC(F)(F)FZZUYEGMKIIREEE-UHFFFAOYSA-N0.000description10
- 101100225582Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) nip-1 geneProteins0.000description9
- DNIAPMSPPWPWGF-UHFFFAOYSA-NPropylene glycolChemical compoundCC(O)CODNIAPMSPPWPWGF-UHFFFAOYSA-N0.000description9
- 239000000556agonistSubstances0.000description9
- 239000007864aqueous solutionSubstances0.000description9
- 230000001186cumulative effectEffects0.000description9
- 239000012528membraneSubstances0.000description9
- 238000010979pH adjustmentMethods0.000description9
- 239000002953phosphate buffered salineSubstances0.000description9
- 210000001525retinaAnatomy0.000description9
- 239000000126substanceSubstances0.000description9
- 210000001519tissueAnatomy0.000description9
- 230000002792vascularEffects0.000description9
- 230000004888barrier functionEffects0.000description8
- 235000019441ethanolNutrition0.000description8
- 235000011187glycerolNutrition0.000description8
- 239000004615ingredientSubstances0.000description8
- 238000003780insertionMethods0.000description8
- 230000037431insertionEffects0.000description8
- 239000000825pharmaceutical preparationSubstances0.000description8
- 239000011148porous materialSubstances0.000description8
- 239000007787solidSubstances0.000description8
- 210000000795conjunctivaAnatomy0.000description7
- 235000013773glyceryl triacetateNutrition0.000description7
- 239000001087glyceryl triacetateSubstances0.000description7
- 238000001556precipitationMethods0.000description7
- 229960002622triacetinDrugs0.000description7
- KRKNYBCHXYNGOX-UHFFFAOYSA-Ncitric acidChemical compoundOC(=O)CC(O)(C(O)=O)CC(O)=OKRKNYBCHXYNGOX-UHFFFAOYSA-N0.000description6
- 239000003889eye dropSubstances0.000description6
- 150000004673fluoride saltsChemical class0.000description6
- 238000002347injectionMethods0.000description6
- 239000007924injectionSubstances0.000description6
- 230000000670limiting effectEffects0.000description6
- 239000000546pharmaceutical excipientSubstances0.000description6
- 239000002244precipitateSubstances0.000description6
- 239000000725suspensionSubstances0.000description6
- 238000013268sustained releaseMethods0.000description6
- 239000012730sustained-release formSubstances0.000description6
- NZAQRZWBQUIBSF-UHFFFAOYSA-N4-(4-sulfobutoxy)butane-1-sulfonic acidChemical compoundOS(=O)(=O)CCCCOCCCCS(O)(=O)=ONZAQRZWBQUIBSF-UHFFFAOYSA-N0.000description5
- 206010012688Diabetic retinal oedemaDiseases0.000description5
- MBBZMMPHUWSWHV-BDVNFPICSA-NN-methylglucamineChemical compoundCNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)COMBBZMMPHUWSWHV-BDVNFPICSA-N0.000description5
- 208000022873Ocular diseaseDiseases0.000description5
- 108010073929Vascular Endothelial Growth Factor AProteins0.000description5
- 102000009524Vascular Endothelial Growth Factor AHuman genes0.000description5
- 239000004480active ingredientSubstances0.000description5
- 239000000654additiveSubstances0.000description5
- 229940097362cyclodextrinsDrugs0.000description5
- 201000011190diabetic macular edemaDiseases0.000description5
- LOKCTEFSRHRXRJ-UHFFFAOYSA-Idipotassium trisodium dihydrogen phosphate hydrogen phosphate dichlorideChemical compoundP(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+]LOKCTEFSRHRXRJ-UHFFFAOYSA-I0.000description5
- 238000004090dissolutionMethods0.000description5
- 239000003937drug carrierSubstances0.000description5
- 125000002791glucosyl groupChemical groupC1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)*0.000description5
- HNDVDQJCIGZPNO-UHFFFAOYSA-NhistidineNatural productsOC(=O)C(N)CC1=CN=CN1HNDVDQJCIGZPNO-UHFFFAOYSA-N0.000description5
- QYRFJLLXPINATB-UHFFFAOYSA-Nhydron;2,4,5,6-tetrafluorobenzene-1,3-diamine;dichlorideChemical compoundCl.Cl.NC1=C(F)C(N)=C(F)C(F)=C1FQYRFJLLXPINATB-UHFFFAOYSA-N0.000description5
- XMBWDFGMSWQBCA-UHFFFAOYSA-MiodideChemical compound[I-]XMBWDFGMSWQBCA-UHFFFAOYSA-M0.000description5
- 229960003194meglumineDrugs0.000description5
- 229920000642polymerPolymers0.000description5
- 239000001267polyvinylpyrrolidoneSubstances0.000description5
- 229920000036polyvinylpyrrolidonePolymers0.000description5
- 235000013855polyvinylpyrrolidoneNutrition0.000description5
- 238000000527sonicationMethods0.000description5
- 238000003260vortexingMethods0.000description5
- VEXZGXHMUGYJMC-UHFFFAOYSA-MChloride anionChemical compound[Cl-]VEXZGXHMUGYJMC-UHFFFAOYSA-M0.000description4
- HNDVDQJCIGZPNO-YFKPBYRVSA-NL-histidineChemical compoundOC(=O)[C@@H](N)CC1=CN=CN1HNDVDQJCIGZPNO-YFKPBYRVSA-N0.000description4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-NSodium azideChemical compound[Na+].[N-]=[N+]=[N-]PXIPVTKHYLBLMZ-UHFFFAOYSA-N0.000description4
- 230000009286beneficial effectEffects0.000description4
- 210000004204blood vesselAnatomy0.000description4
- 150000003842bromide saltsChemical class0.000description4
- 230000008878couplingEffects0.000description4
- 238000010168coupling processMethods0.000description4
- 238000005859coupling reactionMethods0.000description4
- 239000006185dispersionSubstances0.000description4
- 230000000694effectsEffects0.000description4
- 229940012356eye dropsDrugs0.000description4
- 239000011521glassSubstances0.000description4
- 230000001976improved effectEffects0.000description4
- 230000014759maintenance of locationEffects0.000description4
- 239000003002pH adjusting agentSubstances0.000description4
- 229920001223polyethylene glycolPolymers0.000description4
- 230000000699topical effectEffects0.000description4
- 150000004917tyrosine kinase inhibitor derivativesChemical class0.000description4
- 229920001353DextrinPolymers0.000description3
- 239000004375DextrinSubstances0.000description3
- 239000001856Ethyl celluloseSubstances0.000description3
- ZZSNKZQZMQGXPY-UHFFFAOYSA-NEthyl celluloseChemical compoundCCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1ZZSNKZQZMQGXPY-UHFFFAOYSA-N0.000description3
- 206010030113OedemaDiseases0.000description3
- 241000283973Oryctolagus cuniculusSpecies0.000description3
- 239000002202Polyethylene glycolSubstances0.000description3
- 102000001253Protein KinaseHuman genes0.000description3
- 206010038934Retinopathy proliferativeDiseases0.000description3
- FAPWRFPIFSIZLT-UHFFFAOYSA-MSodium chlorideChemical group[Na+].[Cl-]FAPWRFPIFSIZLT-UHFFFAOYSA-M0.000description3
- 108091008605VEGF receptorsProteins0.000description3
- DPXJVFZANSGRMM-UHFFFAOYSA-Nacetic acid;2,3,4,5,6-pentahydroxyhexanal;sodiumChemical compound[Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=ODPXJVFZANSGRMM-UHFFFAOYSA-N0.000description3
- 239000008186active pharmaceutical agentSubstances0.000description3
- 238000004458analytical methodMethods0.000description3
- 230000008901benefitEffects0.000description3
- 239000001768carboxy methyl celluloseSubstances0.000description3
- 230000001684chronic effectEffects0.000description3
- 230000002939deleterious effectEffects0.000description3
- 235000019425dextrinNutrition0.000description3
- 235000019325ethyl celluloseNutrition0.000description3
- 229920001249ethyl cellulosePolymers0.000description3
- 238000001914filtrationMethods0.000description3
- 230000006870functionEffects0.000description3
- IXCSERBJSXMMFS-UHFFFAOYSA-Nhcl hclChemical compoundCl.ClIXCSERBJSXMMFS-UHFFFAOYSA-N0.000description3
- 238000004128high performance liquid chromatographyMethods0.000description3
- 230000006872improvementEffects0.000description3
- 238000001727in vivoMethods0.000description3
- 208000014674injuryDiseases0.000description3
- 238000012986modificationMethods0.000description3
- 230000004048modificationEffects0.000description3
- 150000007530organic basesChemical class0.000description3
- 239000002245particleSubstances0.000description3
- 230000000149penetrating effectEffects0.000description3
- 230000002265preventionEffects0.000description3
- 230000002035prolonged effectEffects0.000description3
- 230000000069prophylactic effectEffects0.000description3
- 235000013772propylene glycolNutrition0.000description3
- 108060006633protein kinaseProteins0.000description3
- 235000018102proteinsNutrition0.000description3
- 102000004169proteins and genesHuman genes0.000description3
- 108090000623proteins and genesProteins0.000description3
- 235000019812sodium carboxymethyl celluloseNutrition0.000description3
- 229920001027sodium carboxymethylcellulosePolymers0.000description3
- 210000002435tendonAnatomy0.000description3
- 230000004304visual acuityEffects0.000description3
- HDTRYLNUVZCQOY-UHFFFAOYSA-Nα-D-glucopyranosyl-α-D-glucopyranosideNatural productsOC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1HDTRYLNUVZCQOY-UHFFFAOYSA-N0.000description2
- MTCFGRXMJLQNBG-REOHCLBHSA-N(2S)-2-Amino-3-hydroxypropansäureChemical compoundOC[C@H](N)C(O)=OMTCFGRXMJLQNBG-REOHCLBHSA-N0.000description2
- LNAZSHAWQACDHT-XIYTZBAFSA-N(2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxaneChemical compoundCO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COCLNAZSHAWQACDHT-XIYTZBAFSA-N0.000description2
- IXPNQXFRVYWDDI-UHFFFAOYSA-N1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamideChemical compoundCN1CC(C(N)=N)C(=O)NC1=OIXPNQXFRVYWDDI-UHFFFAOYSA-N0.000description2
- 206010007766Cataract traumaticDiseases0.000description2
- 208000017667Chronic DiseaseDiseases0.000description2
- FBPFZTCFMRRESA-FSIIMWSLSA-ND-GlucitolNatural productsOC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)COFBPFZTCFMRRESA-FSIIMWSLSA-N0.000description2
- FBPFZTCFMRRESA-JGWLITMVSA-ND-glucitolChemical compoundOC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)COFBPFZTCFMRRESA-JGWLITMVSA-N0.000description2
- KDXKERNSBIXSRK-YFKPBYRVSA-NL-lysineChemical compoundNCCCC[C@H](N)C(O)=OKDXKERNSBIXSRK-YFKPBYRVSA-N0.000description2
- 241001465754MetazoaSpecies0.000description2
- FXHOOIRPVKKKFG-UHFFFAOYSA-NN,N-DimethylacetamideChemical compoundCN(C)C(C)=OFXHOOIRPVKKKFG-UHFFFAOYSA-N0.000description2
- SECXISVLQFMRJM-UHFFFAOYSA-NN-MethylpyrrolidoneChemical compoundCN1CCCC1=OSECXISVLQFMRJM-UHFFFAOYSA-N0.000description2
- GXCLVBGFBYZDAG-UHFFFAOYSA-NN-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amineChemical compoundCN(CCC1=CNC2=C1C=CC=C2)CC=CGXCLVBGFBYZDAG-UHFFFAOYSA-N0.000description2
- 208000002193PainDiseases0.000description2
- 208000034038Pathologic NeovascularizationDiseases0.000description2
- 108091000080PhosphotransferaseProteins0.000description2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-NPotassiumChemical class[K]ZLMJMSJWJFRBEC-UHFFFAOYSA-N0.000description2
- 102000009516Protein Serine-Threonine KinasesHuman genes0.000description2
- 108010009341Protein Serine-Threonine KinasesProteins0.000description2
- 206010038848Retinal detachmentDiseases0.000description2
- MTCFGRXMJLQNBG-UHFFFAOYSA-NSerineNatural productsOCC(N)C(O)=OMTCFGRXMJLQNBG-UHFFFAOYSA-N0.000description2
- 229920002385Sodium hyaluronatePolymers0.000description2
- CZMRCDWAGMRECN-UGDNZRGBSA-NSucroseChemical compoundO[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1CZMRCDWAGMRECN-UGDNZRGBSA-N0.000description2
- HDTRYLNUVZCQOY-WSWWMNSNSA-NTrehaloseNatural productsO[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1HDTRYLNUVZCQOY-WSWWMNSNSA-N0.000description2
- 102100033177Vascular endothelial growth factor receptor 2Human genes0.000description2
- 238000002441X-ray diffractionMethods0.000description2
- 230000001154acute effectEffects0.000description2
- 238000012382advanced drug deliveryMethods0.000description2
- HDTRYLNUVZCQOY-LIZSDCNHSA-Nalpha,alpha-trehaloseChemical compoundO[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1HDTRYLNUVZCQOY-LIZSDCNHSA-N0.000description2
- 239000003125aqueous solventSubstances0.000description2
- 229920001400block copolymerPolymers0.000description2
- 210000000746body regionAnatomy0.000description2
- 239000007853buffer solutionSubstances0.000description2
- 238000006243chemical reactionMethods0.000description2
- 229940124447delivery agentDrugs0.000description2
- 238000013461designMethods0.000description2
- 239000003085diluting agentSubstances0.000description2
- 239000002552dosage formSubstances0.000description2
- 239000013583drug formulationSubstances0.000description2
- 230000004406elevated intraocular pressureEffects0.000description2
- 239000003995emulsifying agentSubstances0.000description2
- 206010014801endophthalmitisDiseases0.000description2
- 150000002148estersChemical class0.000description2
- MMXKVMNBHPAILY-UHFFFAOYSA-Nethyl laurateChemical compoundCCCCCCCCCCCC(=O)OCCMMXKVMNBHPAILY-UHFFFAOYSA-N0.000description2
- 230000002349favourable effectEffects0.000description2
- GDSRMADSINPKSL-HSEONFRVSA-Ngamma-cyclodextrinChemical compoundOC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COGDSRMADSINPKSL-HSEONFRVSA-N0.000description2
- 239000003102growth factorSubstances0.000description2
- 230000036541healthEffects0.000description2
- 239000000017hydrogelSubstances0.000description2
- 239000001866hydroxypropyl methyl celluloseSubstances0.000description2
- 235000010979hydroxypropyl methyl celluloseNutrition0.000description2
- 229920003088hydroxypropyl methyl cellulosePolymers0.000description2
- UFVKGYZPFZQRLF-UHFFFAOYSA-Nhydroxypropyl methyl celluloseChemical compoundOC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1UFVKGYZPFZQRLF-UHFFFAOYSA-N0.000description2
- 230000002401inhibitory effectEffects0.000description2
- 150000007529inorganic basesChemical class0.000description2
- 230000010354integrationEffects0.000description2
- 230000003993interactionEffects0.000description2
- 230000004410intraocular pressureEffects0.000description2
- 239000003446ligandSubstances0.000description2
- 238000011068loading methodMethods0.000description2
- 239000012931lyophilized formulationSubstances0.000description2
- 230000007246mechanismEffects0.000description2
- 229920000609methyl cellulosePolymers0.000description2
- 239000001923methylcelluloseSubstances0.000description2
- 235000010981methylcelluloseNutrition0.000description2
- LXCFILQKKLGQFO-UHFFFAOYSA-NmethylparabenChemical compoundCOC(=O)C1=CC=C(O)C=C1LXCFILQKKLGQFO-UHFFFAOYSA-N0.000description2
- 150000004682monohydratesChemical class0.000description2
- 210000003205muscleAnatomy0.000description2
- 239000002090nanochannelSubstances0.000description2
- 231100000252nontoxicToxicity0.000description2
- 230000003000nontoxic effectEffects0.000description2
- 210000000056organAnatomy0.000description2
- 150000002898organic sulfur compoundsChemical class0.000description2
- 230000036407painEffects0.000description2
- MQHIQUBXFFAOMK-UHFFFAOYSA-Npazopanib hydrochlorideChemical groupCl.C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1MQHIQUBXFFAOMK-UHFFFAOYSA-N0.000description2
- 125000002467phosphate groupChemical group[H]OP(=O)(O[H])O[*]0.000description2
- 102000020233phosphotransferaseHuman genes0.000description2
- 239000003880polar aprotic solventSubstances0.000description2
- 239000005373porous glassSubstances0.000description2
- 230000000750progressive effectEffects0.000description2
- 230000002062proliferating effectEffects0.000description2
- QELSKZZBTMNZEB-UHFFFAOYSA-NpropylparabenChemical compoundCCCOC(=O)C1=CC=C(O)C=C1QELSKZZBTMNZEB-UHFFFAOYSA-N0.000description2
- HNJBEVLQSNELDL-UHFFFAOYSA-Npyrrolidin-2-oneChemical compoundO=C1CCCN1HNJBEVLQSNELDL-UHFFFAOYSA-N0.000description2
- 230000002829reductive effectEffects0.000description2
- 230000004264retinal detachmentEffects0.000description2
- 210000003583retinal pigment epitheliumAnatomy0.000description2
- 238000012552reviewMethods0.000description2
- 238000004062sedimentationMethods0.000description2
- 235000010413sodium alginateNutrition0.000description2
- 239000000661sodium alginateSubstances0.000description2
- 229940005550sodium alginateDrugs0.000description2
- 229940010747sodium hyaluronateDrugs0.000description2
- YWIVKILSMZOHHF-QJZPQSOGSA-Nsodium;(2s,3s,4s,5r,6r)-6-[(2s,3r,4r,5s,6r)-3-acetamido-2-[(2s,3s,4r,5r,6r)-6-[(2r,3r,4r,5s,6r)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-Chemical compound[Na+].CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1YWIVKILSMZOHHF-QJZPQSOGSA-N0.000description2
- 239000012439solid excipientSubstances0.000description2
- 235000010356sorbitolNutrition0.000description2
- 239000000600sorbitolSubstances0.000description2
- 238000001179sorption measurementMethods0.000description2
- 241000894007speciesSpecies0.000description2
- 230000006641stabilisationEffects0.000description2
- 238000011105stabilizationMethods0.000description2
- 231100001274therapeutic indexToxicity0.000description2
- 238000012546transferMethods0.000description2
- 230000008733traumaEffects0.000description2
- DSDAICPXUXPBCC-MWDJDSKUSA-Ntrimethyl-β-cyclodextrinChemical compoundCOC[C@H]([C@H]([C@@H]([C@H]1OC)OC)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COC)[C@H]([C@@H]([C@H]3OC)OC)O[C@H]3O[C@H](COC)[C@H]([C@@H]([C@H]3OC)OC)O[C@H]3O[C@H](COC)[C@H]([C@@H]([C@H]3OC)OC)O[C@H]3O[C@H](COC)[C@H]([C@@H]([C@H]3OC)OC)O3)[C@H](OC)[C@H]2OC)COC)O[C@@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@@H]3O[C@@H]1COCDSDAICPXUXPBCC-MWDJDSKUSA-N0.000description2
- 239000002525vasculotropin inhibitorSubstances0.000description2
- 239000003981vehicleSubstances0.000description2
- 230000000007visual effectEffects0.000description2
- 238000004876x-ray fluorescenceMethods0.000description2
- BJEPYKJPYRNKOW-REOHCLBHSA-N(S)-malic acidChemical compoundOC(=O)[C@@H](O)CC(O)=OBJEPYKJPYRNKOW-REOHCLBHSA-N0.000description1
- QCQCHGYLTSGIGX-GHXANHINSA-N4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acidChemical compoundN([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1QCQCHGYLTSGIGX-GHXANHINSA-N0.000description1
- QTBSBXVTEAMEQO-UHFFFAOYSA-MAcetateChemical compoundCC([O-])=OQTBSBXVTEAMEQO-UHFFFAOYSA-M0.000description1
- 208000030090Acute DiseaseDiseases0.000description1
- 229920001817AgarPolymers0.000description1
- GUBGYTABKSRVRQ-XLOQQCSPSA-NAlpha-LactoseChemical compoundO[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1OGUBGYTABKSRVRQ-XLOQQCSPSA-N0.000description1
- USFZMSVCRYTOJT-UHFFFAOYSA-NAmmonium acetateChemical compoundN.CC(O)=OUSFZMSVCRYTOJT-UHFFFAOYSA-N0.000description1
- 239000005695Ammonium acetateSubstances0.000description1
- 206010002091AnaesthesiaDiseases0.000description1
- 241000416162Astragalus gummiferSpecies0.000description1
- IJGRMHOSHXDMSA-UHFFFAOYSA-NAtomic nitrogenChemical compoundN#NIJGRMHOSHXDMSA-UHFFFAOYSA-N0.000description1
- 206010006784Burning sensationDiseases0.000description1
- 241000700198CaviaSpecies0.000description1
- 229920001661ChitosanPolymers0.000description1
- 229920002261Corn starchPolymers0.000description1
- 241000195493CryptophytaSpecies0.000description1
- FBPFZTCFMRRESA-KVTDHHQDSA-ND-MannitolChemical compoundOC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)COFBPFZTCFMRRESA-KVTDHHQDSA-N0.000description1
- FEWJPZIEWOKRBE-JCYAYHJZSA-NDextrotartaric acidChemical compoundOC(=O)[C@H](O)[C@@H](O)C(O)=OFEWJPZIEWOKRBE-JCYAYHJZSA-N0.000description1
- LVGKNOAMLMIIKO-UHFFFAOYSA-NElaidinsaeure-aethylesterNatural productsCCCCCCCCC=CCCCCCCCC(=O)OCCLVGKNOAMLMIIKO-UHFFFAOYSA-N0.000description1
- 102000004190EnzymesHuman genes0.000description1
- 108090000790EnzymesProteins0.000description1
- IAYPIBMASNFSPL-UHFFFAOYSA-NEthylene oxideChemical compoundC1CO1IAYPIBMASNFSPL-UHFFFAOYSA-N0.000description1
- 108010010803GelatinProteins0.000description1
- WQZGKKKJIJFFOK-GASJEMHNSA-NGlucoseNatural productsOC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1OWQZGKKKJIJFFOK-GASJEMHNSA-N0.000description1
- 241000282412HomoSpecies0.000description1
- 101600082430Homo sapiens Vascular endothelial growth factor A (isoform VEGF165)Proteins0.000description1
- UFHFLCQGNIYNRP-UHFFFAOYSA-NHydrogenChemical compound[H][H]UFHFLCQGNIYNRP-UHFFFAOYSA-N0.000description1
- 206010021143HypoxiaDiseases0.000description1
- 206010061218InflammationDiseases0.000description1
- 206010022998IrritabilityDiseases0.000description1
- 235000019766L-LysineNutrition0.000description1
- GUBGYTABKSRVRQ-QKKXKWKRSA-NLactoseNatural productsOC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1OGUBGYTABKSRVRQ-QKKXKWKRSA-N0.000description1
- 239000004472LysineSubstances0.000description1
- 206010025415Macular oedemaDiseases0.000description1
- 241000124008MammaliaSpecies0.000description1
- 229930195725MannitolNatural products0.000description1
- RZCHTMXTKQHYDT-UHFFFAOYSA-NN-Lactoyl ethanolamineChemical compoundCC(O)C(=O)NCCORZCHTMXTKQHYDT-UHFFFAOYSA-N0.000description1
- 229910019142PO4Inorganic materials0.000description1
- 150000004931Pazopanib derivativesChemical class0.000description1
- 235000019483Peanut oilNutrition0.000description1
- 229920003171Poly (ethylene oxide)Polymers0.000description1
- 241000288906PrimatesSpecies0.000description1
- GOOHAUXETOMSMM-UHFFFAOYSA-NPropylene oxideChemical compoundCC1CO1GOOHAUXETOMSMM-UHFFFAOYSA-N0.000description1
- 208000008709Retinal TelangiectasisDiseases0.000description1
- 208000017442Retinal diseaseDiseases0.000description1
- 206010038923RetinopathyDiseases0.000description1
- KEAYESYHFKHZAL-UHFFFAOYSA-NSodiumChemical class[Na]KEAYESYHFKHZAL-UHFFFAOYSA-N0.000description1
- 229920002472StarchPolymers0.000description1
- 229930006000SucroseNatural products0.000description1
- 235000019486Sunflower oilNutrition0.000description1
- FEWJPZIEWOKRBE-UHFFFAOYSA-NTartaric acidNatural products[H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=OFEWJPZIEWOKRBE-UHFFFAOYSA-N0.000description1
- RTAQQCXQSZGOHL-UHFFFAOYSA-NTitaniumChemical compound[Ti]RTAQQCXQSZGOHL-UHFFFAOYSA-N0.000description1
- 229920001615TragacanthPolymers0.000description1
- 102000009484Vascular Endothelial Growth Factor ReceptorsHuman genes0.000description1
- 102300041083Vascular endothelial growth factor A isoform VEGF165Human genes0.000description1
- 206010047571Visual impairmentDiseases0.000description1
- 208000027418Wounds and injuryDiseases0.000description1
- GCSPRLPXTPMSTL-IBDNADADSA-N[(2s,3r,4s,5s,6r)-2-[(2s,3s,4s,5r)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl] dodecanoateChemical compoundCCCCCCCCCCCC(=O)O[C@@]1([C@]2(CO)[C@H]([C@H](O)[C@@H](CO)O2)O)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1OGCSPRLPXTPMSTL-IBDNADADSA-N0.000description1
- 230000002378acidificating effectEffects0.000description1
- 230000004913activationEffects0.000description1
- 230000006978adaptationEffects0.000description1
- 239000008272agarSubstances0.000description1
- 235000010443alginic acidNutrition0.000description1
- 239000000783alginic acidSubstances0.000description1
- 229920000615alginic acidPolymers0.000description1
- 229960001126alginic acidDrugs0.000description1
- 150000004781alginic acidsChemical class0.000description1
- BJEPYKJPYRNKOW-UHFFFAOYSA-Nalpha-hydroxysuccinic acidNatural productsOC(=O)C(O)CC(O)=OBJEPYKJPYRNKOW-UHFFFAOYSA-N0.000description1
- WNROFYMDJYEPJX-UHFFFAOYSA-Kaluminium hydroxideChemical compound[OH-].[OH-].[OH-].[Al+3]WNROFYMDJYEPJX-UHFFFAOYSA-K0.000description1
- 235000019257ammonium acetateNutrition0.000description1
- 229940043376ammonium acetateDrugs0.000description1
- 238000005280amorphizationMethods0.000description1
- 230000037005anaesthesiaEffects0.000description1
- 230000003444anaesthetic effectEffects0.000description1
- 230000000202analgesic effectEffects0.000description1
- 210000003484anatomyAnatomy0.000description1
- 230000033115angiogenesisEffects0.000description1
- 238000013459approachMethods0.000description1
- 239000012062aqueous bufferSubstances0.000description1
- 238000010420art techniqueMethods0.000description1
- WQZGKKKJIJFFOK-VFUOTHLCSA-Nbeta-D-glucoseChemical compoundOC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1OWQZGKKKJIJFFOK-VFUOTHLCSA-N0.000description1
- 230000002457bidirectional effectEffects0.000description1
- 239000011230binding agentSubstances0.000description1
- 230000008275binding mechanismEffects0.000description1
- 239000000560biocompatible materialSubstances0.000description1
- 230000004071biological effectEffects0.000description1
- 230000015572biosynthetic processEffects0.000description1
- 230000000740bleeding effectEffects0.000description1
- 210000004155blood-retinal barrierAnatomy0.000description1
- 230000004378blood-retinal barrierEffects0.000description1
- 230000037396body weightEffects0.000description1
- 230000003139buffering effectEffects0.000description1
- 238000004364calculation methodMethods0.000description1
- 230000015556catabolic processEffects0.000description1
- 230000003197catalytic effectEffects0.000description1
- 229920002678cellulosePolymers0.000description1
- 239000001913celluloseSubstances0.000description1
- 229920002301cellulose acetatePolymers0.000description1
- 230000008859changeEffects0.000description1
- 239000003153chemical reaction reagentSubstances0.000description1
- 229940045110chitosanDrugs0.000description1
- UETQVDZZPKAQIC-UHFFFAOYSA-NchloraneChemical compoundCl.Cl.Cl.ClUETQVDZZPKAQIC-UHFFFAOYSA-N0.000description1
- 239000011248coating agentSubstances0.000description1
- 238000000576coating methodMethods0.000description1
- 229940110456cocoa butterDrugs0.000description1
- 235000019868cocoa butterNutrition0.000description1
- 230000000052comparative effectEffects0.000description1
- 239000000306componentSubstances0.000description1
- 235000005687corn oilNutrition0.000description1
- 239000002285corn oilSubstances0.000description1
- 239000008120corn starchSubstances0.000description1
- 235000012343cottonseed oilNutrition0.000description1
- 239000002385cottonseed oilSubstances0.000description1
- 230000006378damageEffects0.000description1
- 230000003247decreasing effectEffects0.000description1
- 230000007812deficiencyEffects0.000description1
- 238000006731degradation reactionMethods0.000description1
- 238000002716delivery methodMethods0.000description1
- 230000008021depositionEffects0.000description1
- 238000011161developmentMethods0.000description1
- 206010012601diabetes mellitusDiseases0.000description1
- 229910001873dinitrogenInorganic materials0.000description1
- 208000002173dizzinessDiseases0.000description1
- 229940126534drug productDrugs0.000description1
- 238000001035dryingMethods0.000description1
- 238000010828elutionMethods0.000description1
- LVGKNOAMLMIIKO-QXMHVHEDSA-Nethyl oleateChemical compoundCCCCCCCC\C=C/CCCCCCCC(=O)OCCLVGKNOAMLMIIKO-QXMHVHEDSA-N0.000description1
- 229940093471ethyl oleateDrugs0.000description1
- 229940117927ethylene oxideDrugs0.000description1
- 208000030533eye diseaseDiseases0.000description1
- 230000004438eyesightEffects0.000description1
- 210000003195fasciaAnatomy0.000description1
- 239000000945fillerSubstances0.000description1
- 239000006260foamSubstances0.000description1
- 239000007789gasSubstances0.000description1
- 239000008273gelatinSubstances0.000description1
- 229920000159gelatinPolymers0.000description1
- 235000019322gelatineNutrition0.000description1
- 235000011852gelatine dessertsNutrition0.000description1
- 239000008103glucoseSubstances0.000description1
- 150000002334glycolsChemical class0.000description1
- 230000002650habitual effectEffects0.000description1
- LNEPOXFFQSENCJ-UHFFFAOYSA-Nhaldol DecanoateNatural productsC1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1LNEPOXFFQSENCJ-UHFFFAOYSA-N0.000description1
- 229960003878haloperidolDrugs0.000description1
- 125000005842heteroatomChemical group0.000description1
- 239000012456homogeneous solutionSubstances0.000description1
- 239000001257hydrogenSubstances0.000description1
- 229910052739hydrogenInorganic materials0.000description1
- 230000005661hydrophobic surfaceEffects0.000description1
- 206010020718hyperplasiaDiseases0.000description1
- 230000007954hypoxiaEffects0.000description1
- 238000000338in vitroMethods0.000description1
- 230000001939inductive effectEffects0.000description1
- 230000004054inflammatory processEffects0.000description1
- 239000003112inhibitorSubstances0.000description1
- 230000000977initiatory effectEffects0.000description1
- 230000003834intracellular effectEffects0.000description1
- 238000010253intravenous injectionMethods0.000description1
- 150000004694iodide saltsChemical class0.000description1
- 230000001788irregularEffects0.000description1
- 239000008101lactoseSubstances0.000description1
- 239000012669liquid formulationSubstances0.000description1
- 230000007774longtermEffects0.000description1
- 201000010230macular retinal edemaDiseases0.000description1
- VTHJTEIRLNZDEV-UHFFFAOYSA-Lmagnesium dihydroxideChemical compound[OH-].[OH-].[Mg+2]VTHJTEIRLNZDEV-UHFFFAOYSA-L0.000description1
- 239000000347magnesium hydroxideSubstances0.000description1
- 229910001862magnesium hydroxideInorganic materials0.000description1
- 238000009115maintenance therapyMethods0.000description1
- 239000001630malic acidSubstances0.000description1
- 235000011090malic acidNutrition0.000description1
- 239000000594mannitolSubstances0.000description1
- 235000010355mannitolNutrition0.000description1
- 229910052751metalInorganic materials0.000description1
- 239000002184metalSubstances0.000description1
- 239000004292methyl p-hydroxybenzoateSubstances0.000description1
- 235000010270methyl p-hydroxybenzoateNutrition0.000description1
- 125000000250methylamino groupChemical group[H]N(*)C([H])([H])[H]0.000description1
- 229960002216methylparabenDrugs0.000description1
- 239000000693micelleSubstances0.000description1
- PJUIMOJAAPLTRJ-UHFFFAOYSA-NmonothioglycerolChemical compoundOCC(O)CSPJUIMOJAAPLTRJ-UHFFFAOYSA-N0.000description1
- 239000002105nanoparticleSubstances0.000description1
- 239000003921oilSubstances0.000description1
- 235000019198oilsNutrition0.000description1
- 239000004006olive oilSubstances0.000description1
- 235000008390olive oilNutrition0.000description1
- 230000003287optical effectEffects0.000description1
- 238000001139pH measurementMethods0.000description1
- 238000002638palliative careMethods0.000description1
- 230000036961partial effectEffects0.000description1
- 230000037361pathwayEffects0.000description1
- 239000000312peanut oilSubstances0.000description1
- 230000035515penetrationEffects0.000description1
- 230000002085persistent effectEffects0.000description1
- 239000008177pharmaceutical agentSubstances0.000description1
- 229940124531pharmaceutical excipientDrugs0.000description1
- NBIIXXVUZAFLBC-UHFFFAOYSA-KphosphateChemical compound[O-]P([O-])([O-])=ONBIIXXVUZAFLBC-UHFFFAOYSA-K0.000description1
- 239000010452phosphateSubstances0.000description1
- 239000008055phosphate buffer solutionSubstances0.000description1
- 150000003904phospholipidsChemical class0.000description1
- 230000004962physiological conditionEffects0.000description1
- 230000035479physiological effects, processes and functionsEffects0.000description1
- 229920003023plasticPolymers0.000description1
- 239000004033plasticSubstances0.000description1
- 229920005862polyolPolymers0.000description1
- 150000003077polyolsChemical class0.000description1
- 229920001451polypropylene glycolPolymers0.000description1
- 229950008882polysorbateDrugs0.000description1
- 229920000136polysorbatePolymers0.000description1
- 229920001592potato starchPolymers0.000description1
- 125000002924primary amino groupChemical group[H]N([H])*0.000description1
- 201000007914proliferative diabetic retinopathyDiseases0.000description1
- 238000011321prophylaxisMethods0.000description1
- 239000004405propyl p-hydroxybenzoateSubstances0.000description1
- 235000010232propyl p-hydroxybenzoateNutrition0.000description1
- 229960003415propylparabenDrugs0.000description1
- 230000001681protective effectEffects0.000description1
- 239000000376reactantSubstances0.000description1
- 108091008598receptor tyrosine kinasesProteins0.000description1
- 102000027426receptor tyrosine kinasesHuman genes0.000description1
- 108020003175receptorsProteins0.000description1
- 102000005962receptorsHuman genes0.000description1
- 238000012827research and developmentMethods0.000description1
- 230000002207retinal effectEffects0.000description1
- 230000002441reversible effectEffects0.000description1
- JQXXHWHPUNPDRT-WLSIYKJHSA-NrifampicinChemical compoundO([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1JQXXHWHPUNPDRT-WLSIYKJHSA-N0.000description1
- 229960001225rifampicinDrugs0.000description1
- 230000037390scarringEffects0.000description1
- 230000035807sensationEffects0.000description1
- 230000035945sensitivityEffects0.000description1
- 230000001953sensory effectEffects0.000description1
- 239000008159sesame oilSubstances0.000description1
- 235000011803sesame oilNutrition0.000description1
- 230000019491signal transductionEffects0.000description1
- 230000011664signalingEffects0.000description1
- 238000004513sizingMethods0.000description1
- 150000003384small moleculesChemical group0.000description1
- 239000011780sodium chlorideSubstances0.000description1
- 239000001488sodium phosphateSubstances0.000description1
- 229910000162sodium phosphateInorganic materials0.000description1
- 230000003381solubilizing effectEffects0.000description1
- 239000003549soybean oilSubstances0.000description1
- 235000012424soybean oilNutrition0.000description1
- 238000010561standard procedureMethods0.000description1
- 235000019698starchNutrition0.000description1
- 239000005720sucroseSubstances0.000description1
- 235000000346sugarNutrition0.000description1
- 150000008163sugarsChemical class0.000description1
- 239000002600sunflower oilSubstances0.000description1
- 239000013589supplementSubstances0.000description1
- 239000000829suppositorySubstances0.000description1
- 239000004094surface-active agentSubstances0.000description1
- 238000003786synthesis reactionMethods0.000description1
- 230000009885systemic effectEffects0.000description1
- 239000000454talcSubstances0.000description1
- 229910052623talcInorganic materials0.000description1
- 239000011975tartaric acidSubstances0.000description1
- 235000002906tartaric acidNutrition0.000description1
- 238000011287therapeutic doseMethods0.000description1
- 230000004797therapeutic responseEffects0.000description1
- 239000002562thickening agentSubstances0.000description1
- 125000000341threoninyl groupChemical group[H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O0.000description1
- 239000010936titaniumSubstances0.000description1
- 229910052719titaniumInorganic materials0.000description1
- 235000010487tragacanthNutrition0.000description1
- 239000000196tragacanthSubstances0.000description1
- 229940116362tragacanthDrugs0.000description1
- 230000001131transforming effectEffects0.000description1
- 230000001052transient effectEffects0.000description1
- 108091008578transmembrane receptorsProteins0.000description1
- 102000027257transmembrane receptorsHuman genes0.000description1
- UFTFJSFQGQCHQW-UHFFFAOYSA-NtriforminChemical compoundO=COCC(OC=O)COC=OUFTFJSFQGQCHQW-UHFFFAOYSA-N0.000description1
- RYFMWSXOAZQYPI-UHFFFAOYSA-Ktrisodium phosphateChemical compound[Na+].[Na+].[Na+].[O-]P([O-])([O-])=ORYFMWSXOAZQYPI-UHFFFAOYSA-K0.000description1
- 238000011144upstream manufacturingMethods0.000description1
- 229940124676vascular endothelial growth factor receptorDrugs0.000description1
- 230000008728vascular permeabilityEffects0.000description1
- 208000029257vision diseaseDiseases0.000description1
- 230000004393visual impairmentEffects0.000description1
- 239000011800void materialSubstances0.000description1
Images






Classifications
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
- A61K47/183—Amino acids, e.g. glycine, EDTA or aspartame
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/20—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing sulfur, e.g. dimethyl sulfoxide [DMSO], docusate, sodium lauryl sulfate or aminosulfonic acids
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/40—Cyclodextrins; Derivatives thereof
- A61K47/4823—
- A61K47/48969—
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/61—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule the organic macromolecular compound being a polysaccharide or a derivative thereof
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6949—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit inclusion complexes, e.g. clathrates, cavitates or fullerenes
- A61K47/6951—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit inclusion complexes, e.g. clathrates, cavitates or fullerenes using cyclodextrin
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0048—Eye, e.g. artificial tears
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0048—Eye, e.g. artificial tears
- A61K9/0051—Ocular inserts, ocular implants
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M5/00—Devices for bringing media into the body in a subcutaneous, intra-vascular or intramuscular way; Accessories therefor, e.g. filling or cleaning devices, arm-rests
- A61M5/14—Infusion devices, e.g. infusing by gravity; Blood infusion; Accessories therefor
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Ophthalmology & Optometry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Inorganic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Heart & Thoracic Surgery (AREA)
- Vascular Medicine (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Anesthesiology (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Translated fromKorean
Description
Translated fromKorean관련 출원Related application
본 출원은 2014년 8월 8일에 출원된 미국 특허 가출원 62/035274를 우선권 주장하며, 이 가출원의 개시내용은 그 전문이 참조로 포함된다.This application claims priority to U.S. Provisional Patent Application No. 62/035274, filed Aug. 8, 2014, the disclosure of which is incorporated herein by reference in its entirety.
기술분야Technical field
본 개시내용은 수용체 티로신 키나제 억제제 (TKI), 예를 들어 파조파닙의 안정한 제제; 그의 제조 방법; 및 활성 작용제의 표적 부위로의 서방성 전달에서의 개시된 제제의 용도에 관한 것이다. 본 개시내용은 추가로 TKI의 하나의 다형체 형태를 또 다른 다형체 형태 및/또는 무정형 형태로 전환시키는 방법에 관한 것이다.The present disclosure relates to receptor tyrosine kinase inhibitors (TKI), for example stabilizers of pazopanib; Its preparation method; And the use of the disclosed agents in sustained delivery to the target site of an active agent. The present disclosure further relates to a method of converting one polymorphic form of TKI to another polymorphic form and / or amorphous form.
낮은 수중 용해도를 갖는 치료제 제제의 제조 및 치료제의 표적 조직으로의 전달은 약리학자 및 치료제 전달 과학자에게 주요한 난제였다. 문헌 [Gaudana R. et al.,Ocular Therapeutic agent Delivery, AAPS J., 12(3): 348-360 (2010)]을 참조한다. 안구 질환 또는 장애의 치료에 있어서 눈의 특이한 해부학 및 생리학과 치료제의 낮은 수용해도의 조합된 영향으로 인해 이들 치료제의 눈의 목적하는 표적 부위로의 전달이 어려워졌다. 가우다나(Gaudana) 문헌을 참조한다. 따라서, 치료제의 높은 용해도를 가능하게 하고 표적 조직에서의 안정성 및 효능을 개선할 제제 및 전달 시스템이 요구된다.The preparation of therapeutic agents with low water solubility and delivery of therapeutic agents to target tissues has been a major challenge for pharmacologists and drug delivery scientists. See Gaudana R. et al.,Ocular Therapeutic Agent Delivery , AAPS J., 12 (3): 348-360 (2010). The combination of the specific anatomy of the eye and the physiology of the eye and the low water solubility of the therapeutic agent in the treatment of an eye disease or disorder has made it difficult to deliver these therapeutic agents to the intended target site of the eye. See the Gaudana literature. Accordingly, there is a need for agents and delivery systems that enable high solubility of therapeutic agents and improve stability and efficacy in target tissues.
단백질 키나제는 연령-관련 황반 변성 (이하, "AMD"), 당뇨병성 황반 부종 및 증식성 당뇨병성 망막병증을 비제한적으로 포함하는 안구 질환과 관련있다. 막횡단 수용체 단백질 키나제는 리간드 결합 능력이 있는 세포외 도메인을 나타낸다. 이러한 리간드 결합 메카니즘은 키나제 촉매 도메인의 활성화를 촉발시키고, 이는 세포내 기능을 제어하는 신호의 캐스케이드를 개시한다.Protein kinases are associated with ocular diseases that include, but are not limited to, age-related macular degeneration (hereinafter "AMD"), diabetic macular edema, and proliferative diabetic retinopathy. Transmembrane receptor protein kinases represent extracellular domains with ligand binding capacity. This ligand binding mechanism triggers the activation of the kinase catalytic domain, which initiates a cascade of signals that control intracellular function.
수용체 단백질 키나제의 예는 성장 인자, 예컨대 EGF, FGF, VEGF, PDGF 및 IGF이다. 가용성 성장 인자, 예컨대 혈관 내피 성장 인자-A (VEGF)의 상승된 수준이 병리학적 안구 혈관신생을 갖는 환자에게서 제거된 안구 조직 및 안액에서 확인되었다. 감각신경 망막 및 망막 색소 상피 (RPE)를 포함하는 다양한 안구 조직은 혈액-망막 장벽 붕괴 (즉, 증대된 혈관 투과성 및 세포외 부종) 및/또는 병리학적 신생혈관화 (NV)를 유도할 수 있는 VEGF 발현의 증가를 통해 저산소증, 염증, 및 외상에 반응하는 것으로 공지되어 있다.Examples of receptor protein kinases are growth factors such as EGF, FGF, VEGF, PDGF and IGF. Elevated levels of soluble growth factors, such as vascular endothelial growth factor-A (VEGF), have been identified in ocular tissues and eye fluids removed from patients with pathological ocular angiogenesis. Various ocular tissues, including the sensory and retinal pigment epithelium (RPE), are known to be capable of inducing blood-retinal barrier disruption (i.e., increased vascular permeability and extracellular edema) and / or pathological neovascularization (NV) It is known to respond to hypoxia, inflammation, and trauma through increased expression of VEGF.
눈에서의 치료제 전달은 난제이다. 만성적인 유지 요법에서 필요한 반복되는 유리체내 주사 때문에 현재 통용되는 전달 수단에는 주요 단점이 존재한다. 반복되는 유리체내 주사는 환자에게 위험하기도 하고 또한 부담이 되기도 한다. 안내염, 망막 박리, 외상성 백내장, 및 안내압 상승 (IOP)은 모두 유리체내 투여 경로의 잠재적인 시력-위협 후유증이다. 게다가, 한 달에 한 번의 치료 또는 심지어 한 달에 한 번의 모니터링도, 특히 치료가 환자의 평생 동안 계속될 필요가 있을 수 있음을 고려하면, 환자, 환자의 간병인 및 의료진에게 상당한 부담이 된다. 대략 환자의 1/3이 특정 생물학적 VEGF 억제제의 반복되는 유리체내 주사로 치료를 받으면 시력 개선을 경험하지만, 대부분의 환자는 단지 저하된 시력의 안정화를 경험한다.Delivery of remedies from the eye is a challenge. There are major disadvantages to currently available delivery methods due to repeated intravesicular injections required in chronic maintenance therapy. Repeated intravitreal injections are both dangerous and burdensome to the patient. Endophthalmitis, retinal detachment, traumatic cataract, and elevated intraocular pressure (IOP) are both potential eye-threatening sequelae of the vitreous route of administration. In addition, once a month, or even once a month, a considerable burden is placed on the patient, the patient's caregiver, and the medical staff, especially considering that the treatment may need to be continued throughout the patient's lifetime. Approximately one-third of patients experience visual improvement when treated with repeated intra-vitals injection of a specific biological VEGF inhibitor, but most patients experience only a stabilization of the deteriorated visual acuity.
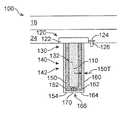
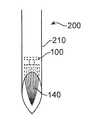
제제는 적어도 일부 경우에 치료 장치에 주입되었을 때 하나 이상의 방식으로 이상적이지 않은 안정성을 제공할 수 있다. 예를 들어, 주입된 제제의 완충제가 적어도 일부 경우에 장치에서 유리체 내로 방출될 수 있다. 또한, 저장소와 유리체 사이에서의 수소 이온 및 히드록시드 이온의 확산이 장치 내의 제제의 pH에 영향을 줄 수 있다.The formulation may provide stability that is not ideal in one or more ways when injected into the treatment device, at least in some instances. For example, the buffer of the injected preparation may be released into the vitreous in the device, at least in some cases. Also, diffusion of hydrogen and hydroxide ions between the reservoir and the vitreous can affect the pH of the formulation in the device.
적어도 일부 경우에, 생리학적 pH를 갖는 유리액과 같은 안액의 완충액이 장치에 들어가 장치 내의 제제의 pH에 영향을 줄 수 있으므로, 치료제의 안정성이 적어도 일부 경우에 이상적이지 않을 수 있다.In at least some cases, the stability of the therapeutic agent may not be ideal, at least in some cases, because an aqueous buffer solution, such as a glass liquid having a physiological pH, can enter the device and affect the pH of the formulation in the device.
적어도 일부 경우에, 치료제의 용해도를 증가시키기 위해 첨가된 제제화 성분이 치료제와 매우 강력하게 결합할 수 있어, 표적 조직에서의 효능이 적어도 일부 경우에 이상적이지 않을 수 있다.At least in some cases, the added formulation components to increase the solubility of the therapeutic agent can bind very strongly to the therapeutic agent, so that efficacy in the target tissue may not be ideal in at least some cases.
상기에 비추어 볼 때, 공지된 제제의 상기 결함 중 적어도 일부를 극복한 치료 장치를 위한 치료제의 개선된 제제, 예를 들어 이식되었을 때 연장된 시간 동안 유지가능한 개선된 치료제 방출을 갖는 제제를 제공하는 것이 바람직하다.In view of the above, it would be desirable to provide an improved formulation of a therapeutic agent for a therapeutic device that overcomes at least some of the deficiencies of a known formulation, for example, an agent with improved therapeutic agent release that can be maintained for extended periods of time when implanted .
달리 정의되지 않는 한, 본원에 사용된 모든 기술 및 과학 용어는 본 발명이 속하는 기술분야의 통상의 기술자에게 일반적으로 이해되는 것과 동일한 의미를 갖는다. 본원에 기재된 것과 유사한 또는 등가의 방법 및 물질이 본 발명을 실시하거나 또는 시험할 때 사용될 수 있지만, 적합한 방법 및 물질이 하기에 기재되어 있다. 본원에 언급된 모든 공보, 특허 출원, 특허 및 다른 참고문헌은 그 전문이 참조로 포함된다. 상충하는 경우에는, 정의를 포함하여 본 명세서가 우선할 것이다. 또한, 물질, 방법 및 실시예는 단지 예시일 뿐이며 제한하려는 것이 아니다.Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood to one of ordinary skill in the art to which this invention belongs. Although suitable or equivalent methods and materials similar to those described herein can be used in the practice or testing of the present invention, suitable methods and materials are described below. All publications, patent applications, patents, and other references mentioned herein are incorporated by reference in their entirety. In case of conflict, the present specification, including definitions, will control. In addition, the materials, methods, and examples are illustrative only and not intended to be limiting.
본 발명의 다른 특징 및 장점이 하기 상세한 설명 및 청구범위로부터 자명할 것이다.Other features and advantages of the invention will be apparent from the following detailed description and claims.
본 개시내용은 일반적으로 낮은 수중 용해도를 갖는 치료제의 제제에 관한 것이다. 수용체 티로신 키나제 억제제, 예를 들어 파조파닙 제제 및 제조 방법 및 안과 질환 및/또는 장애의 치료 및 완화에 있어서의 용도가 본원에 개시되어 있다.The present disclosure relates generally to formulations of therapeutic agents having low water solubility. Receptor tyrosine kinase inhibitors, such as pachopanim preparations and methods of manufacture and their use in the treatment and amelioration of ocular diseases and / or disorders are disclosed herein.
본 개시내용은 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염, 및 1종 이상의 제제화 작용제의 안정한 제약 제제(들)를 제공하고, 여기서 제약상 허용되는 염은 1가 또는 2가 염이고, 1종 이상의 제제화 작용제는 착물화제, 가용화제, 및 임의로 완충제를 포함하고; 치료제의 염은 제제에 용해되어 있다. 치료제는 파조파닙이다.The disclosure provides a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility and a stable pharmaceutical formulation (s) of one or more formulatory agents wherein the pharmaceutically acceptable salt is a monovalent or divalent salt, Wherein the at least one formulation agent comprises a complexing agent, a solubilizing agent, and optionally a buffering agent; The salt of the therapeutic agent is dissolved in the preparation. The therapeutic agent is Pazopanib.
본 개시내용은 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염, 및 1종 이상의 제제화 작용제의 안정한 제약 제제(들)를 제공하고, 여기서 제약상 허용되는 염은 1가 또는 2가 염이고, 1종 이상의 제제화 작용제는 착물화제, 가용화제, 및 완충제를 포함하고; 치료제의 염은 제제에 용해되어 있다. 치료제는 파조파닙이다.The disclosure provides a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility and a stable pharmaceutical formulation (s) of one or more formulatory agents wherein the pharmaceutically acceptable salt is a monovalent or divalent salt, Wherein the at least one formulation agent comprises a complexing agent, a solubilizing agent, and a buffering agent; The salt of the therapeutic agent is dissolved in the preparation. The therapeutic agent is Pazopanib.
본 개시내용은 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염, 및 1종 이상의 제제화 작용제의 안정한 제약 제제(들)를 제공하고, 여기서 제약상 허용되는 염은 1가 또는 2가 염이고, 1종 이상의 제제화 작용제는 착물화제, 가용화제를 포함하나, 완충제는 포함하지 않고; 치료제의 염은 제제에 용해되어 있다. 치료제는 파조파닙이다.The disclosure provides a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility and a stable pharmaceutical formulation (s) of one or more formulatory agents wherein the pharmaceutically acceptable salt is a monovalent or divalent salt, More than one species of formulation agent includes complexing agents, solubilizing agents, but not buffering agents; The salt of the therapeutic agent is dissolved in the preparation. The therapeutic agent is Pazopanib.
제약상 허용되는 염은 1가 또는 2가 할라이드 염이다. 염은 클로라이드 염이다. 1가 염은 제제에서 약 60 mg/mL의 농도까지 안정하다. 2가 염은 제제에서 약 70 mg/mL의 농도까지 안정하다. 제제화 전의 2가 염 결정 구조는 XRPD에 의해 결정된 형태 XIV이다. 제제에서의 1가 염의 안정성은 제제화 작용제가 포함된 용액에서의 가용화전에 유기 용매로부터의 치료제의 동결건조를 수행함으로써 증가한다. 유기 용매는 디메틸 술폭시드 (DMSO) 또는 트리플루오로 에탄올 (TFE)이다. DMSO로부터의 동결건조는 치료제의 하나의 결정질 상 형태를 또 다른 형태로 전환시킨다. DMSO로부터의 동결건조는 XRPD에 의해 결정될 때, 결정질 상 형태 A를 적어도 약 70%의 결정질 상 형태 G를 함유하는 물질로 전환시킨다. TFE로부터의 동결건조는 결정질 상 형태 A를 부분적으로 또는 완전히 무정형 상으로 전환시킨다. pH는 치료제의 제제화 중에 조정되거나, 또는 pH는 치료제의 제제화 중에 조정되지 않는다.A pharmaceutically acceptable salt is a monovalent or divalent halide salt. The salt is a chloride salt. Monovalent salts are stable up to a concentration of about 60 mg / mL in the formulation. The divalent salt is stable up to a concentration of about 70 mg / mL in the formulation. The divalent salt crystal structure before formulation is form XIV determined by XRPD. The stability of the monovalent salt in the formulation is increased by performing lyophilization of the therapeutic agent from the organic solvent prior to solubilization in the solution containing the formulation agent. The organic solvent is dimethylsulfoxide (DMSO) or trifluoroethanol (TFE). Lyophilization from DMSO converts one crystalline form of the therapeutic into another. Lyophilization from DMSO converts crystalline phase A to a material containing at least about 70% crystalline phase G when determined by XRPD. Freeze-drying from TFE converts the crystalline phase A partially or completely into an amorphous phase. The pH is adjusted during the formulation of the therapeutic agent, or the pH is not adjusted during the formulation of the therapeutic agent.
본 개시내용의 제제화 및 제제의 제조 방법에서 가용화제는 중합체, 예를 들어 폴리(비닐 피롤리돈) (PVP)이고; 완충제는, 존재할 경우에, 히스티딘 HCl이고; 착물화제는 시클로덱스트린: 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 또는 이들의 임의의 조합(들)이며; 치료제는 파조파닙 (5-[[4-[(2,3-디메틸-2H-인다졸-6-일)메틸아미노]-2-피리미디닐]아미노]-2-메틸벤졸술폰아미드) 염, 예를 들어 파조파닙 1HCl 또는 파조파닙 2HCl이다.In the formulation of the present disclosure and the method of preparation of the formulation, the solubilizing agent is a polymer, for example poly (vinylpyrrolidone) (PVP); The buffer, if present, is histidine HCl; The complexing agent may be selected from the group consisting of cyclodextrin: 2-hydroxypropyl-beta-cyclodextrin, methyl-beta-cyclodextrin, random methylated- beta -cyclodextrin, ethylated- beta -cyclodextrin, triacetyl-beta-cyclodextrin, Beta-cyclodextrin, carboxymethyl- beta -cyclodextrin, hydroxyethyl-beta-cyclodextrin, 2-hydroxy-3- (trimethylammonio) propyl- beta -cyclodextrin, glucosyl- beta -cyclodextrin Cyclodextrin, branched-beta-cyclodextrin, hydroxypropyl-gamma -cyclodextrin, random methylated-gamma -cyclodextrin, trimethyl-gamma -cyclodextrin, Or any combination (s) thereof; The therapeutic agent is selected from the group consisting of pazopanib (5 - [[4- [(2,3-dimethyl-2H-indazol-6-yl) methylamino] -2- pyrimidinyl] amino] -2- For example, Pazopapenib HC1 or Pazopapanib 2HCl.
본 개시내용은 (a) 염을, 착물화제, 가용화제, 및 임의로 완충제를 포함하는 1종 이상의 제제화 작용제의 용액에 용해시키고, (b) 염을 제제화 작용제에 용해시킨 후에 pH를 최적의 값으로 조정하는 것을 포함하는, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 2가 염이다. 본 개시내용은 (a) 염을, 착물화제, 가용화제, 및 완충제를 포함하는 1종 이상의 제제화 작용제의 용액에 용해시키고, (b) 염을 제제화 작용제에 용해시킨 후에 pH를 최적의 값으로 조정하는 것을 포함하는, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 2가 염이다. 본 개시내용은 (a) 염을, 착물화제 및 가용화제를 포함하나, 완충제는 포함하지 않는 1종 이상의 제제화 작용제의 용액에 용해시키고, (b) 염을 제제화 작용제에 용해시킨 후에 pH를 최적의 값으로 조정하는 것을 포함하는, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 2가 염이다.The present disclosure relates to a process for the preparation of a pharmaceutical composition comprising: (a) dissolving a salt in a solution of at least one formulation agent comprising a complexing agent, a solubilizing agent, and optionally a buffering agent; (b) A stable solution of a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility, wherein the salt is a divalent salt. The present disclosure relates to a process for the preparation of a pharmaceutical composition comprising: (a) dissolving a salt in a solution of at least one formulation agent comprising a complexing agent, a solubilizing agent and a buffer; (b) A stable solution of a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility, wherein the salt is a divalent salt. (A) dissolving the salt in a solution of one or more formulation agents, including complexing agents and solubilizing agents but not buffering agents, and (b) dissolving the salt in the formulation agent, Of a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility, wherein the salt is a divalent salt.
본 개시내용은 (a) 염을 염기로 처리하고; (b) 염기 처리된 염을, 착물화제, 가용화제, 및 임의로 완충제를 포함하는 1종 이상의 제제화 작용제의 용액에 용해시키고, (c) pH를 산을 사용하여 약 2 이하의 pH로 조정하는 것을 포함하고, 여기서 염기 처리는 제제에서의 총 염 함량을 증가시키고 산을 사용하는 pH 조정은 제제에서의 염의 용해도를 증가시키는 것인, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 1가 염이다.This disclosure discloses a process for the preparation of (a) treating a salt with a base; (b) dissolving the base treated salt in a solution of one or more formulating agents, including complexing agents, solubilizing agents, and optionally buffering agents, and (c) adjusting the pH to a pH of about 2 or less Wherein the base treatment increases the total salt content in the formulation and the pH adjustment using the acid increases the solubility of the salt in the formulation. A stable solution of a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility, Wherein the salt is a monovalent salt.
본 개시내용은 (a) 유기 용매 중의 염의 용액을 제조하고; (b) 용액을 동결건조시켜 치료제의 동결건조 염을 제조하고; (c) 가용화제 및 완충제를 물에 용해시켜 용액을 제조하고; (d) 착물화제를 용액에 용해시키고; (e) 약 주위 온도 이상에서 동결건조 염을 용액에 첨가하고 혼합하여, 염을 용액에 용해시키는 것을 포함하고; 여기서 제제의 pH는 임의로 조정되는 것인, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 1가 염이다.The present disclosure provides a process for the preparation of (a) a solution of a salt in an organic solvent; (b) lyophilizing the solution to prepare a lyophilized salt of the therapeutic; (c) dissolving the solubilizing agent and the buffer in water to prepare a solution; (d) dissolving the complexing agent in the solution; (e) adding a lyophilized salt to the solution at a temperature above the ambient temperature and mixing to dissolve the salt in the solution; Wherein the pH of the formulation is arbitrarily adjusted, wherein the salt is a monovalent salt, wherein the pH of the formulation is arbitrarily adjusted, wherein the salt is a monovalent salt.
본 개시내용은 (a) 유기 용매 (예를 들어, 트리플루오로 에탄올, 트리플루오로 에탄올-물 혼합물, 또는 디메틸 술폭시드) 중의 염의 용액을 제조하고; (b) 용액을 동결건조시켜 치료제의 동결건조 염을 제조하고; (c) 가용화제 및 완충제를 물에 용해시켜 용액을 제조하고; (d) 일정량의 착물화제를 용액에 용해시켜 저 점도 용액을 제조하고; (e) 약 주위 온도 이상에서 (약 37℃ - 약 50℃) 동결건조 염을 저 점도 용액에 첨가하고 혼합하여 용액에 용해시키고; 저 점도 용액의 pH를 조정하고; (f) 약 2x 더 많은 양의 착물화제를 저 점도 용액에 첨가하여 용해시키는 것을 포함하는, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 1가 염이다.The present disclosure provides a process for preparing a solution of a salt in an organic solvent (e.g., trifluoroethanol, trifluoroethanol-water mixture, or dimethylsulfoxide); (b) lyophilizing the solution to prepare a lyophilized salt of the therapeutic; (c) dissolving the solubilizing agent and the buffer in water to prepare a solution; (d) dissolving a quantity of complexing agent in solution to produce a low viscosity solution; (e) adding a lyophilized salt to the low viscosity solution at a temperature above the ambient temperature (about 37 ° C to about 50 ° C) and mixing and dissolving in the solution; Adjusting the pH of the low viscosity solution; (f) a stable solution of a pharmaceutically acceptable salt of a therapeutically active agent having a low water solubility, which comprises adding and dissolving about 2x greater amount of a complexing agent to a low viscosity solution, wherein the salt is Monovalent salts.
극성 비양성자성 용매로부터의 동결건조는 XRPD에 의해 결정될 때, 결정질 상 형태 A를 적어도 약 70%의 파조파닙의 형태 G를 함유하는 물질로 전환시킨다. 동결건조 염은 약 37℃ 내지 약 50℃의 온도에서 용액에 용해된다. 유기황 화합물로부터의 동결건조는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 약 70% 이하의 또는 적어도 약 70%의 파조파닙 1HCl의 형태 G를 함유하는 물질로 전환시킨다. 디메틸 술폭시드 (DMSO)로부터의 동결건조는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 약 70% 이하의 또는 적어도 약 70%의 파조파닙 1HCl의 형태 G를 함유하는 물질로 전환시킨다. 디메틸 술폭시드 (DMSO)로부터의 동결건조는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 약 100%의 파조파닙 1HCl의 형태 G를 함유하는 물질로 전환시킨다. 알콜로부터의 동결건조 단계는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 파조파닙 1HCl의 무정형 (또는 미세결정질) 물질 형태로 전환시킨다. 알콜, 예를 들어 트리플루오로에탄올 (TFE)로부터의 동결건조는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 파조파닙 1HCl의 무정형 (또는 미세결정질) 물질 형태로 전환시킨다. 파조파닙 1HCl의 형태 A는 알콜, 예를 들어 TFE, 또는 TFE/물 혼합물에 용해된 다음, 용액이 동결건조된다. 동결건조 염은 약 37℃ 내지 약 50℃의 온도에서 용액에 용해된다.Lyophilization from a polar aprotic solvent converts crystalline phase A to a material containing at least about 70% of the Pazopanib form G, as determined by XRPD. The lyophilized salt is dissolved in the solution at a temperature of about 37 캜 to about 50 캜. Freeze drying from the organosulfur compounds converts the crystalline phase A of the ripopanib 1HCl to a material containing form G of about 70% or less, or at least about 70% of the ripopanib HCl, as determined by XRPD. Lyophilization from dimethylsulfoxide (DMSO), when determined by XRPD, involves crystallizing crystalline phase A of Pazopapanil 1HCl to a material containing form G of about 70% or less, or at least about 70% of PazopapenHCI . Lyophilization from dimethylsulfoxide (DMSO) converts the crystalline phase A of PazopapanipHCl to a material containing Form G of about 100% of PazopapanipHCl, as determined by XRPD. The lyophilization step from alcohol converts the crystalline phase A of PazopapanipHCl to the amorphous (or microcrystalline) material form of PazopapinHCI, as determined by XRPD. Lyophilization from an alcohol, such as trifluoroethanol (TFE), converts the crystalline phase A of PazopapanipHCl to the amorphous (or microcrystalline) material form of PazopapinHCI, as determined by XRPD. Form A of rapamphanipHCl is dissolved in an alcohol, such as TFE, or a TFE / water mixture, and the solution is lyophilized. The lyophilized salt is dissolved in the solution at a temperature of about 37 캜 to about 50 캜.
본 개시내용은 (a) 유기 용매 (예를 들어, 트리플루오로 에탄올, 트리플루오로 에탄올-물 혼합물, 또는 디메틸 술폭시드) 중의 염의 용액을 제조하고; (b) 용액을 동결건조시켜 치료제의 동결건조 염을 제조하고; (c) 약 주위 온도 이상에서 (예를 들어, 약 37℃ 내지 약 50℃) 물을 첨가하면서, 적어도 가용화제, 완충제, 착물화제, 및 치료제의 동결건조 염, 예를 들어 파조파닙 1HCl을 연속적으로 혼합하는 것을 포함하는, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 1가 염, 예를 들어 파조파닙 1HCl이다. 제제의 pH는 염기를 사용하여 약 6 내지 약 7로 조정된다.The present disclosure provides a process for preparing a solution of a salt in an organic solvent (e.g., trifluoroethanol, trifluoroethanol-water mixture, or dimethylsulfoxide); (b) lyophilizing the solution to prepare a lyophilized salt of the therapeutic; (c) adding at least one solubilizing agent, buffering agent, complexing agent, and lyophilized salt of the therapeutic agent, such as Pazopapenib 1HCl, at a temperature above the ambient temperature (e.g., from about 37 ° C to about 50 ° C) A stable solution of a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility, comprising continuously mixing, wherein the salt is a monovalent salt such as Pazopanib HCI. The pH of the formulation is adjusted to between about 6 and about 7 using a base.
본 개시내용은 (a) 유기 용매 (예를 들어, 트리플루오로 에탄올, 트리플루오로 에탄올-물 혼합물, 또는 디메틸 술폭시드) 중의 염의 용액을 제조하고; (b) 용액을 동결건조시켜 치료제의 동결건조 염을 제조하고; (c) 약 주위 온도 이상에서 (예를 들어, 약 37℃ 내지 약 50℃) 물을 첨가하면서, 적어도 가용화제, 완충제, 착물화제, 및 치료제의 동결건조 염, 예를 들어 파조파닙 1HCl을 연속적으로 혼합하는 것을 포함하는, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 1가 염, 예를 들어 파조파닙 1HCl이다. 상기 방법에 의해 제조된 제제의 pH는 조정되지 않는다.The present disclosure provides a process for preparing a solution of a salt in an organic solvent (e.g., trifluoroethanol, trifluoroethanol-water mixture, or dimethylsulfoxide); (b) lyophilizing the solution to prepare a lyophilized salt of the therapeutic; (c) adding at least one solubilizing agent, buffering agent, complexing agent, and lyophilized salt of the therapeutic agent, such as Pazopapenib 1HCl, at a temperature above the ambient temperature (e.g., from about 37 ° C to about 50 ° C) A stable solution of a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility, comprising continuously mixing, wherein the salt is a monovalent salt such as Pazopanib HCI. The pH of the formulation prepared by this method is not adjusted.
본 개시내용은 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하는, 파조파닙의 결정 형태 A를 적어도 약 70%의 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다. 본 개시내용은 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하는, 파조파닙의 결정 형태 A를 약 100%의 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다.This disclosure discloses a method of converting crystalline Form A of pazopopanib to a substance containing crystalline Form G of at least about 70% of the wave pan nip, including dissolving Form A in DMSO and lyophilizing the resulting solution to provide. This disclosure provides a method for converting crystalline Form A of pazopopanib to a substance containing crystalline form G of about 100% pazopanib, including dissolving Form A in DMSO and lyophilizing the resulting solution do.
본 개시내용은 대상체 눈의 망막에서의 혈관 누수 또는 신생혈관화 (NV)를 특징으로 하는 장애를 치료하거나, 그의 진행을 예방하거나, 또는 그의 증상을 완화시키는 방법에서의 본 개시내용의 제제(들)의 용도를 제공한다.The present disclosure relates to the preparation of the present disclosure in a method of treating a disorder characterized by vascular leakage or neovascularization (NV) in the retina of a subject's eye, preventing its progression, ). ≪ / RTI >
본 개시내용은 대상체 눈의 망막에서의 혈관 누수 또는 신생혈관화 (NV)를 특징으로 하는 장애를 치료하거나, 그의 진행을 예방하거나, 또는 그의 증상을 완화시키는 방법에 사용하기 위한 의약의 제조에 있어서의 본 개시내용의 제제(들)의 용도를 제공한다.The present disclosure relates to the preparation of a medicament for use in a method of treating a disorder characterized by vascular leakage or neovascularization (NV) in the retina of a subject's eye, preventing the progression thereof, or alleviating its symptoms (S) of this disclosure.
본 개시내용은 치료 장치의 저장소 챔버에 함유된 본 개시내용의 안정한 제제(들)를 포함하고, 여기서 저장소 챔버는 눈의 유리체에서의 치료제의 제어 방출을 위해 다공성 구조에 커플링된 것인 키트를 제공한다.The present disclosure includes a stable formulation (s) of the present disclosure contained in a reservoir chamber of a treatment device, wherein the reservoir chamber is coupled to a porous structure for controlled release of a therapeutic agent in the vitreous of the eye to provide.
본 개시내용은 눈의 유리체에서의 치료제의 제어 방출을 위해 치료제 전달 시스템 내의 다공성 구조에 커플링된 저장소 챔버에 함유된 본 개시내용의 약물 전달 제제(들)를 제공하고; 여기서 제제의 다공성 구조로부터의 제어 방출은 저장소 챔버에서의 치료제 농도보다 적어도 두 자릿수가 더 낮은 유리체에서의 치료제 농도를 발생시킨다.The present disclosure provides a drug delivery formulation (s) of the present disclosure contained in a reservoir chamber coupled to a porous structure in a therapeutic delivery system for controlled release of a therapeutic agent in the vitreous of the eye; Wherein the controlled release from the porous structure of the formulation results in a therapeutic agent concentration in the vitreous which is at least two orders of magnitude lower than the therapeutic agent concentration in the reservoir chamber.
본 개시내용의 제제(들)는 안구 약물 전달 방법에 사용된다. 본 개시내용의 제제(들)는 유리체내 전달 제제이다. 본 개시내용의 제제(들)는 점안제가 아니다. 본 개시내용의 제제(들)는 국소 전달 제제가 아니다. 본 개시내용의 제제(들)는 경구 전달 제제 또는 비경구 전달 제제가 아니다. 본 개시내용의 제제(들)는 눈주위 전달 제제가 아니다.The formulation (s) of the present disclosure are used in ocular drug delivery methods. The formulation (s) of the present disclosure are intravesicular delivery formulations. The formulation (s) of this disclosure are not eye drops. The formulation (s) of the present disclosure are not topical delivery formulations. The formulation (s) of the present disclosure are not oral or parenteral delivery formulations. The formulation (s) of this disclosure is not an ocular delivery formulation.
본 개시내용은 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염, 및 1종 이상의 제제화 작용제의 안정한 제약 제제를, 다공성 구조에 커플링된 저장소 챔버를 포함하는 유리체내 전달 장치로부터 전달하는 것을 포함하고, 여기서 제제는 장치의 저장소에 함유되어 있으며, 제제의 다공성 구조를 통한 저장소로부터의 제어 방출은 유리체에서의 치료제의 반감기를 증가시키고; 제약상 허용되는 염은 1가 또는 2가 염이고, 1종 이상의 제제화 작용제는 착물화제, 가용화제, 및 완충제를 포함하고; 치료제의 염은 제제에 용해되어 있는 것인, 후안부의 안과 질환 또는 장애를 치료 및/또는 완화시키는 방법을 제공한다. 저장소 챔버는 재충전가능하고 장치가 눈에 삽입된 후에 제제가 재충전된다.The present disclosure includes delivering a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility and a stable pharmaceutical formulation of one or more formulation agents from a delivery system within the vitreous body comprising a reservoir chamber coupled to the porous structure Wherein the formulation is contained in a reservoir of the device and controlled release from the reservoir through the porous structure of the formulation increases the half-life of the therapeutic agent in the vitreous; Pharmaceutically acceptable salts are monovalent or divalent salts, wherein at least one formulation agent comprises a complexing agent, a solubilizing agent, and a buffering agent; Wherein the salt of the therapeutic agent is dissolved in the formulation. The reservoir chamber is rechargeable and the formulation is recharged after the device is inserted into the eye.
저장소 챔버는 장치가 눈에 30 - 90일 동안, 또는 최대 6개월 동안 위치한 후에 제제가 재충전된다.The reservoir chamber is recharged after the device has been placed in the eye for 30-90 days, or for up to 6 months.
본 개시내용의 제제(들)로 치료 및/또는 완화되는 안과 질환 또는 장애는 당뇨병성 망막병증, 연령-관련 황반 변성 (AMD), 병리학적 맥락막 신생혈관화 (CNV), 병리학적 망막 신생혈관화, 포도막염, 망막 정맥 폐쇄, 안구 외상, 수술 유래 부종, 수술 유래 신생혈관화, 낭포양 황반 부종, 안구 허혈, 미숙아 망막병증, 코츠병(Coat's disease), 겸상 적혈구 망막병증, 및/또는 신생혈관 녹내장이다.Ocular diseases or disorders that are treated and / or alleviated by the agent (s) of this disclosure include diabetic retinopathy, age-related macular degeneration (AMD), pathological choroidal neovascularization (CNV), pathological retinal neovascularization Ocular ischemia, retinopathy of prematurity, Coat's disease, sickle cell retinopathy, and / or neovascular glaucoma, and / or neovascular glaucoma, and / or neovascular glaucoma to be.
본 개시내용은 결정 형태를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 적어도 약 70%의 파조파닙의 형태 G가 형성되는 것인, 파조파닙, 예를 들어 파조파닙 1HCl의 결정 형태를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다. 본 개시내용은 결정 형태를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고, 여기서 약 100%의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다. 본 개시내용은 결정 형태를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 약 70% 내지 약 100% (예를 들어, 약 70%, 약 71%, 약 72%, 약 73%, 약 74%, 약 75%, 약 76%, 약 77%, 약 78%, 약 79%, 약 80%, 약 81%, 약 82%, 약 83%, 약 84%, 약 85%, 약 86%, 약 87%, 약 88%, 약 89%, 약 90%, 약 91%, 약 92%, 약 93%, 약 94%, 약 95%, 약 96%, 약 97%, 약 98%, 약 99%, 약 100%)의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다.This disclosure includes dissolving the crystal form in DMSO and lyophilizing the resulting solution; Wherein a form G of at least about 70% of the wave pan nip is formed, wherein the crystalline form of the wave pan nip, e.g., wave pan nip HCl, is converted to a material containing crystal form G of the wave pan nip do. The present disclosure relates to the determination of the crystal form of the ripopanib, which comprises dissolving the crystalline form in DMSO and lyophilizing the resulting solution, wherein a form G of about 100% of the ripopanib is formed, Lt; RTI ID = 0.0 > G < / RTI > This disclosure includes dissolving the crystal form in DMSO and lyophilizing the resulting solution; Wherein about 70% to about 100% (e. G., About 70%, about 71%, about 72%, about 73%, about 74%, about 75% About 87%, about 87%, about 88%, about 89%, about 90%, about 91%, about 80%, about 80%, about 81%, about 82% , About 92%, about 93%, about 94%, about 95%, about 96%, about 97%, about 98%, about 99%, about 100% Provides a method of converting the crystal form of the pazopanib into the material containing the crystalline form G of the pazapanip.
본 개시내용은 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고, 여기서 적어도 약 70%의 파조파닙의 형태 G가 형성되는 것인, 파조파닙, 예를 들어 파조파닙 1HCl의 결정 형태 A를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다. 본 개시내용은 결정 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 약 100%의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태 A를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다. 본 개시내용은 결정 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 약 70% 내지 약 100% (예를 들어, 약 70%, 약 71%, 약 72%, 약 73%, 약 74%, 약 75%, 약 76%, 약 77%, 약 78%, 약 79%, 약 80%, 약 81%, 약 82%, 약 83%, 약 84%, 약 85%, 약 86%, 약 87%, 약 88%, 약 89%, 약 90%, 약 91%, 약 92%, 약 93%, 약 94%, 약 95%, 약 96%, 약 97%, 약 98%, 약 99%, 약 100%)의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태 A를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다.The present disclosure relates to a method of dissolving Form A in DMSO and lyophilizing the resulting solution, wherein a form G of at least about 70% of the ripopanib is formed, such as a wave pan nip, e.g., ≪ / RTI > of crystalline form A of the papazopub. The present disclosure includes dissolving crystalline Form A in DMSO and lyophilizing the resulting solution; Wherein a form G of about 100% of the ripopanib is formed, wherein the crystalline form A of the pazopapanip is converted to a material containing crystalline form G of the papazopub. The present disclosure includes dissolving crystalline Form A in DMSO and lyophilizing the resulting solution; Wherein about 70% to about 100% (e. G., About 70%, about 71%, about 72%, about 73%, about 74%, about 75% About 87%, about 87%, about 88%, about 89%, about 90%, about 91%, about 80%, about 80%, about 81%, about 82% , About 92%, about 93%, about 94%, about 95%, about 96%, about 97%, about 98%, about 99%, about 100% To provide a method of converting crystal form A of the pazopanip to a material containing crystal form G of the pazapanip.
한 측면에서, 본 개시내용은 전달 장치로부터의 유리체내 전달을 위한, 착물화제, 가용화제, 및 임의로 완충제를 포함하는 파조파닙 1HCl의 안정한 제약 제제를 제공한다. 용해되기 전에, 파조파닙 1HCl은 DMSO 중에서 동결건조되어, 적어도 약 70%의 파조파닙 1HCl의 결정질 상 형태 A가 결정질 상 형태 G로 전환되고 안정성이 증가된다. 이렇게 형성된 제제 중의 파조파닙 1HCl은 희석될 때 및/또는 유리체 내로의 전달 중에 또는 전달 시에 적어도 50일 동안 침전되지 않는다.In one aspect, the present disclosure provides stable pharmaceutical formulations of PachopapanipHCl comprising complexing agents, solubilizing agents, and optionally buffers, for delivery into the vitreous body from a delivery device. Before dissolving, Pazopapenib 1HCl is lyophilized in DMSO, converting at least about 70% of the crystalline phase A of the ripopanib 1HCI to crystalline phase G and increasing its stability. The rapamycin N HCI in the formulation thus formed is not precipitated during dilution and / or during delivery into the vitreous or during transfer for at least 50 days.
한 측면에서, 본 개시내용은 전달 장치로부터의 유리체내 전달을 위한, 착물화제, 가용화제, 및 임의로 완충제를 포함하는 파조파닙 1HCl의 안정한 제약 제제를 제공한다. 용해되기 전에, 파조파닙 1HCl은 트리플루오로 에탄올 (TFE) 중에서 동결건조되어, 파조파닙 1HCl의 결정질 상 형태 A가 부분적으로 또는 완전히 무정형 및/또는 미세결정질 상으로 전환된다. 이렇게 형성된 제제 중의 파조파닙 1HCl은 희석될 때 및/또는 유리체 내로의 전달 중에 또는 전달 시에 적어도 50일 동안 침전되지 않는다.In one aspect, the present disclosure provides stable pharmaceutical formulations of PachopapanipHCl comprising complexing agents, solubilizing agents, and optionally buffers, for delivery into the vitreous body from a delivery device. Before dissolving, Pazopapanib 1HCI is lyophilized in trifluoroethanol (TFE) to convert the crystalline phase A of PazopapanipHCl to a partially or fully amorphous and / or microcrystalline phase. The rapamycin N HCI in the formulation thus formed is not precipitated during dilution and / or during delivery into the vitreous or during transfer for at least 50 days.
본 개시내용은 결정 형태를 TFE에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 96% 이하의 또는 적어도 96%의 무정형 파조파닙이 형성되는 것인, 파조파닙의 결정 형태를 파조파닙의 무정형 형태로 전환시키는 방법을 제공한다.The present disclosure includes dissolving a crystalline form in TFE and lyophilizing the resulting solution; Wherein at least 96% or at least 96% of the amorphous ripple nip is formed, wherein the crystalline form of the ripopanip is converted to the amorphous form of the ripopanib.
본 발명을 이해하고 실제로 수행될 수 있는 방식을 입증하기 위해, 첨부 도면을 참조하여, 단지 비제한적 예시 방식으로, 이제부터 실시양태가 기재되고, 여기서:
도 1은 캅티솔(CAPTISOL)®과 함께 제제화된 파조파닙의 다양한 형태가 충전된 이식물 (4.5 sccm 기체 유량)로부터의 시험관내 약물 방출의 선 그래프를 도시한다. 측정된 방출 데이터는 확산 모델로부터 예측된 방출과 함께 도시된다.
도 2a는 본원에 기재된 변형예에 따른, 망막을 치료하기 위해 눈의 유리액으로 치료제를 방출하는, 결막 아래에 이식되어 공막을 통해 연장되는 치료 장치를 도시한다.
도 2b는 본원에 기재된 변형예에 따른, 도 2a에서와 같이 눈에 위치하도록 구성된 치료 장치의 구조를 도시한다.
도 2c는 본원에 기재된 변형예에 따른, 삽입 캐뉼라에 로딩된 치료 장치를 도시하고, 여기서 장치는 공막으로의 삽입을 위해 신장된 협폭 형상을 포함하고, 또한 장치는 적어도 부분적으로 공막에서의 보유를 위해 제2의 신장된 광폭 형상으로 팽창되도록 구성된다.
도 2d는 본원에 기재된 변형예에 따른, 캐뉼라에 로딩하기에 적합한 저장소를 포함하는 치료 장치를 도시한다.
도 2e는 본원에 기재된 변형예에 따른, 도 2a에서와 같이 눈에 위치하도록 구성된 치료 장치를 도시한다.
도 2f는 치료 장치(100)와의 통합을 위해 적합한 접속 포트(180)를 도시한다.
도 3은 제1 단부에 배치된 관통성 장벽을 갖는 저장소, 연장된 기간 동안 치료제를 방출하는 제2 단부에 배치된 다공성 구조, 및 공막 및 결막에의 커플링을 위해 용기로부터 바깥쪽으로 돌출된 연장부를 포함하는 보유 구조를 포함하는 치료 장치를 도시한다.In order to understand the invention and to demonstrate how it may be practiced, reference is made to the accompanying drawings, in which, by way of nonlimiting example only, an embodiment is now described, in which:
BRIEF DESCRIPTION OF THE DRAWINGS Figure 1 shows a line graph of in vitro drug release from implants (4.5 sccm gas flow rate) filled with various forms of pachoparamips formulated with CAPTISOL®. The measured emission data is shown with the emission predicted from the diffusion model.
Figure 2a illustrates a therapeutic device that is implanted under the conjunctiva and extends through the sclera, releasing the therapeutic agent into the vitreous fluid of the eye to treat the retina, according to a variant described herein.
Figure 2b shows the structure of a treatment device configured to be placed in the eye as in Figure 2a, according to a variant described herein.
Figure 2c illustrates a therapeutic device loaded into an insertion cannula, according to a variant described herein, wherein the device comprises a narrowed configuration elongated for insertion into the sclera, and the device further comprises a retention device in the sclera at least partially To expand into a second elongated wide configuration for the first portion.
Figure 2D illustrates a treatment device comprising a reservoir suitable for loading into a cannula, according to a variant described herein.
Figure 2e depicts a treatment device configured to be positioned in the eye as in Figure 2a, according to a variant described herein.
FIG. 2F illustrates an
Figure 3 shows a reservoir having a penetrating barrier disposed at a first end, a porous structure disposed at a second end releasing the therapeutic agent for an extended period of time, and an elongated protrusion outwardly from the vessel for coupling to the sclera and conjunctiva ≪ RTI ID = 0.0 > a < / RTI >
본원에 기재된 물질, 화합물, 조성물, 물품 및 방법은 개시된 대상의 구체적 측면에 대한 하기 상세한 설명 및 그에 포함되는 실시예를 참조하면 보다 용이하게 이해할 수 있다. 본 발명의 물질, 화합물, 조성물, 물품, 장치 및 방법이 개시되고 기재되기 전에, 하기에 기재된 측면이 구체적 방법 또는 구체적 시약으로 제한되지 않으므로, 달라질 수 있음을 이해하여야 한다. 또한, 본원에 사용된 용어들은 단지 특정 측면을 기재하기 위한 것이며 제한하려는 것이 아님을 이해하여야 한다.The materials, compounds, compositions, articles, and methods described herein may be more readily understood by reference to the following detailed description of specific aspects of the disclosed subject matter and the examples included therein. Before the materials, compounds, compositions, articles, apparatus and methods of the present invention are disclosed and described, it is to be understood that the aspects described below are not limited to specific methods or reagents, and may vary. It is also to be understood that the terminology used herein is for the purpose of describing particular aspects only and is not intended to be limiting.
또한, 본 명세서 전체에서, 다양한 공보가 언급된다. 이들 공보의 개시내용은 그 전문이, 반대로 특정되지 않는 한, 개시된 대상이 관련된 최신 기술을 보다 자세히 기재하기 위해 본 출원에 참조로 포함된다. 개시된 참고문헌은 또한 문장 중에 논의된 그에 함유된 물질에 대하여 개별적으로 그리고 구체적으로 본원에 참조로 포함되고, 여기서 참고문헌은 신뢰받는다.In addition, throughout the present specification, various publications are mentioned. The disclosures of these publications are incorporated by reference into the present application to more fully describe the state of the art to which the disclosed subject matter relates, unless the context requires otherwise. The disclosed references are also incorporated herein by reference, individually and specifically, for materials contained therein as discussed in the context, where references are relied upon.
낮은 용해도의 화합물 또는 활성 작용제 (본 개시내용에서 상호교환하여 언급됨)의 약물 전달 제제는, 활성 작용제가 제제 중에서 안정할 뿐만 아니라, 목적하는 치료 기간 동안 활성이고 효과적인 속도로 방출되도록 보장하기 위해 연구 및 개발에 상당한 투자를 필요로 한다. 따라서, 질환 및/또는 장애의 치료 목적에 따라, 목적하는 치료 목적을 충족시키는 제제의 개발이 바람직하다. 후안부의 질환 및/또는 장애를 치료하거나 또는 완화시키기 위한 활성 작용제의 전달은, 예를 들어 점안제에 의한 활성제의 국소 전달이, 가능하다 하더라도 아주 드물게, 최적의 양의 활성 작용제가 표적 부위에 전달되도록 하기 때문에, 특히 난제이다. 게다가, 후안부 표적 부위에 효과적인 양의 활성제를 전달하기 위해 필요한 농도는 종종 고 농도의 활성제여야 하므로, 이는 적어도 탈표적 효과로 인한 일부 전신성 부작용에 기여한다. 주사를 통한 약물 제제의 전달은 점안제의 문제점을 피한다. 그러나, 반복되는 유리체내 주사는 환자에게 위험하기도 하고 또한 부담이 되기도 한다. 안내염, 망막 박리, 외상성 백내장, 및 안내압 상승 (IOP)은 모두 유리체내 투여 경로의 잠재적인 시력-위협 후유증이다. 게다가, 한 달에 한 번의 치료 또는 심지어 한 달에 한 번의 모니터링도, 특히 치료가 환자의 평생 동안 계속될 필요가 있을 수 있음을 고려하면, 환자, 환자의 간병인 및 의료진에게 상당한 부담이 된다. 대략 환자의 1/3이 특정 생물학적 VEGF 억제제의 반복되는 유리체내 주사로 치료를 받으면 시력 개선을 경험하지만, 대부분의 환자는 단지 저하된 시력의 안정화를 경험한다.Drug delivery formulations of low solubility compounds or active agents (referred to interchangeably in this disclosure) should be studied to ensure that the active agent is stable in the formulation as well as active and effective at the desired therapeutic duration and released at an effective rate. And significant investment in development. Accordingly, it is desirable to develop an agent that meets the intended therapeutic objective, depending on the therapeutic purpose of the disease and / or disorder. Delivery of an active agent to treat or ameliorate the disease and / or disorder of the posterior segment can be achieved, for example, by local delivery of the active agent by eye drops, although very possibly, This is especially a challenge. In addition, the concentration required to deliver an effective amount of active agent to the posterior segment target site often must be a high concentration of active agent, which contributes to some systemic side effects, at least due to de-targeting effects. Delivery of the drug formulation via injection avoids the problem of eye drops. However, repeated intravitreal injections are both dangerous and burdensome to the patient. Endophthalmitis, retinal detachment, traumatic cataract, and elevated intraocular pressure (IOP) are both potential eye-threatening sequelae of the vitreous route of administration. In addition, once a month, or even once a month, a considerable burden is placed on the patient, the patient's caregiver, and the medical staff, especially considering that the treatment may need to be continued throughout the patient's lifetime. Approximately one-third of patients experience visual improvement when treated with repeated intra-vitals injection of a specific biological VEGF inhibitor, but most patients experience only a stabilization of the deteriorated visual acuity.
WO2010/088548에 기재된 바와 같이, 유리체내 전달 장치를 통한 전달을 위해 개발된 약물 전달 제제는, 활성 작용제가 장치로부터 방출되기 전과 방출되는 동안에 용해되어 있을 뿐만 아니라 (즉, 침전 및/또는 응집되지 않음), 또한 활성 작용제가 그 기간 동안 안정할 것을 요구한다. 또한 활성제가 목적하는 치료 목적을 충족시키기 위해 연장된 기간 동안 후안부의 표적 부위에 전달되도록 보장하기 위해, 활성제는 수일 동안, 수주 동안, 또한 수개월 동안 전달되어야 한다. 상기 목적을 달성하는 것은 수년간의 연구 및 개발, 및 여러 기관에 의한 상당한 재정 투자에도 불구하고, 후안부의 질환 및/또는 장애에 대한 치료 옵션이 부족한 것으로 증명되는 바와 같이, 어려운 주문이다.As described in WO2010 / 088548, the drug delivery formulations developed for delivery via the intravesicular delivery device are not only dissolved before the active agent is released from the device and during release (i.e., not precipitated and / or aggregated ), And also requires that the active agent be stable during that period. In order to ensure that the active agent is delivered to the target site of the posterior segment for extended periods of time to meet the desired therapeutic purpose, the active agent must be delivered for days, weeks, and months. Achieving this goal is a difficult order, as evidenced by lack of treatment options for diseases and / or disorders of the posterior segment, despite years of research and development, and considerable financial investment by various agencies.
본 개시내용은 활성 작용제를 후안부에 전달하기 위한 제제를 제공한다. 본 개시내용의 활성 작용제 제제는 연장된 기간 동안의 약물 전달을 위한 안정한 제제이다. 본 개시내용은 또한 수용액에서 불용성이거나 또는 낮은 용해도를 갖는 활성 작용제의 전달을 위한 약물 전달 제제의 제조 (및/또는 생산) 방법을 제공한다. 본 개시내용의 방법에 의해 제조 (및/또는 생산)된 제제는 본 개시내용의 유리체내 전달 장치로부터 전달된다.The present disclosure provides formulations for delivering active agents to the posterior segment. The active agent formulations of this disclosure are stable agents for drug delivery for extended periods of time. The present disclosure also provides a method of manufacturing (and / or producing) a drug delivery formulation for delivery of an active agent that is insoluble or has low solubility in aqueous solution. Formulations prepared (and / or produced) by the methods of the present disclosure are delivered from a delivery device in the body of the present disclosure.
확산 제어 장치로부터의 치료제 전달은 용해된 치료제 농도의 에너지가 표적 조직에서의 치료제 농도보다 높은, 치료제의 공급원을 필요로 한다. 일부 치료제의 전달은 용해된 치료제 농도 및 장치에 로딩된 공급원 제제로 달성가능한 열역학 에너지에 의해 제한된다.Therapeutic delivery from a diffusion control device requires a source of therapeutic agent where the energy of the dissolved therapeutic agent concentration is higher than the therapeutic agent concentration in the target tissue. Delivery of some therapeutic agents is limited by the dissolved therapeutic agent concentration and the thermodynamic energy achievable with the source formulation loaded into the device.
예를 들어 3개월의 기간 동안, 치료 수준의 치료제를 전달하는 것이 바람직하다. 이는 특히, 조직에서 치료적이기 위해 필요한 수준보다 훨씬 높지 않은 수용해도를 갖는 치료제의 경우에 난제이다. 예를 들어, 티로신 키나제 억제제를 포함하는 대다수의 치료제가 그러한 것처럼 수용액에서의 치료제 용해도가 1 - 10 ㎍/mL 이하라면, 유리체에서의 약 0.1 - 10 ㎍/mL의 표적 농도는 확산 제어 치료 장치 이식물로부터 달성가능하지 않다.For example, for a period of 3 months, it is desirable to deliver therapeutic levels of the therapeutic agent. This is particularly a challenge in the case of therapeutic agents having a water solubility not much higher than that required to be therapeutic in the tissue. For example, if the therapeutic agent solubility in aqueous solution is less than or equal to 1 - 10 [mu] g / mL, as in most therapeutic agents including tyrosine kinase inhibitors, a target concentration of about 0.1 - 10 [ It is not achievable from water.
추가적으로, 일부 제제 접근법은 고체 형태가 아닌 제제 중에서 치료제의 양을 증가시키나, 용액으로 제제화된 개체는 크기가 크고 개별적으로 용해된 치료제 분자보다 느린 확산 속도를 갖는다. 예를 들어, 여러 개의 치료제 분자가 단일 치료제 분자보다 한 자릿수가 더 큰 크기 및 한 자릿수가 더 느린 확산 속도를 갖는 미셀과 같은 구조로 회합되거나 또는 자기-조립될 수 있다. 추가적으로, 확산 화학종의 크기는 재현가능한 또는 재현불가능한 방식으로 시간이 지날수록 증가하여, 시간이 지날수록 떨어지고 연장된 시간 동안의 서방성 전달 표적 프로파일을 충족시키지 못한 확산 제어 장치로부터의 전달 속도 프로파일을 초래한다.Additionally, some formulation approaches increase the amount of the therapeutic agent in the non-solid form, but the individual formulated in solution has a slower diffusion rate than the larger, individually dissolved therapeutic agent molecule. For example, multiple therapeutic agent molecules can be assembled or self-assembled into a structure such as a micelle that has one digit larger size and one order of magnitude slower diffusion rate than a single therapeutic agent molecule. Additionally, the size of the diffusion species increases over time in a reproducible or non-reproducible manner, resulting in a delivery rate profile from the diffusion controller that fails over time and does not meet the sustained delivery target profile for extended periods of time .
본 개시내용은 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염, 및 1종 이상의 제제화 작용제의 안정한 제약 제제를 제공한다. 제약상 허용되는 염은 1가 또는 2가 염이고, 1종 이상의 제제화 작용제는 적어도 착물화제, 가용화제 및 완충제를 포함한다. 치료제의 염은 본 개시내용의 제약 제제에 용해되어 있다. 본 개시내용에서 제공된 제제는 파조파닙 제제이다. 제제는 제약상 허용되는 1가 또는 2가 염, 예를 들어 할라이드 염이다. 본 개시내용은 파조파닙 모노- 및/또는 디-클로라이드 염의 안정한 제약 제제를 제공한다.The present disclosure provides pharmaceutically acceptable salts of therapeutic agents having low water solubility and stable pharmaceutical formulations of one or more formulation agents. Pharmaceutically acceptable salts are monovalent or divalent salts, and at least one formulating agent includes at least a complexing agent, a solubilizing agent, and a buffering agent. Salts of therapeutic agents are soluble in the pharmaceutical preparations of this disclosure. The formulation provided in the present disclosure is aripipap nip formulation. The agent is a pharmaceutically acceptable monovalent or divalent salt, for example a halide salt. The present disclosure provides stable pharmaceutical formulations of paraphanipip mono-and / or di-chloride salts.
본 개시내용은 염 분자 (예를 들어, 1HCl 또는 2HCl)의 개수 및 다형체 형태가 제제에서의 치료제의 용해도 및/또는 안정성에 영향을 준다는 것을 제공한다. 1HCl이 존재하는지 또는 2HCl이 존재하는지의 여부 및/또는 염의 다형체 형태에 따라, 치료제의 제약상 허용되는 염의 안정하고/거나 가용성인 제제를 달성하는 것은 제제의 상이한 제조 방법을 필요로 한다. 게다가, 본 개시내용은 치료제의 제약상 허용되는 염의 하나의 다형체 형태를 또 다른 다형체 형태로 변형시키는 방법을 제공한다. 상기 방법은 제제에서 치료제의 제약상 허용되는 염의 보다 높은 용해도를 갖는 변형된 다형체 형태를 제공한다. 상기 방법은 부분적 및/또는 완전한 무정형 형태로서 활성 작용제를 제제에 용해시키는 것을 제공한다. 따라서, 본 개시내용은 수용액에서 낮은 수용해도를 갖거나 또는 불용성인 것으로서 특징화되는 치료제의 제약상 허용되는 염의 다형체 형태의 안정하고/거나 고 가용성인 제제를 제공한다.The present disclosure provides that the number of salt molecules (e.g., 1 HCl or 2HCl) and the polymorphic form affect the solubility and / or stability of the therapeutic agent in the formulation. Achieving a stable and / or soluble formulation of a pharmaceutically acceptable salt of a therapeutic agent, depending on whether 1 HCl is present or 2HCl is present and / or depending on the polymorphic form of the salt, requires different preparation methods of the preparation. In addition, the present disclosure provides a method of transforming one polymorphic form of a pharmaceutically acceptable salt of a therapeutic agent into another polymorphic form. The method provides a modified polymorphic form having a higher solubility of the pharmaceutically acceptable salt of the therapeutic agent in the formulation. The method provides for dissolving the active agent in the formulation as a partial and / or complete amorphous form. Accordingly, the disclosure provides a formulation that is stable and / or highly soluble in the polymorphic form of a pharmaceutically acceptable salt of a therapeutic agent characterized as having a low water solubility in aqueous solution or being insoluble.
물 또는 수성 용매에서의 치료제의 용해도는 조금 녹는 것(sparingly soluble) (용질 1부를 위해 필요한 용매 30 내지 100부), 녹기 어려운 것(slightly soluble) (용질 1부를 위해 필요한 용매 100 내지 1000부), 매우 녹기 어려운 것(very slightly soluble) (용질 1부를 위해 필요한 용매 1000 내지 10,000부), 및 거의 녹지 않는 것(practically insoluble) 또는 불용성인 것 (≥10,000)으로 다양할 수 있다. 본 발명의 치료제는 수중 난용성 또는 낮은 수용성의 화합물일 수 있다. 본원에 언급된 바와 같이, 수중 난용성 또는 낮은 수용성의 화합물은, 예를 들어 1 mg/mL 미만 또는 0.01 mg/mL 미만의 용해도를 가질 수 있다.The solubility of the therapeutic agent in water or an aqueous solvent is determined by sparingly soluble (30-100 parts solvent required for 1 part solute), slightly soluble (100-1000 parts solvent required for 1 part solute) Very slightly soluble (1000 to 10,000 parts solvent required for one solute), and practically insoluble or insoluble (> = 10,000). The therapeutic agent of the present invention may be a water-insoluble or water-insoluble compound. As mentioned herein, a poorly water-soluble or low-water-soluble compound may have a solubility of, for example, less than 1 mg / mL or less than 0.01 mg / mL.
제제Formulation
본 개시내용의 제제는 수용성이 아니거나 또는 수중 난용성인 것으로서 특징화되는 치료제의 고 농도 (약 1 mg/mL - 약 300 mg/mL)를 달성하도록 제제화된다.The formulations of this disclosure are formulated to achieve a high concentration of the therapeutic agent (about 1 mg / mL to about 300 mg / mL) characterized as being non-aqueous or non-aqueous.
생체 막을 용이하게 통과하지 않고 치료제의 PK 특성에 영향을 주지 않는 착물화제, 예컨대 시클로덱스트린이 본 개시내용의 치료 장치의 저장소에서의 작용제의 수성 농도를 증가시키기 위해 사용된다. 본 개시내용의 착물화제, 예를 들어 시클로덱스트린 제제는 용해된 치료제의 농도를 800,000배까지 증가시켜, 10 mg/mL 이하의 수용해도를 갖는 치료제, 예를 들어 약 0.1 ㎍/mL 이하의 수용해도를 갖는 치료제의 경우에 농도가 약 10 mg/mL 내지 약 100 mg/mL 정도로 높다.Complexing agents that do not readily pass through the biological membrane and do not affect the PK characteristics of the therapeutic agent, such as cyclodextrin, are used to increase the aqueous concentration of the agent in the reservoir of the therapeutic device of the present disclosure. The complexing agents of this disclosure, such as cyclodextrin formulations, can increase the concentration of the dissolved therapeutic agent by up to 800,000 times to provide a therapeutic agent having a water solubility of less than or equal to 10 mg / mL, The concentration is as high as about 10 mg / mL to about 100 mg / mL.
본 개시내용은 치료제, 예를 들어 파조파닙 모노- 또는 디-히드로클로라이드의 제제를 제공하고, 여기서 장치 및/또는 전달 시 표적에서의 농도는 약 10 mg/mL 내지 약 70 mg/mL 이하 (예를 들어, 약 10 mg/mL, 약 11 mg/mL, 약 12 mg/mL, 약 13 mg/mL, 약 14 mg/mL, 약 15 mg/mL, 약 16 mg/mL, 약 17 mg/mL, 약 18 mg/mL, 약 19 mg/mL, 약 20 mg/mL, 약 21 mg/mL, 약 22 mg/mL, 약 23 mg/mL, 약 24 mg/mL, 약 25 mg/mL, 약 26 mg/mL, 약 27 mg/mL, 약 28 mg/mL, 약 29 mg/mL, 약 30 mg/mL, 약 31 mg/mL, 약 32 mg/mL, 약 33 mg/mL, 약 34 mg/mL, 약 35 mg/mL, 약 36 mg/mL, 약 37 mg/mL, 약 38 mg/mL, 약 39 mg/mL, 약 40 mg/mL, 약 41 mg/mL, 약 42 mg/mL, 약 43 mg/mL, 약 44 mg/mL, 약 45 mg/mL, 약 46 mg/mL, 약 47 mg/mL, 약 48 mg/mL, 약 49 mg/mL, 약 50 mg/mL, 약 51 mg/mL, 약 52 mg/mL, 약 53 mg/mL, 약 54 mg/mL, 약 55 mg/mL, 약 56 mg/mL, 약 57 mg/mL, 약 58 mg/mL, 약 59 mg/mL, 약 60 mg/mL, 약 61 mg/mL, 약 62 mg/mL, 약 63 mg/mL, 약 64 mg/mL, 약 65 mg/mL, 약 66 mg/mL, 약 67 mg/mL, 약 68 mg/mL, 약 69 mg/mL, 또는 약 70 mg/mL)이다. 본 개시내용은 제제 중의 약 30 mg/mL 내지 약 50 mg/mL의 파조파닙을 제공한다.The present disclosure provides a formulation of a therapeutic agent, for example pachopanip mono-or di-hydrochloride, wherein the concentration in the target during device and / or delivery is from about 10 mg / mL to about 70 mg / mL or less For example, about 10 mg / mL, about 11 mg / mL, about 12 mg / mL, about 13 mg / mL, about 14 mg / mL, about 15 mg / mL, about 19 mg / mL, about 20 mg / mL, about 21 mg / mL, about 22 mg / mL, about 23 mg / mL, about 24 mg / About 29 mg / mL, about 30 mg / mL, about 31 mg / mL, about 32 mg / mL, about 33 mg / mL, about 34 mg / 37 mg / mL, about 38 mg / mL, about 39 mg / mL, about 40 mg / mL, about 41 mg / mL, about 42 mg / about 45 mg / mL, about 46 mg / mL, about 47 mg / mL, about 48 mg / mL, about 49 mg / mL, about 50 mg / mL, about 43 mg / mL, about 44 mg / About 55 mg / mL, about 52 mg / mL, about 53 mg / mL, about 54 mg / mL, about 55 mg / mL, about 56 mg / mg / mL, about 60 mg / mL, about 61 mg / mL, about 62 mg / mL, about 63 m about 66 mg / mL, about 67 mg / mL, about 68 mg / mL, about 69 mg / mL, or about 70 mg / mL. The present disclosure provides a pachoparamip from about 30 mg / mL to about 50 mg / mL in the formulation.
측정된 농도는 약 10 mg/mL 내지 약 70 mg/mL 이하 (예를 들어, 약 10 mg/mL, 약 11 mg/mL, 약 12 mg/mL, 약 13 mg/mL, 약 14 mg/mL, 약 15 mg/mL, 약 16 mg/mL, 약 17 mg/mL, 약 18 mg/mL, 약 19 mg/mL, 약 20 mg/mL, 약 21 mg/mL, 약 22 mg/mL, 약 23 mg/mL, 약 24 mg/mL, 약 25 mg/mL, 약 26 mg/mL, 약 27 mg/mL, 약 28 mg/mL, 약 29 mg/mL, 약 30 mg/mL, 약 31 mg/mL, 약 32 mg/mL, 약 33 mg/mL, 약 34 mg/mL, 약 35 mg/mL, 약 36 mg/mL, 약 37 mg/mL, 약 38 mg/mL, 약 39 mg/mL, 약 40 mg/mL, 약 41 mg/mL, 약 42 mg/mL, 약 43 mg/mL, 약 44 mg/mL, 약 45 mg/mL, 약 46 mg/mL, 약 47 mg/mL, 약 48 mg/mL, 약 49 mg/mL, 약 50 mg/mL, 약 51 mg/mL, 약 52 mg/mL, 약 53 mg/mL, 약 54 mg/mL, 약 55 mg/mL, 약 56 mg/mL, 약 57 mg/mL, 약 58 mg/mL, 약 59 mg/mL, 약 60 mg/mL, 약 61 mg/mL, 약 62 mg/mL, 약 63 mg/mL, 약 64 mg/mL, 약 65 mg/mL, 약 66 mg/mL, 약 67 mg/mL, 약 68 mg/mL, 약 69 mg/mL, 또는 약 70 mg/mL)이다.The measured concentrations may be from about 10 mg / mL to about 70 mg / mL (e.g., about 10 mg / mL, about 11 mg / mL, about 12 mg / mL, about 13 mg / About 15 mg / mL, about 16 mg / mL, about 17 mg / mL, about 18 mg / mL, about 19 mg / mL, about 20 mg / mL, about 21 mg / mL, about 22 mg / About 25 mg / mL, about 26 mg / mL, about 27 mg / mL, about 28 mg / mL, about 29 mg / mL, about 30 mg / mL, about 31 mg / mL, about 24 mg / about 38 mg / mL, about 33 mg / mL, about 34 mg / mL, about 35 mg / mL, about 36 mg / mL, about 37 mg / About 40 mg / mL, about 41 mg / mL, about 42 mg / mL, about 43 mg / mL, about 44 mg / mL, about 45 mg / mL, about 46 mg / About 48 mg / mL, about 49 mg / mL, about 50 mg / mL, about 51 mg / mL, about 52 mg / mL, about 53 mg / mL, about 54 mg / about 59 mg / mL, about 59 mg / mL, about 60 mg / mL, about 61 mg / mL, about 62 mg / mL, about 63 mg / mL, about 64 mg / mL, about 57 mg / About 65 mg / mL, about 66 mg / mL, about 67 mg / mL, about 68 mg / mL, about 69 mg / mL, or about 70 mg / mL).
본 개시내용은 전달 장치에서의 치료제, 예를 들어 파조파닙 1HCL 또는 2HCL의 충전 농도가 약 10 mg/mL 내지 약 70 mg/mL 이하 (예를 들어, 약 10 mg/mL, 약 11 mg/mL, 약 12 mg/mL, 약 13 mg/mL, 약 14 mg/mL, 약 15 mg/mL, 약 16 mg/mL, 약 17 mg/mL, 약 18 mg/mL, 약 19 mg/mL, 약 20 mg/mL, 약 21 mg/mL, 약 22 mg/mL, 약 23 mg/mL, 약 24 mg/mL, 약 25 mg/mL, 약 26 mg/mL, 약 27 mg/mL, 약 28 mg/mL, 약 29 mg/mL, 약 30 mg/mL, 약 31 mg/mL, 약 32 mg/mL, 약 33 mg/mL, 약 34 mg/mL, 약 35 mg/mL, 약 36 mg/mL, 약 37 mg/mL, 약 38 mg/mL, 약 39 mg/mL, 약 40 mg/mL, 약 41 mg/mL, 약 42 mg/mL, 약 43 mg/mL, 약 44 mg/mL, 약 45 mg/mL, 약 46 mg/mL, 약 47 mg/mL, 약 48 mg/mL, 약 49 mg/mL, 약 50 mg/mL, 약 51 mg/mL, 약 52 mg/mL, 약 53 mg/mL, 약 54 mg/mL, 약 55 mg/mL, 약 56 mg/mL, 약 57 mg/mL, 약 58 mg/mL, 약 59 mg/mL, 약 60 mg/mL, 약 61 mg/mL, 약 62 mg/mL, 약 63 mg/mL, 약 64 mg/mL, 약 65 mg/mL, 약 66 mg/mL, 약 67 mg/mL, 약 68 mg/mL, 약 69 mg/mL, 또는 약 70 mg/mL)인 것을 제공한다.This disclosure is based on the finding that the charge concentration of the therapeutic agent in the delivery device, for example rapamycin NIPHCL or 2HCL, is about 10 mg / mL to about 70 mg / mL or less (e.g., about 10 mg / mL, mL, about 13 mg / mL, about 14 mg / mL, about 15 mg / mL, about 16 mg / mL, about 17 mg / mL, about 18 mg / mL, about 19 mg / About 24 mg / mL, about 25 mg / mL, about 26 mg / mL, about 27 mg / mL, about 28 mg / mL, about 22 mg / about 35 mg / mL, about 36 mg / mL, about 29 mg / mL, about 30 mg / mL, about 31 mg / mL, about 32 mg / mL, about 38 mg / mL, about 39 mg / mL, about 40 mg / mL, about 41 mg / mL, about 42 mg / mL, about 43 mg / About 47 mg / mL, about 48 mg / mL, about 49 mg / mL, about 50 mg / mL, about 51 mg / mL, about 52 mg / mL, about 53 mg / mL, about 46 mg / about 55 mg / mL, about 55 mg / mL, about 57 mg / mL, about 58 mg / mL, about 59 mg / mL, about 60 mg / mL, about 63 mg / mL, about 64 mg / mL, about 65 mg / mL, about 66 mg / mL, about 67 mg / About 69 mg / mL, or about 70 mg / mL).
본 개시내용은 전달 장치에서의 개시된 제제 중에서, 약 10 mg/ml, 약 15 mg/ml, 약 20 mg/ml, 약 25 mg/ml, 약 30 mg/ml, 약 35 mg/ml, 약 45 mg/ml, 약 50 mg/ml, 약 55 mg/ml, 약 60 mg/ml, 약 65 mg/ml, 약 70 mg/ml, 약 75 mg/ml, 또는 약 80 mg/ml일 때 안정한 파조파닙의 1가 할라이드 염, 예를 들어 클로라이드 염을 제공한다. 본 개시내용은 전달 장치에서의 개시된 제제 중에서, 약 10 mg/ml 내지 약 15 mg/ml 이하, 약 15 mg/ml 내지 약 20 mg/ml 이하, 약 20 mg/ml 내지 약 25 mg/ml 이하, 약 25 mg/ml 내지 약 30 mg/ml 이하, 약 30 mg/ml 내지 약 35 mg/ml 이하, 약 35 mg/ml 내지 약 45 mg/ml 이하, 약 45 mg/ml 내지 약 50 mg/ml 이하, 약 50 mg 내지 약 55 mg/ml 이하, 약 55 mg/ml 내지 약 60 mg/ml 이하, 약 60 mg/ml 내지 약 65 mg/ml 이하, 약 65 mg/ml 내지 약 70 mg/ml 이하, 약 70 mg/ml 내지 약 75 mg/ml 이하, 또는 약 75 mg/ml 내지 약 80 mg/ml 이하일 때 안정한 파조파닙의 1가 할라이드 염, 예를 들어 클로라이드 염을 제공한다. 파조파닙의 1가 할라이드 염, 예를 들어 클로라이드 염은 개시된 제제 중에서 약 40 mg/ml 내지 약 60 mg/ml 이하일 때 안정하다.The disclosure provides a pharmaceutical composition comprising about 10 mg / ml, about 15 mg / ml, about 20 mg / ml, about 25 mg / ml, about 30 mg / ml, about 35 mg / ml, about 45 When a stable wave is generated at a concentration of about 50 mg / ml, about 55 mg / ml, about 60 mg / ml, about 65 mg / ml, about 70 mg / ml, about 75 mg / ml, or about 80 mg / Provides monovalent halide salts of zapanib, such as chloride salts. The disclosure provides a pharmaceutical composition comprising a compound of the invention in a dosage form comprising from about 10 mg / ml to about 15 mg / ml or less, from about 15 mg / ml to about 20 mg / ml or less, from about 20 mg / ml to about 25 mg / , About 25 mg / ml to about 30 mg / ml, about 30 mg / ml to about 35 mg / ml or less, about 35 mg / ml to about 45 mg / ml or less, about 50 mg to about 55 mg / ml, about 55 mg / ml to about 60 mg / ml or less, about 60 mg / ml to about 65 mg / ml, less than about 70 mg / ml to less than about 75 mg / ml, or less than about 75 mg / ml to less than about 80 mg / ml, to provide stable monophoryl halide salts, such as chloride salts. Monovalent halide salts of pazopanib, such as the chloride salt, are stable when from about 40 mg / ml to about 60 mg / ml in the disclosed formulation.
본 개시내용은 전달 장치에서의 개시된 제제 중에서, 약 10 mg/ml, 약 15 mg/ml, 약 20 mg/ml, 약 25 mg/ml, 약 30 mg/ml, 약 35 mg/ml, 약 45 mg/ml, 약 50 mg/ml, 약 55 mg/ml, 약 60 mg/ml, 약 65 mg/ml, 약 70 mg/ml, 약 75 mg/ml, 또는 약 80 mg/ml일 때 안정한 파조파닙의 2가 할라이드 염, 예를 들어 클로라이드 염을 제공한다. 본 개시내용은 전달 장치에서의 개시된 제제 중에서, 약 10 mg/ml 내지 약 15 mg/ml, 약 15 mg/ml 내지 약 20 mg/ml, 약 20 mg/ml 내지 약 25 mg/ml, 약 25 mg/ml 내지 약 30 mg/ml, 약 30 mg/ml 내지 약 35 mg/ml, 약 35 mg/ml 내지 약 45 mg/ml, 약 45 mg/ml 내지 약 50 mg/ml, 약 50 mg 내지 약 55 mg/ml, 약 55 mg/ml 내지 약 60 mg/ml, 약 60 mg/ml 내지 약 65 mg/ml, 약 65 mg/ml 내지 약 70 mg/ml, 약 70 mg/ml 내지 약 75 mg/ml, 또는 약 75 mg/ml 내지 약 80 mg/ml일 때 안정한 파조파닙의 2가 할라이드 염, 예를 들어 클로라이드 염을 제공한다. 본 개시내용의 2가 파조파닙 할라이드 염, 예를 들어 클로라이드 염은 개시된 제제 중에서 약 60 mg/ml일 때 안정하다.The disclosure provides a pharmaceutical composition comprising about 10 mg / ml, about 15 mg / ml, about 20 mg / ml, about 25 mg / ml, about 30 mg / ml, about 35 mg / ml, about 45 When a stable wave is generated at a concentration of about 50 mg / ml, about 55 mg / ml, about 60 mg / ml, about 65 mg / ml, about 70 mg / ml, about 75 mg / ml, or about 80 mg / Provides a divalent halide salt of zopanub, such as a chloride salt. The disclosure provides a pharmaceutical composition comprising about 10 mg / ml to about 15 mg / ml, about 15 mg / ml to about 20 mg / ml, about 20 mg / ml to about 25 mg / ml, about 25 from about 30 mg / ml to about 35 mg / ml, from about 35 mg / ml to about 45 mg / ml, from about 45 mg / ml to about 50 mg / ml, About 55 mg / ml, about 55 mg / ml to about 60 mg / ml, about 60 mg / ml to about 65 mg / ml, about 65 mg / ml to about 70 mg / mg / ml, or from about 75 mg / ml to about 80 mg / ml, a stable haloperidol halide salt, such as a chloride salt, is provided. The divalent ripopanib halide salts of this disclosure, such as chloride salts, are stable at about 60 mg / ml in the disclosed formulation.
착물화제는 술포부틸 에테르-β-시클로덱스트린 ("SBEβCD") 또는 캅티솔®이다. 본 개시내용의 유리체내 전달 제제는 캅티솔®과의 착물로 치료제 파조파닙 모노- 또는 디-히드로클로라이드를 포함한다. 치료제 파조파닙 모노- 또는 디-히드로클로라이드와 캅티솔®의 회합은 작용제의 수용해도를 10 내지 25,000배 증가시킨다. 치료제 파조파닙 모노- 또는 디-히드로클로라이드와 캅티솔®의 상호작용은 캅티솔®의 친유성 공동에 치료제를 위한 유익한 보호 환경을 제공하고, 반면에 캅티솔®의 소수성 표면이 효과적인 수용해도를 제공하므로, 그에 따라 치료제의 용해도와 안정성을 둘다 증가시킨다. 추가적으로, 치료제와 캅티솔®의 상호작용은 수성 환경에서의 잠재적인 반응물로부터 불안정한 영역을 보호함으로써 작용제의 분해를 감소시킨다.The complexing agent is sulfobutyl ether-beta-cyclodextrin ("SBE beta CD") or capricol®. The intravitreal delivery formulations of this disclosure include the therapeutic Pazopanip mono-or di-hydrochloride in complex with Captisol®. The association of the therapeutic Pazopanip mon mono- or di-hydrochloride with capricol increases the water solubility of the agent by 10-25,000 times. The interaction of the therapeutic Pazopanip mon mono- or di-hydrochloride with Captisol® provides a beneficial protective environment for the therapeutic agent in the lipophilic cavity of Capsicol®, while the hydrophobic surface of Capsicol® provides an effective water solubility , Thereby increasing both the solubility and the stability of the therapeutic agent. In addition, the interaction of the therapeutic with capricol® reduces the degradation of the agonist by protecting the unstable region from potential reactants in the aqueous environment.
본 개시내용의 제제는 착물화제, 예를 들어 시클로덱스트린 ("CD"), 즉 본원의 목록으로 제한되지 않는 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 또는 이들의 임의의 조합(들)과 회합된 파조파닙 또는 파조파닙 모노- 또는 디-히드로클로라이드를 포함한다. 제제 중의 CD는 약 9:1, 약 8:1, 약 7:1, 약 6:1, 약 5:1, 약 4:1, 약 3:1, 또는 약 2:1의 치료제에 대한 비로 존재한다. CD:치료제의 비는 약 2.5:1이다. CD:치료제의 비는 약 2.2:1; 약 2.5:1; 약 3.7:1; 약 5:1; 약 8:1; 또는 약 9:1이다.The formulations of the present disclosure may also be formulated with complexing agents, for example, cyclodextrins ("CD"), ie, 2-hydroxypropyl-β-cyclodextrin, methyl-β-cyclodextrin, random methylated- -Cyclodextrin, ethyl-beta-cyclodextrin, triacetyl-beta-cyclodextrin, peracetylated-beta-cyclodextrin, carboxymethyl- beta -cyclodextrin, hydroxyethyl- beta -cyclodextrin, Beta-cyclodextrin, maltosyl- beta -cyclodextrin, sulfobutyl ether-beta-cyclodextrin, branched-beta-cyclodextrin, hydroxy-3- (trimethylammonio) propyl- beta -cyclodextrin, glucosyl- Cyclodextrin, trimethyl-gamma -cyclodextrin, or any combination (s) thereof, or a combination thereof, such as, for example, cyclodextrin, propyl- gamma -cyclodextrin, random methylated-gamma -cyclodextrin, trimethyl- do. The CD in the formulation is present in a ratio to the therapeutic agent of about 9: 1, about 8: 1, about 7: 1, about 6: 1, about 5: 1, about 4: 1, about 3: do. CD: Therapeutic ratio is about 2.5: 1. CD: the therapeutic ratio is about 2.2: 1; About 2.5: 1; About 3.7: 1; About 5: 1; About 8: 1; Or about 9: 1.
장치에서의 치료제 농도의 증가는 망막에서의 혈관 누수 및 신생혈관화 (NV)의 효과적인 치료, 진행 예방 또는 완화를 위해 유리체에서 필요한 농도보다 약 100x 더 높다. 혈관 누수 및 신생혈관화 (NV)의 효과적인 치료, 진행 예방, 또는 완화를 위해 유리체에서 필요한 농도가 치료제의 용해도 한계값보다 높기 때문에, 본 개시내용의 실시양태는 작용제의 본래의 수용해도보다 약 1000x 또는 그 초과의 증가한 치료제 용해도를 제공한다.The increase in therapeutic agent concentration in the device is about 100x higher than the required concentration in the vitreous for effective treatment, prophylactic or palliative treatment of vascular leakage and neovascularization (NV) in the retina. Because the concentration required in the vitreous is greater than the solubility limit of the therapeutic for effective treatment, prophylactic, or palliation of vascular leakage and neovascularization (NV), embodiments of the present disclosure may provide a dose of about 1000 x Or more of the therapeutic agent solubility.
본 개시내용의 제제는 착물화제로서 캅티솔®을 포함한다. 캅티솔®의 존재 하에 치료제의 농도는 약물 전달 작용제 및/또는, 전달 시에, 유리체에서 0.5 mg/ml 내지 약 90 mg/mL이다. 예를 들어, 캅티솔®의 존재 하에 파조파닙 모노- 또는 디-히드로클로라이드의 농도는 약 20 mg/mL, 약 30 mg/mL, 약 40 mg/mL, 약 50 mg/mL, 약 60 mg/mL, 약 70 mg/mL, 약 80 mg/mL, 또는 약 90 mg/mL이다.The preparations of this disclosure include Capsytol® as complexing agent. The concentration of therapeutic agent in the presence of Capsytol® is from 0.5 mg / ml to about 90 mg / ml in the vitreous body at the time of drug delivery agent and / or delivery. For example, the concentration of rapamycin nip mono- or di-hydrochloride in the presence of Capsytol may be about 20 mg / mL, about 30 mg / mL, about 40 mg / mL, about 50 mg / mL, about 60 mg / mL, about 70 mg / mL, about 80 mg / mL, or about 90 mg / mL.
제제의 추가 성분은 트레할로스, 메틸셀룰로스, 에틸셀룰로스, 소듐 카르복시메틸셀룰로스, 히드록시프로필메틸셀룰로스, 소듐 히알루로네이트, 소듐 알기네이트, 키토산 및 그의 유도체, 폴리에틸렌 글리콜, 글리세린, 프로필렌 글리콜, 트리아세틴, N,N-디메틸아세트아미드, 피롤리돈, 디메틸 술폭시드, 에탄올, N-(-베타-히드록시에틸)-락트아미드, 1-메틸-2-피롤리디논, 트리글리세리드, 모노티오글리세롤, 소르비톨, 레시틴, 메틸파라벤, 프로필파라벤, 폴리소르베이트, 에틸렌 옥시드 및 프로필렌 옥시드의 블록 공중합체, 폴리에틸렌 옥시드 및 폴리프로필렌 옥시드의 디-블록 중합체 또는 트리-블록 공중합체, 에톡실화된 유화제, 폴리에틸렌 글리콜 에스테르, 수크로스 라우레이트, 토코페롤-PEG-숙시네이트, 인지질 및 그의 유도체, 또는 다른 비-이온성 자기-유화제이다.Additional components of the formulation may be selected from the group consisting of trehalose, methylcellulose, ethylcellulose, sodium carboxymethylcellulose, hydroxypropylmethylcellulose, sodium hyaluronate, sodium alginate, chitosan and its derivatives, polyethylene glycol, glycerin, propylene glycol, triacetin, N N-dimethylacetamide, pyrrolidone, dimethylsulfoxide, ethanol, N - (- beta -hydroxyethyl) -lactamide, 1-methyl-2-pyrrolidinone, triglyceride, monothioglycerol, sorbitol, , Block copolymers of methylparaben, propylparaben, polysorbate, ethylene oxide and propylene oxide, di-block polymers or tri-block copolymers of polyethylene oxide and polypropylene oxide, ethoxylated emulsifiers, polyethylene glycols Ester, sucrose laurate, tocopherol-PEG-succinate, phospholipids and derivatives thereof, or other non-ionic It is a self-emulsifying agent.
본 개시내용의 제제 중의 가용화제는 트레할로스, 메틸셀룰로스, 에틸셀룰로스, 소듐 카르복시메틸셀룰로스, 소듐 히알루로네이트, 소듐 알기네이트, 폴리에틸렌 글리콜, 글리세린, 프로필렌 글리콜, 트리아세틴, N,N-디메틸아세트아미드, 폴리(비닐 피롤리돈), 피롤리돈, 또는 이들의 임의의 조합(들)을 포함하나, 이들은 제한적인 예가 아니다. 본 개시내용의 제제의 제조에 사용되는 가용화제는 폴리(비닐 피롤리돈) (PVP)이다. 예를 들어, 본 개시내용의 제제는 약 0.2% 내지 약 1%의 PVP를 포함한다. 본 개시내용은 약 5 mg/mL의 PVP 내지 약 30 mg/mL의 PVP를 갖는 제제를 제공한다.The solubilizing agent in the preparations of the present disclosure is selected from the group consisting of trehalose, methylcellulose, ethylcellulose, sodium carboxymethylcellulose, sodium hyaluronate, sodium alginate, polyethylene glycol, glycerin, propylene glycol, triacetin, N, N-dimethylacetamide, Poly (vinyl pyrrolidone), pyrrolidone, or any combination (s) thereof, but these are not limiting examples. The solubilizing agent used in the preparation of the preparations of this disclosure is poly (vinylpyrrolidone) (PVP). For example, the formulation of the present disclosure comprises from about 0.2% to about 1% PVP. The present disclosure provides formulations with about 5 mg / mL of PVP to about 30 mg / mL of PVP.
본 개시내용의 제제에 첨가되는 가용화제는 약 0.1% 내지 약 5.0% (예를 들어, 약 0.1%, 약 0.2%, 약 0.3%, 약 0.4%, 약 0.5%, 약 0.6%, 약 0.7%, 약 0.8%, 약 0.9%, 약 1.0%, 약 1.1%, 약 1.2%, 약 1.3%, 약 1.4%, 약 1.5%, 약 1.6%, 약 1.7%, 약 1.8%, 약 1.9%, 약 2.0%, 약 2.1%, 약 2.2%, 약 2.3%, 약 2.4%, 약 2.5%, 약 2.6%, 약 2.7%, 약 2.8%, 약 2.9%, 약 3.0%, 약 3.1%, 약 3.2%, 약 3.3%, 약 3.4%, 약 3.5%, 약 3.6%, 약 3.7%, 약 3.8%, 약 3.9%, 약 4.0%, 약 4.1%, 약 4.2%, 약 4.3%, 약 4.4%, 약 4.5%, 약 4.6%, 약 4.7%, 약 4.8%, 약 4.9%, 약 5.0%)의 PVP를 포함한다. 본 개시내용의 제제는 예를 들어, 약 1%의 PVP를 포함한다.The solubilizer added to the formulation of the present disclosure may contain from about 0.1% to about 5.0% (e.g., about 0.1%, about 0.2%, about 0.3%, about 0.4%, about 0.5%, about 0.6% , About 0.8%, about 0.9%, about 1.0%, about 1.1%, about 1.2%, about 1.3%, about 1.4%, about 1.5%, about 1.6%, about 1.7%, about 1.8% About 2.1%, about 2.2%, about 2.3%, about 2.4%, about 2.5%, about 2.6%, about 2.7%, about 2.8%, about 2.9%, about 3.0%, about 3.1% , About 3.3%, about 3.4%, about 3.5%, about 3.6%, about 3.7%, about 3.8%, about 3.9%, about 4.0%, about 4.1%, about 4.2%, about 4.3% 4.5%, about 4.6%, about 4.7%, about 4.8%, about 4.9%, about 5.0%). The formulation of the present disclosure includes, for example, about 1% PVP.
본 개시내용의 제제는 완충제, 예를 들어 히스티딘 HCl을 포함한다. 제제는 약 5 mg/ml 내지 약 30 mg/mL의 완충제, 예를 들어 히스티딘 HCl (예를 들어, 약 5 mg/mL, 약 6 mg/mL, 약 7 mg/mL, 약 8 mg/mL, 약 9 mg/mL, 약 10 mg/mL, 약 11 mg/mL, 약 12 mg/mL, 약 13 mg/mL, 약 14 mg/mL, 약 15 mg/mL, 약 16 mg/mL, 약 17 mg/mL, 약 18 mg/mL, 약 19 mg/mL, 약 20 mg/mL, 약 21 mg/mL, 약 22 mg/mL, 약 23 mg/mL, 약 24 mg/mL, 약 25 mg/mL, 약 26 mg/mL, 약 27 mg/mL, 약 28 mg/mL, 약 29 mg/mL, 또는 약 30 mg/mL)을 포함한다.Formulations of the present disclosure include buffering agents such as histidine HCl. The formulation may contain a buffering agent such as about 5 mg / ml to about 30 mg / ml of a buffer such as histidine HCl (e.g., about 5 mg / ml, about 6 mg / ml, about 7 mg / ml, about 8 mg / About 10 mg / mL, about 11 mg / mL, about 12 mg / mL, about 13 mg / mL, about 14 mg / mL, about 15 mg / mL, about 16 mg / about 18 mg / mL, about 19 mg / mL, about 20 mg / mL, about 21 mg / mL, about 22 mg / mL, about 23 mg / mL, about 24 mg / mL, about 26 mg / mL, about 27 mg / mL, about 28 mg / mL, about 29 mg / mL, or about 30 mg / mL).
제제의 pH는 약 1.0 - 약 7.0이다 (예를 들어, 약 1.0, 약 1.1, 약 1.2, 약 1.3, 약 1.4, 약 1.5, 약 1.6, 약 1.7, 약 1.9, 약 2.0, 약 2.1, 약 2.2, 약 2.3, 약 2.4, 약 2.5, 약 2.6, 약 2.7, 약 2.8, 약 2.9, 약 3.0, 약 3.1, 약 3.2, 약 3.3, 약 3.4, 약 3.5, 약 3.6, 약 3.7, 약 3.8, 약 3.9, 약 4.0, 약 4.1, 약 4.2, 약 4.3, 약 4.4, 약 4.5, 약 4.6, 약 4.7, 약 4.8, 약 4.9, 약 5.0, 약 5.1, 약 5.2, 약 5.3, 약 5.4, 약 5.5, 약 5.6, 약 5.7, 약 5.8, 약 5.9, 약 6.0, 약 6.1, 약 6.2, 약 6.3, 약 6.4, 약 6.5, 약 6.6, 약 6.7, 약 6.8, 약 6.9, 또는 약 7.0의 pH).The pH of the formulation is about 1.0 to about 7.0 (e.g., about 1.0, about 1.1, about 1.2, about 1.3, about 1.4, about 1.5, about 1.6, about 1.7, about 1.9, about 2.0, about 2.1, About 2.3, about 2.4, about 2.5, about 2.6, about 2.7, about 2.8, about 2.9, about 3.0, about 3.1, about 3.2, about 3.3, about 3.4, about 3.5, about 3.6, about 3.7, About 4.0, about 4.1, about 4.2, about 4.3, about 4.4, about 4.5, about 4.6, about 4.7, about 4.8, about 4.9, about 5.0, about 5.1, about 5.2, about 5.3, about 5.4, about 5.5, A pH of about 5.6, about 5.7, about 5.8, about 5.9, about 6.0, about 6.1, about 6.2, about 6.3, about 6.4, about 6.5, about 6.6, about 6.7, about 6.8, about 6.9, or about 7.0.
본 개시내용의 제제에 포함되는 추가 첨가제는 트리아세틴 (치료제의 약 1x 몰량), L-리신 (약 25 mg/mL), 암모늄 아세테이트 약 0.1% - 약 5% (w/v) (예를 들어, 약 2% (w/v)), 또는 글리세롤 약 0.1% - 약 5% (w/v) (예를 들어, 약 2% (w/v))이나, 이들은 제한적인 예가 아니다.Additional excipients included in the preparations of this disclosure include triacetin (about 1x molar of therapeutic agent), L-lysine (about 25 mg / mL), ammonium acetate about 0.1% to about 5% (w / v) , About 2% (w / v)), or glycerol about 0.1% to about 5% (w / v) (e.g., about 2% (w / v)).
본 개시내용의 제제는 치료 장치에서의 제제의 완충 능력을 증가시키기 위한 pH 조정을 위해 1종 또는 2종의 작용제를 포함한다. 1종 또는 2종의 pH 조정 작용제는 수산화나트륨, 염산, 시트르산, 말산, 아세테이트, 타르타르산, 히스티딘, 포스페이트, 또는 이들의 임의의 조합(들)으로부터 선택되나, 이들은 제한적인 예가 아니다. 한 실시양태에서, 제제는 pH 조정을 위한 작용제를 포함하나, 착물화제는 포함하지 않는다. 1종 또는 2종의 pH 조정 작용제는 시트르산 및/또는 히스티딘이다.The formulations of the present disclosure include one or two agents for adjusting the pH to increase the buffering capacity of the formulation in a therapeutic device. The one or two pH adjusting agents are selected from sodium hydroxide, hydrochloric acid, citric acid, malic acid, acetate, tartaric acid, histidine, phosphate, or any combination (s) thereof, but these are not limiting examples. In one embodiment, the agent comprises an agonist for pH adjustment, but does not include a complexing agent. One or two pH adjusting agents are citric acid and / or histidine.
본 개시내용의 제제는 장성 조정 작용제를 포함한다. 예를 들어, 장성 조정 작용제는 소듐 클로라이드, 소듐 포스페이트, 또는 이들의 임의의 조합(들)이나, 이들은 제한적인 예가 아니다.Formulations of the present disclosure include wall thickening agents. For example, the wall-thickening agonist is sodium chloride, sodium phosphate, or any combination (s) thereof, but these are not limiting examples.
본 개시내용의 제제는 PDS 이식물의 사용 시간 동안 높은 안정성을 갖는다. 예를 들어, 제제는 생리학적 조건 하의 37℃의 PDS 저장소 챔버에서 적어도 6개월 동안 안정하다. 예를 들어, 제제는 유리체로부터 확산되는 유리체 성분의 존재 하에 PDS에서 안정하다.The preparations of this disclosure have a high stability during the time of use of the PDS implant. For example, the formulation is stable for at least 6 months in a PDS storage chamber at 37 DEG C under physiological conditions. For example, the formulation is stable in PDS in the presence of a vitreous component diffusing from the vitreous.
본 개시내용의 제제는 안구 약물 전달 방법에 사용된다. 본 개시내용의 제제는 유리체내 전달 제제이다. 본 개시내용의 제제는 점안제로서 제제화되지 않는다. 본 개시내용의 제제는 국소 전달용으로 제제화되지 않는다. 본 개시내용의 제제는 경구 전달 또는 비경구 전달용으로 제제화되지 않는다. 본 개시내용의 제제는 눈주위 전달용으로 제제화되지 않는다.The preparations of this disclosure are used in ocular drug delivery methods. The formulation of the present disclosure is a delivery system in the vitreous body. The formulation of the present disclosure is not formulated as an eye drop. The formulations of this disclosure are not formulated for topical delivery. The formulations of this disclosure are not formulated for oral or parenteral delivery. The formulation of the present disclosure is not formulated for ocular delivery.
제조 방법Manufacturing method
본 개시내용은 파조파닙의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 및/또는 생산 방법을 제공한다.The present disclosure provides methods of making and / or producing a stable pharmaceutical pharmaceutical formulation of a pharmaceutically acceptable salt of pachoparamip.
본 개시내용은 활성 작용제의 샘플, 및 그에 따라 주어진 샘플에서의 활성제의 특징에 따라 좌우되는 약물 제제화 방법을 제공한다. 본 개시내용은 활성 작용제의 염 형태 및 결정화도에 따라 좌우되는 제제화 방법을 제공한다. 표 1, 2 및 3은 본 개시내용의 활성 파조파닙의 특징을 요약하여 제공한다.The present disclosure provides a method of drug formulation depending on a sample of the active agent, and thus on the characteristics of the active agent in a given sample. The present disclosure provides a formulation method that depends on the salt form and the degree of crystallinity of the active agent. Tables 1, 2 and 3 summarize the features of the active rippip nip of this disclosure.
<표 1><Table 1>
유사한 조성을 갖는 파조파닙/캅티솔® 용액의 pH 비교:Comparison of the pH of Pazopanib / Capsytol® solutions with similar composition:

*XRD 패턴에 기반하여, 샘플-2는 약물 분자 당 1개의 HCl을 함유하는, 형태-A의 결정 구조를 갖는다.* Based on the XRD pattern, Sample-2 has a crystalline structure of Form-A containing one HCl per drug molecule.
클로라이드 함량 비교: 표 2는 X선 형광법 (XRF)에 의해 측정된, 다양한 API 샘플의 클로라이드 함량을 제공한다. 문헌 [Evans Analytical Laboratories, X-RAY FLUORESCENCE (XRF) ANALYSIS REPORT, 21 Feb 2014, JOB NUMBER C0ELG412]을 참조한다.Comparison of chloride content: Table 2 provides the chloride content of various API samples, as determined by X-ray fluorescence (XRF). See Evans Analytical Laboratories, X-RAY FLUORESCENCE (XRF) ANALYSIS REPORT, 21 Feb 2014, JOB NUMBER C0ELG412.
<표 2><Table 2>

pH 및 클로라이드 함량의 분석 결과로부터, 약물 분자 당 단지 1개의 HCl을 함유하는 샘플-2의 활성제와 비교하여, 샘플-1의 제약 성분은 각각의 파조파닙 분자 당 2개의 HCl을 함유한다는 결론을 내렸다.Analysis of the pH and chloride content showed that the pharmaceutical ingredient of Sample-1 contained two HCl per each ripopanip molecule, as compared to the active ingredient of Sample-2 containing only one HCl per drug molecule I fell.
2가 염Divalent salt
본 개시내용은 파조파닙의 2가 염의 제제의 제조 방법을 제공한다. 상기 방법은 (a) 파조파닙의 2가 염을 1종 이상의 제제화 작용제의 용액에 용해시키는 단계를 포함한다. 상기 방법에 사용되는 제제화 작용제는 착물화제, 가용화제 및 완충제를 포함하나, 이들로 제한되지는 않는다. 2가 염을 제제화 작용제 용액에 용해시킨 후에 용액의 pH는 방출 전 및/또는 후에 제제 중의 2가 염의 안정성, 방출 전 및/또는 후에 제제 중의 2가 염의 용해도, 전달 장치로부터의 제제 중의 2가 염의 방출 속도, 및/또는 후안부로의 방출 시에 파조파닙의 치료 효능을 유지하기 위해 최적의 pH로 조정된다. 제약상 허용되는 염은 파조파닙의 2가 할라이드 염, 예를 들어 파조파닙의 2가 클로라이드 염, 파조파닙의 2가 브로마이드 염, 파조파닙의 2가 아이오다이드 염, 또는 파조파닙의 2가 플루오라이드 염이다. 본 개시내용은 파조파닙의 2가 클로라이드 염의 제조 방법을 제공한다.The present disclosure provides a method for preparing a divalent salt formulation of pazopanib. The method comprises (a) dissolving the diphasic salt of the pachopanip in a solution of at least one formulation agent. The formulation agents used in the method include, but are not limited to, complexing agents, solubilizing agents, and buffers. The pH of the solution after dissolving the divalent salt in the formulation agent solution may be determined by the stability of the divalent salt in the formulation before and / or after the release, the solubility of the divalent salt in the formulation before and / or after release, The rate of release, and / or the optimal pH to maintain the therapeutic efficacy of the wave pan nip upon release to the fascia. Pharmaceutically acceptable salts include, but are not limited to, divalent halide salts of pazopanib, such as the divalent chloride salt of pazopanib, the dibasic bromide salt of pazopanib, the divalent iodide salt of pazopanib, Is a divalent fluoride salt of the nip. The present disclosure provides a process for preparing a divalent chloride salt of pazopanib.
본 개시내용의 방법에 의해 제조된 파조파닙 제제 중의 2가 클로라이드 염은 2θ로 7.8, 12.4, 22.9, 23.6 및 26.9 ± 0.2도에서 XRPD 회절 피크를 갖는 다형체 형태 XIV (추가로 2θ로 14.8, 17.5, 19.0, 25.3 및 27.4 ± 0.2도에서의 피크에 의해 특징화됨); 또는 2θ로 6.7, 7.4, 12.0, 14.8, 23.6 ± 0.2도에서 XRPD 회절 피크를 갖는 형태 I (추가로 2θ로 13.3, 14.8, 19.0, 26.6 ± 0.2도에서의 피크에 의해 특징화됨); 또는 2θ로 6.9, 12.1, 23.6, 26.8 및 27.4 ± 0.2도에서 XRPD 회절 피크를 갖는 형태 XV (추가로 2θ로 15.7, 19.4, 23.3, 및 25.7 ± 0.2도에서의 피크에 의해 특징화됨)이다. 본 개시내용은 2θ로 7.8, 12.4, 22.9, 23.6 및 26.9 ± 0.2도에서 XRPD 회절 피크를 갖는 파조파닙 디히디로클로라이드의 다형체 형태 XIV (추가로 2θ로 14.8, 17.5, 19.0, 25.3 및 27.4 ± 0.2도에서의 피크에 의해 특징화됨)의 제제의 제조 방법을 제공한다.The divalent chloride salts in the ripshave nip formulations prepared by the methods of this disclosure were characterized by the polymorphic form XIV with XRPD diffraction peaks at 7.8, 12.4, 22.9, 23.6 and 26.9 + 0.2 degrees in 2θ (further 14.8, 17.5, 19.0, 25.3 and 27.4 +/- 0.2 degrees); Or characterized by peaks at 13.3, 14.8, 19.0, 26.6 ± 0.2 degrees in 2θ with an XRPD diffraction peak at 6.7, 7.4, 12.0, 14.8, 23.6 ± 0.2 degrees in 2θ; (Further characterized by peaks at 15.7, 19.4, 23.3, and 25.7 ± 0.2 degrees in 2θ) with XRPD diffraction peaks at 6.9, 12.1, 23.6, 26.8 and 27.4 ± 0.2 degrees at 2θ. This disclosure describes polymorphic forms XIV of X-ray diffraction peaks having XRPD diffraction peaks at 7.8, 12.4, 22.9, 23.6 and 26.9 +/- 0.2 degrees in 2 [theta] (further 14.8, 17.5, 19.0, 25.3 and 27.4 Lt; RTI ID = 0.0 > +/- 0.2 < / RTI > degrees).
파조파닙의 2가 염 제제의 제조 방법에 사용되는 착물화제는 시클로덱스트린, 예를 들어 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 또는 이들의 임의의 조합(들)이다.The complexing agent used in the process for preparing the divalent salt of pazopanib may be cyclodextrins such as 2-hydroxypropyl- beta -cyclodextrin, methyl- beta -cyclodextrin, random methylated-beta-cyclodextrin, ethyl Cyclodextrin, carboxymethyl-? -Cyclodextrin, hydroxyethyl-? -Cyclodextrin, 2-hydroxy-3- (trimethyl-β-cyclodextrin, triacetyl-β-cyclodextrin, Aminonio) propyl-beta-cyclodextrin, glucosyl-beta-cyclodextrin, maltosyl- beta -cyclodextrin, sulfobutyl ether-beta-cyclodextrin, branched-beta-cyclodextrin, hydroxypropyl- Dextrin, random methylated-gamma -cyclodextrin, trimethyl-gamma-cyclodextrin, or any combination (s) thereof.
본 개시내용의 파조파닙 2가 염의 제제의 제조 방법에 사용되는 가용화제는 폴리(비닐 피롤리돈) (PVP)이다. 본 개시내용의 파조파닙 2가 염의 제제의 제조 방법에 사용되는 완충제는 히스티딘 HCl이다.The solubilizing agent used in the process of making the present invention is a poly (vinylpyrrolidone) (PVP). The buffer used in the process for preparing the rapamycin nanoparticulate salt formulation of the present disclosure is histidine HCl.
본 개시내용의 방법은 약 60 mg/ml 이하의 약물 농도로 제제화되는 각각의 파조파닙 분자 당 2개의 HCl의 제제를 제공한다. 약물은 캅티솔® 용액에 용해된 다음, 제제화 작용제가 첨가되고, 그 후에 pH가 조정된다.The method of the present disclosure provides a formulation of two HCl per molecule of each ripple nip that is formulated at a drug concentration of about 60 mg / ml or less. The drug is dissolved in the Captisol solution and then the formulation agent is added, after which the pH is adjusted.
제제는 필요한 양의 파조파닙 2HCl 염을 수중의 시클로덱스트린, 산, 및 제제화 작용제에 용해시킴으로써 제조된다. 파조파닙 2HCl은 첨가되어 용해될 때까지 혼합된다. 그 후에, 최종 pH에 도달할 때까지 수산화나트륨이 첨가된다. 제제는 여과된 다음, PDS 이식물에 주입되어, 치료제 방출 시험을 수행한다.The preparation is prepared by dissolving the required amount of rapamycin nipHCl salt in water in a cyclodextrin, acid, and formulation agent. The wave pan nip 2HCl is added and mixed until dissolved. Thereafter, sodium hydroxide is added until the final pH is reached. The formulation is filtered and then injected into a PDS implant to perform a therapeutic release test.
본 개시내용은 치료제, 예를 들어 파조파닙 디-히드로클로라이드 (2HCl)의 제제 및 그의 제조 방법을 제공하고, 여기서 활성 작용제는 제제에서 연장된 기간 동안 (즉, 60일 초과 및/또는 90일 초과) 안정하다. 안정한 활성 작용제, 예를 들어 파조파닙 2HCl을 갖는 제제는 약 70 mg/mL 이하의 활성 작용제를 갖는다. 본 개시내용은 약 10 mg/mL 이하, 약 11 mg/mL 이하, 약 12 mg/mL 이하, 약 13 mg/mL 이하, 약 14 mg/mL 이하, 약 15 mg/mL 이하, 약 16 mg/mL 이하, 약 17 mg/mL 이하, 약 18 mg/mL 이하, 약 19 mg/mL 이하, 약 20 mg/mL 이하, 약 21 mg/mL 이하, 약 22 mg/mL 이하, 약 23 mg/mL 이하, 약 24 mg/mL 이하, 약 25 mg/mL 이하, 약 26 mg/mL 이하, 약 27 mg/mL 이하, 약 28 mg/mL 이하, 약 29 mg/mL 이하, 약 30 mg/mL 이하, 약 31 mg/mL 이하, 약 32 mg/mL 이하, 약 33 mg/mL 이하, 약 34 mg/mL 이하, 약 35 mg/mL 이하, 약 36 mg/mL 이하, 약 37 mg/mL 이하, 약 38 mg/mL 이하, 약 39 mg/mL 이하, 약 40 mg/mL 이하, 약 41 mg/mL 이하, 약 42 mg/mL 이하, 약 43 mg/mL 이하, 약 44 mg/mL 이하, 약 45 mg/mL 이하, 약 46 mg/mL 이하, 약 47 mg/mL 이하, 약 48 mg/mL 이하, 약 49 mg/mL 이하, 약 50 mg/mL 이하, 약 51 mg/mL 이하, 약 52 mg/mL 이하, 약 53 mg/mL 이하, 약 54 mg/mL 이하, 약 55 mg/mL 이하, 약 56 mg/mL 이하, 약 57 mg/mL 이하, 약 58 mg/mL 이하, 약 59 mg/mL 이하, 약 60 mg/mL 이하, 약 61 mg/mL 이하, 약 62 mg/mL 이하, 약 63 mg/mL 이하, 약 64 mg/mL 이하, 약 65 mg/mL 이하, 약 66 mg/mL 이하, 약 67 mg/mL 이하, 약 68 mg/mL 이하, 약 69 mg/mL 이하, 또는 약 70 mg/mL 이하의 안정한 활성제를 갖는 파조파닙 2HCl의 제제를 제공한다.The present disclosure provides formulations of therapeutic agents, such as parapanipid dihydrochloride (2HCl) and methods of preparing the same, wherein the active agent is administered over an extended period of time in the formulation (i. E., Over 60 days and / More stable). Formulations with stable active agents, such as Pazopapenib 2HCl, have an active agent of about 70 mg / mL or less. Less than about 15 mg / mL, less than about 16 mg / mL, less than about 11 mg / mL, less than about 12 mg / mL, less than about 13 mg / mL, less than about 18 mg / mL, less than about 19 mg / mL, less than about 20 mg / mL, less than about 21 mg / mL, less than about 22 mg / mL, less than about 23 mg / mL Less than about 25 mg / mL, less than about 26 mg / mL, less than about 27 mg / mL, less than about 28 mg / mL, less than about 29 mg / mL, less than about 30 mg / mL Less than about 35 mg / mL, less than about 36 mg / mL, less than about 37 mg / mL, less than about 32 mg / mL, less than about 33 mg / Less than about 38 mg / mL, less than about 39 mg / mL, less than about 40 mg / mL, less than about 41 mg / mL, less than about 42 mg / mL, less than about 43 mg / mL, less than about 44 mg / About 45 mg / mL, less than about 47 mg / mL, less than about 48 mg / mL, less than about 49 mg / mL, less than about 50 mg / mL, less than about 51 mg / mg / mL, less than about 53 mg / mL, less than about 54 mg / mL, less than about 55 mg / mL, less than about 56 mg / mL Less than about 59 mg / mL, less than about 60 mg / mL, less than about 61 mg / mL, less than about 62 mg / mL, less than about 63 mg / mL Less than about 65 mg / mL, less than about 66 mg / mL, less than about 67 mg / mL, less than about 68 mg / mL, less than about 69 mg / mL, or less than about 70 mg / mL Of stable active agent.
1가 염Monosodium salt
본 개시내용은 치료제 1가 염의 안정하고/거나 가용성인 제제의 제조 방법을 제공한다. 1가 염은 수용액에서 거의 불용성이며, 저장 중, 및/또는 제제의 표적 부위로의 전달 전 및/또는 후에 침전되는 경향이 있다. 본 개시내용은 용액에 1가 염을 가용화하는 용이함을 증가시키고/거나 용액에서의 1가 염의 안정성을 증가시키는 방법을 제공한다. 상기 방법은 제제 중의 1가 염의 증가한 용해도 및/또는 안정성을 제공하므로, 염이 저장 중, 및/또는 표적 부위로의 전달 전 및/또는 후에 연장된 기간 동안 용해되어 남아있다.The present disclosure provides a method for the preparation of a preparation in which Therapeutic 1 is stable and / or soluble in a salt. Monovalent salts are nearly insoluble in aqueous solutions and tend to settle out during storage and / or before and / or after delivery of the agent to a target site. The present disclosure provides a method of increasing the ease of solubilizing monovalent salts in a solution and / or increasing the stability of monovalent salts in a solution. The method provides increased solubility and / or stability of the monovalent salt in the formulation, so that the salt remains soluble during storage and / or for extended periods of time before and / or after delivery to the target site.
본 개시내용의 한 방법에서 약물, 예를 들어 파조파닙의 모노히드로클로라이드를 용해시키기 위해, 장기간의 가용화 시간 및 과량의 산 첨가가 필요하고; 제제의 pH는 염산 (HCl)으로 약 2 이하의 pH로 조정된다. 약 2 미만의 pH에서 캅티솔® 용액 중의 약물 및 부형제의 장기간의 가용화 공정 (1-3일) 후에, pH가 조정된다.In order to dissolve the drug, e. G. Monohydrochloride of pazopanib, in one method of the present disclosure, a prolonged solubilization time and excess acid addition are required; The pH of the formulation is adjusted to a pH of about 2 or less with hydrochloric acid (HCl). After a prolonged solubilization process (1-3 days) of the drug and excipient in the Capsytol 占 solution at a pH of less than about 2, the pH is adjusted.
약물의 NaOH 전처리 (무정형화)가 또한 낮은 pH에서의 가용화 단계 전에 성공적으로 사용된다. 무정형화 단계는 고 농도 용액을 제조하는데 필요한 시간을 단축시키지만, 이 단계는 또한 제제의 총 염 함량을 상당히 증가시킨다.Pretreatment of the drug with NaOH (amorphous) is also successfully used before the solubilization step at low pH. The amorphization step shortens the time required to make the high concentration solution, but this step also significantly increases the total salt content of the formulation.
본 개시내용은 낮은 수용해도를 갖는 치료제의 제약상 허용되는 1가 염의 안정한 제약 제제의 제조 방법을 제공한다. 치료제 1가 염의 안정한 제약 제제의 제조 방법은 (a) 1가 염을 염기로 처리하고 (무정형화); (b) 염기 처리된 염을 1종 이상의 제제화 작용제 용액에 용해시키고; (c) pH를 산을 사용하여 약 4 이하, 약 3 이하, 약 2 이하, 또는 약 1 이하의 pH로 조정하는 단계를 포함한다. 상기 방법에 사용되는 제제화 작용제는 착물화제, 가용화제 및 완충제를 포함하나, 이들로 제한되지는 않는다. 1가 염의 염기 처리는 제제 중의 총 염 함량을 증가시킨다. 산에 의한 pH 조정은 제제 중의 염의 용해도를 증가시킨다.The present disclosure provides a method for preparing stable pharmaceutical formulations of a pharmaceutically acceptable monovalent salt of a therapeutic agent having low water solubility. Methods for the preparation of pharmaceutical preparations in which the monosial salt is stable include (a) treatment of the monovalent salt with a base (amorphous); (b) dissolving the base treated salt in at least one formulation agent solution; (c) adjusting the pH to less than about 4, less than about 3, less than about 2, or less than about 1 using an acid. The formulation agents used in the method include, but are not limited to, complexing agents, solubilizing agents, and buffers. The base treatment of monovalent salts increases the total salt content in the formulation. The pH adjustment by the acid increases the solubility of the salt in the formulation.
본 개시내용은 파조파닙의 제약상 허용되는 1가 염의 안정한 제약 제제의 제조 방법을 제공한다. 파조파닙의 1가 염,예를 들어 파조파닙의 1가 클로라이드 염, 파조파닙의 1가 브로마이드 염, 파조파닙의 1가 아이오다이드 염, 또는 파조파닙의 1가 플루오라이드 염의 안정한 제약 제제의 제조 방법은 (a) 1가 염을 염기로 처리하고; (b) 염기 처리된 염을 1종 이상의 제제화 작용제 용액에 용해시키고; (c) pH를 산을 사용하여 약 4 이하, 약 3 이하, 약 2 이하, 또는 약 1 이하의 pH로 조정하는 단계를 포함한다. 상기 방법에 사용되는 제제화 작용제는 착물화제, 가용화제 및 완충제를 포함하나, 이들로 제한되지는 않는다. 1가 염의 염기 처리는 제제 중의 총 염 함량을 증가시킨다. 산에 의한 pH 조정은 제제 중의 염의 용해도를 증가시킨다. 염기는 예를 들어, 수산화나트륨 (NaOH)이다. 산은 예를 들어, 염산 (HCl)이다.The present disclosure provides a method for the preparation of stable pharmaceutical preparations of a pharmaceutically acceptable monovalent salt of pazopanib. Monopotassium salt of pazopanib, For example, a process for the preparation of stable pharmaceutical preparations of monovalent chloride salts of pazopanib, monovalent bromide salts of pazopanib, monovalent iodide salts of pazopanib, or monovalent fluoride salts of pazopanib a) treating the monovalent salt with a base; (b) dissolving the base treated salt in at least one formulation agent solution; (c) adjusting the pH to less than about 4, less than about 3, less than about 2, or less than about 1 using an acid. The formulation agents used in the method include, but are not limited to, complexing agents, solubilizing agents, and buffers. The base treatment of monovalent salts increases the total salt content in the formulation. The pH adjustment by the acid increases the solubility of the salt in the formulation. The base is, for example, sodium hydroxide (NaOH). The acid is, for example, hydrochloric acid (HCl).
파조파닙의 1가 염 제제의 제조 방법에 사용되는 착물화제는 시클로덱스트린, 예를 들어 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 또는 이들의 임의의 조합(들)이다.Complexing agents used in the preparation of monopotassium salt of pazopanib include cyclodextrins such as 2-hydroxypropyl- beta -cyclodextrin, methyl- beta -cyclodextrin, random methylated-beta-cyclodextrin, ethyl Cyclodextrin, carboxymethyl-? -Cyclodextrin, hydroxyethyl-? -Cyclodextrin, 2-hydroxy-3- (trimethyl-β-cyclodextrin, triacetyl-β-cyclodextrin, Aminonio) propyl-beta-cyclodextrin, glucosyl-beta-cyclodextrin, maltosyl- beta -cyclodextrin, sulfobutyl ether-beta-cyclodextrin, branched-beta-cyclodextrin, hydroxypropyl- Dextrin, random methylated-gamma -cyclodextrin, trimethyl-gamma-cyclodextrin, or any combination (s) thereof.
본 개시내용의 파조파닙의 1가 염 제제의 제조 방법에 사용되는 가용화제는폴리(비닐 피롤리돈) (PVP)이다. 본 개시내용의 파조파닙의 1가 염 제제의 제조 방법에 사용되는 완충제는 히스티딘 HCl이다.The solubilizing agent used in the process for preparing the monodisperse salt of the present invention is poly (vinylpyrrolidone) (PVP). The buffer used in the process for preparing the monovalent salt formulations of the parasitopin of the present disclosure is histidine HCl.
본 개시내용의 파조파닙의 1가 염 제제의 제조 방법에 사용되는 가용화제는 폴리(비닐 피롤리돈) (PVP)이며, 임의의 완충제는 포함되지 않는다.The solubilizing agent used in the process for preparing the monodisperse salt preparation of the ripopanib of the present disclosure is poly (vinylpyrrolidone) (PVP) and does not include any buffer.
본 개시내용은 약물 농도를 40 mg/mL 미만으로 낮춤으로써 1HCl 파조파닙 제제의 상당히 개선된 안정성을 제공한다.The present disclosure provides significantly improved stability of the 1HCl rifampin nip formulation by lowering the drug concentration to less than 40 mg / mL.
활성제는 고 농도에서 침전되는 경향이 있다.본 개시내용은 낮은 용해도의 활성제, 예를 들어 파조파닙의 안정한 용액 제제의 제조 방법을 제공한다.Active agents tend to precipitate at high concentrations. The present disclosure provides a method for making stable solutions of low solubility active agents, for example pachopanip.
본 개시내용은 치료제, 예를 들어 파조파닙 모노히드로클로라이드 (1HCl)의 제제 및 그의 제조 방법을 제공하고, 여기서 활성 작용제는 제제에서 약 40 mg/mL보다 낮거나 또는 약 40 mg/mL보다 높을 때 연장된 기간 동안 (즉, 60일 초과 및/또는 90일 초과) 안정하다. 안정한 활성 작용제, 예를 들어 파조파닙 1HCl을 갖는 제제는 약 60 mg/mL 이하에서 안정하다. 본 개시내용은 약 10 mg/mL 이하, 약 11 mg/mL 이하, 약 12 mg/mL 이하, 약 13 mg/mL 이하, 약 14 mg/mL 이하, 약 15 mg/mL 이하, 약 16 mg/mL 이하, 약 17 mg/mL 이하, 약 18 mg/mL 이하, 약 19 mg/mL 이하, 약 20 mg/mL 이하, 약 21 mg/mL 이하, 약 22 mg/mL 이하, 약 23 mg/mL 이하, 약 24 mg/mL 이하, 약 25 mg/mL 이하, 약 26 mg/mL 이하, 약 27 mg/mL 이하, 약 28 mg/mL 이하, 약 29 mg/mL 이하, 약 30 mg/mL 이하, 약 31 mg/mL 이하, 약 32 mg/mL 이하, 약 33 mg/mL 이하, 약 34 mg/mL 이하, 약 35 mg/mL 이하, 약 36 mg/mL 이하, 약 37 mg/mL 이하, 약 38 mg/mL 이하, 약 39 mg/mL 이하, 약 40 mg/mL 이하, 약 41 mg/mL 이하, 약 42 mg/mL 이하, 약 43 mg/mL 이하, 약 44 mg/mL 이하, 약 45 mg/mL 이하, 약 46 mg/mL 이하, 약 47 mg/mL 이하, 약 48 mg/mL 이하, 약 49 mg/mL 이하, 약 50 mg/mL 이하, 약 51 mg/mL 이하, 약 52 mg/mL 이하, 약 53 mg/mL 이하, 약 54 mg/mL 이하, 약 55 mg/mL 이하, 약 56 mg/mL 이하, 약 57 mg/mL 이하, 약 58 mg/mL 이하, 약 59 mg/mL 이하, 또는 약 60 mg/mL 이하의 안정한 활성제를 갖는 파조파닙 1HCl의 제제를 제공한다.The present disclosure provides a preparation of a therapeutic agent, for example pachopanip monohydrochloride (1 HCl), and a method of preparation thereof, wherein the active agent is present in the formulation at a level below about 40 mg / mL or higher than about 40 mg / (I.e., greater than 60 days and / or more than 90 days). Agents with stable active agents, such as Pazopapenib HCl, are stable at about 60 mg / mL or less. Less than about 15 mg / mL, less than about 16 mg / mL, less than about 11 mg / mL, less than about 12 mg / mL, less than about 13 mg / mL, less than about 18 mg / mL, less than about 19 mg / mL, less than about 20 mg / mL, less than about 21 mg / mL, less than about 22 mg / mL, less than about 23 mg / mL Less than about 25 mg / mL, less than about 26 mg / mL, less than about 27 mg / mL, less than about 28 mg / mL, less than about 29 mg / mL, less than about 30 mg / mL Less than about 35 mg / mL, less than about 36 mg / mL, less than about 37 mg / mL, less than about 32 mg / mL, less than about 33 mg / Less than about 38 mg / mL, less than about 39 mg / mL, less than about 40 mg / mL, less than about 41 mg / mL, less than about 42 mg / mL, less than about 43 mg / mL, less than about 44 mg / About 45 mg / mL, less than about 47 mg / mL, less than about 48 mg / mL, less than about 49 mg / mL, less than about 50 mg / mL, less than about 51 mg / mg / mL, less than about 53 mg / mL, less than about 54 mg / mL, less than about 55 mg / mL, less than about 56 mg / mL And, provides a formulation of about 57 mg / mL or less, wave harmonics nip 1HCl having a stable active agent of from about 58 mg / mL or less, about 59 mg / mL or less, or about 60 mg / mL or less.
본 개시내용은 가용화 전에 활성 작용제의 동결건조를 수행함으로써 1HCl 파조파닙 제제의 안정성을 개선하는 방법을 제공한다. 본 개시내용은 제제화 작용제 중의 가용화 전에 활성 작용제의 동결건조를 수행함으로써 제제 중의 1HCl 파조파닙의 용해도 및 안정성을 개선하는 제제화 방법 (제제의 제조 방법)을 제공한다.The present disclosure provides a method of improving the stability of 1HCl ripopanim formulations by performing lyophilization of the active agent prior to solubilization. The present disclosure provides a formulation method (preparation method of a preparation) which improves solubility and stability of 1HCl parsopanib in a preparation by performing lyophilization of the active agent before solubilization in the formulation agent.
본 개시내용은 2종의 상이한 용매 (트리플루오로 에탄올 (TFE) 및 디메틸 술폭시드 (DMSO))로부터의 동결건조가, 샘플-2의 활성 작용제의 결정 구조 및/또는 무정형 함량에 미치는 영향을 시험하는 것을 제공한다.The present disclosure examines the effect of lyophilization from two different solvents (trifluoroethanol (TFE) and dimethylsulfoxide (DMSO)) on the crystal structure and / or amorphous content of the active agent of sample-2 .
본 개시내용의 파조파닙 염은 결정질 및/또는 무정형 형태이다. 본 개시내용의 제제에 사용되는 다양한 파조파닙 염에 대한 XRPD 결과의 요약이 표 3에 제공된다.The piropanib salts of the present disclosure are crystalline and / or amorphous. A summary of the XRPD results for the various pirpapin salts used in the preparations of this disclosure is provided in Table 3.
<표 3><Table 3>

동결건조는 트리플루오로 에탄올 (TFE), 트리플루오로 에탄올-물 (90-10) 혼합물 또는 디메틸 술폭시드 (DMSO)로부터, 관련 기술분야의 표준 방법에 의해 수행된다.The lyophilization is carried out by standard methods in the related art from trifluoroethanol (TFE), trifluoroethanol-water (90-10) mixture or dimethylsulfoxide (DMSO).
본 개시내용의 DMSO로부터의 동결건조 조건은 하기 표 4에 기재된 조건을 포함한다.The lyophilization conditions from the DMSO of this disclosure include the conditions listed in Table 4 below.
<표 4><Table 4>

본 개시내용은 낮은 수용해도를 갖는 치료제의 제약상 허용되는 1가 염의 안정한 제약 제제의 제조 방법을 제공한다.The present disclosure provides a method for preparing stable pharmaceutical formulations of a pharmaceutically acceptable monovalent salt of a therapeutic agent having low water solubility.
본 개시내용은 제제화 방법 전에 전처리되는 치료제의 결정질 형태를 제공한다. 본 개시내용은 치료제, 예를 들어 파조파닙 1HCl을 알콜로부터 동걸건조시켜, XRPD에 의해 결정될 때 파조파닙 1HCl의 결정질 상 형태 A를 파조파닙 1HCl의 무정형 (또는 미세결정질) 물질 형태로 전환시키는 것을 제공한다. 알콜, 예를 들어 트리플루오로에탄올 (TFE)로부터의 동결건조는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 파조파닙 1HCl의 무정형 (또는 미세결정질) 물질 형태로 전환시킨다. 파조파닙 1HCl의 형태 A는 알콜, 예를 들어 TFE, 또는 TFE/물 혼합물에 용해된 다음, 용액이 동결건조된다. 동결건조 염은 약 37℃ 내지 약 50℃의 온도에서 용액에 용해된다. 본 개시내용은 결정 형태를 트리플루오로 에탄올 (TFE)에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 96% 이하의 또는 적어도 96%의 무정형 파조파닙이 형성되는 것인, 파조파닙의 결정 형태를 파조파닙의 무정형 (또는 미세결정질) 형태로 전환시키는 방법을 제공한다.This disclosure provides a crystalline form of the therapeutic agent that is pretreated prior to the formulation method. This disclosure discloses the conversion of a therapeutic agent, such as Pazopapenib HCl, from an alcohol to an amorphous (or microcrystalline) material form of Pazopap Nip HCl, as determined by XRPD, . Lyophilization from an alcohol, such as trifluoroethanol (TFE), converts the crystalline phase A of PazopapanipHCl to the amorphous (or microcrystalline) material form of PazopapinHCI, as determined by XRPD. Form A of rapamphanipHCl is dissolved in an alcohol, such as TFE, or a TFE / water mixture, and the solution is lyophilized. The lyophilized salt is dissolved in the solution at a temperature of about 37 캜 to about 50 캜. This disclosure includes dissolving a crystalline form in trifluoroethanol (TFE) and lyophilizing the resulting solution; Wherein at least 96% or at least 96% of the amorphous ripodopipes are formed, wherein the crystalline form of the ripopanib is converted to the amorphous (or microcrystalline) form of the ripopanib.
치료제, 예를 들어 파조파닙 모노히드로클로라이드의 결정질 형태는 동결건조에 의해 전처리된다. 예를 들어, TFE 중의 약 60 mg/mL의 치료제, 예를 들어 파조파닙 모노히드로클로라이드 용액이 제조된다. 약 1% 내지 약 30%의 물 (예를 들어, 약 20%의 물)이 또한 TFE 중의 치료제, 예를 들어 파조파닙 모노히드로클로라이드 용액 중에 첨가된다. 그 후에, 용액은 관련 기술분야의 표준 조건 하에 동결 건조된다 (물의 첨가와 함께 또는 물의 첨가 없이). 용액은 약 35℃ - 약 50℃ (예를 들어, 약 40℃) 하에 약 12시간 내지 약 24시간 동안 또는 약 50℃ - 약 65℃ (예를 들어, 약 60℃)에서 약 4 - 약 8시간 동안 건조된다. TFE로부터의 동결건조는 결정질 상 형태 A를 부분적으로 또는 완전히 (예를 들어, 96% 이하로 또는 적어도 96%) 무정형 상으로 전환시킨다. 무정형 (또는 미세결정질) 파조파닙 모노히드로클로라이드, 즉 TFE 중에서 동결건조된 것은 이어서 본 개시내용에 기재된 바와 같이, 적어도 가용화제, 완충제 및 착물화제를 혼합함으로써 제조된 용액에 용해된다.The crystalline form of the therapeutic agent, for example pachopanip monohydrochloride, is pretreated by lyophilization. For example, about 60 mg / mL of the therapeutic agent in TFE, for example Pazopanip monohydrochloride solution, is prepared. About 1% to about 30% water (e.g., about 20% water) is also added to the therapeutic agent in TFE, such as Pazopanip monohydrochloride solution. Thereafter, the solution is lyophilized (with or without the addition of water) under standard conditions in the relevant art. The solution is heated to a temperature of from about 35 DEG C to about 50 DEG C (e.g., about 40 DEG C) for about 12 hours to about 24 hours or from about 50 DEG C to about 65 DEG C (e.g., about 60 DEG C) Lt; / RTI > Freeze-drying from TFE converts the crystalline phase A partially or fully (e.g., up to 96% or at least 96%) to an amorphous phase. The amorphous (or microcrystalline) ripopanip monohydrochloride, i.e. freeze-dried in TFE, is then dissolved in a solution prepared by mixing at least the solubilizing agent, buffer and complexing agent, as described in the present disclosure.
파조파닙의 결정 형태 (예를 들어, 형태 A)를 무정형 형태를 함유하는 물질로 전환시키는 방법은 약 80% 이하, 약 81% 이하, 약 82% 이하, 약 83% 이하, 약 84% 이하, 약 85% 이하, 약 86% 이하, 약 87% 이하, 약 88% 이하, 약 89% 이하, 약 90% 이하, 약 91% 이하, 약 92% 이하, 약 93% 이하, 약 94% 이하, 약 95% 이하, 또는 약 96% 이하의 무정형 파조파닙 1HCl을 제공한다. 별법으로, 적어도 약 80%, 적어도 약 81%, 적어도 약 82%, 적어도 약 83%, 적어도 약 84%, 적어도 약 85%, 적어도 약 86%, 적어도 약 87%, 적어도 약 88%, 적어도 약 89%, 적어도 약 90%, 적어도 약 91%, 적어도 약 92%, 적어도 약 93%, 적어도 약 94%, 적어도 약 95%, 적어도 약 96%, 적어도 약 97%, 적어도 약 98%, 또는 적어도 약 99%의 무정형 파조파닙 1HCl이 형성되는 방법이 제공된다. 나머지 활성 작용제는 원래의 형태이든지, 또는 원래의 형태 또는 또 다른 형태의 혼합물 (예를 들어, 형태 A 및/또는 형태 G)이든지, 결정질 형태이다.The method of converting the crystalline form of pazopanib (e.g. Form A) into a material containing an amorphous form may include up to about 80%, up to about 81%, up to about 82%, up to about 83%, up to about up to about 84% , About 85%, about 86%, about 87%, about 88%, about 89%, about 90%, about 91%, about 92%, about 93% , About 95% or less, or about 96% or less of amorphous ripopanib HCl. Alternatively at least about 80%, at least about 81%, at least about 82%, at least about 83%, at least about 84%, at least about 85%, at least about 86% At least about 95%, at least about 96%, at least about 97%, at least about 98%, or at least about 95%, at least about 90%, at least about 90% A method is provided wherein about 99% amorphous ripple nip HCI is formed. The remaining active agent is in its original form, or in crystalline form, whether in its original form or in a mixture of other forms (e.g., Form A and / or Form G).
본 개시내용은 XRPD에 의해 결정될 때, 파조파닙 1HCl의 하나의 결정질 상 형태, 예를 들어 결정질 상 형태 A를, 약 70% 이하의 또는 적어도 약 70%의 파조파닙 1HCl의 상이한 결정질 상 형태, 예를 들어 형태 G를 함유하는 물질로 전환시키기 위해 극성 비양성자성 용매로부터 치료제, 예를 들어 파조파닙 1HCl을 동결건조시키는 것을 제공한다. 동결건조 염은 약 37℃ 내지 약 50℃의 온도에서 용액에 용해된다. 유기황 화합물로부터의 동결건조는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 적어도 약 70%의 파조파닙 1HCl의 형태 G를 함유하는 물질로 전환시킨다. 디메틸 술폭시드 (DMSO)로부터의 동결건조는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 약 70% 이하의 또는 적어도 약 70%의 파조파닙 1HCl의 형태 G를 함유하는 물질로 전환시킨다. 디메틸 술폭시드 (DMSO)로부터의 동결건조는 XRPD에 의해 결정될 때, 파조파닙 1HCl의 결정질 상 형태 A를 약 100%의 파조파닙 1HCl의 형태 G를 함유하는 물질로 전환시킨다. 동결건조 염은 약 37℃ 내지 약 50℃의 온도에서 용액에 용해된다. 본 개시내용은 결정 형태를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 적어도 약 70%의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다.The present disclosure contemplates that when determined by XRPD, one crystalline phase, e. G., Crystalline phase A, of the wavefib nip 1 HCl may be present in a different crystalline phase morphology of about 70% or less, or at least about 70% , For example, lyophilization of a therapeutic agent, for example PazopapenHCI, from a polar aprotic solvent to convert it to a material containing Form G. The lyophilized salt is dissolved in the solution at a temperature of about 37 캜 to about 50 캜. Freeze drying from the organosulfur compounds converts the crystalline phase A of PazopapanipHCl to a material containing at least about 70% of the Pazopanib HCl form G, as determined by XRPD. Lyophilization from dimethylsulfoxide (DMSO), when determined by XRPD, involves crystallizing crystalline phase A of Pazopapanil 1HCl to a material containing form G of about 70% or less, or at least about 70% of PazopapenHCI . Lyophilization from dimethylsulfoxide (DMSO) converts the crystalline phase A of PazopapanipHCl to a material containing Form G of about 100% of PazopapanipHCl, as determined by XRPD. The lyophilized salt is dissolved in the solution at a temperature of about 37 캜 to about 50 캜. This disclosure includes dissolving the crystal form in DMSO and lyophilizing the resulting solution; Wherein at least about 70% of the ripopanip forms G are formed, converting the crystal form of the ripopanip into a material containing crystalline form G of the ripopanip.
본 개시내용은 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 약 70% 이하의 또는 적어도 약 70%의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태 A를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다. 본 개시내용은 결정 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 약 70% 내지 약 100% (예를 들어, 약 70%, 약 71%, 약 72%, 약 73%, 약 74%, 약 75%, 약 76%, 약 77%, 약 78%, 약 79%, 약 80%, 약 81%, 약 82%, 약 83%, 약 84%, 약 85%, 약 86%, 약 87%, 약 88%, 약 89%, 약 90%, 약 91%, 약 92%, 약 93%, 약 94%, 약 95%, 약 96%, 약 97%, 약 98%, 약 99%, 약 100%)의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태 A를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법을 제공한다.The present disclosure includes dissolving Form A in DMSO and lyophilizing the resulting solution; Wherein at least about 70% or at least about 70% of the ripopanips of form G are formed, converting the crystalline form A of the ripopanib to a material containing crystalline form G of the ripopanip . The present disclosure includes dissolving crystalline Form A in DMSO and lyophilizing the resulting solution; Wherein about 70% to about 100% (e. G., About 70%, about 71%, about 72%, about 73%, about 74%, about 75% About 87%, about 87%, about 88%, about 89%, about 90%, about 91%, about 80%, about 80%, about 81%, about 82% , About 92%, about 93%, about 94%, about 95%, about 96%, about 97%, about 98%, about 99%, about 100% To provide a method of converting crystal form A of the pazopanip to a material containing crystal form G of the pazapanip.
파조파닙의 결정 형태 (예를 들어, 형태 A)를 결정 형태 G를 함유하는 물질로 전환시키는 방법은 약 50% 이하, 약 51% 이하, 약 52% 이하, 약 53% 이하, 약 54% 이하, 약 55% 이하, 약 56% 이하, 약 57% 이하, 약 58% 이하, 약 59% 이하, 약 60% 이하, 약 61% 이하, 약 62% 이하, 약 63% 이하, 약 64% 이하, 약 65% 이하, 약 66% 이하, 약 67% 이하, 약 68% 이하, 약 69% 이하, 또는 약 70% 이하의 파조파닙의 형태 G가 형성되는 것을 제공한다. 별법으로, 상기 방법은 적어도 약 70%, 적어도 약 71%, 적어도 약 72%, 적어도 약 73%, 적어도 약 74%, 적어도 약 75%, 적어도 약 76%, 적어도 약 77%, 적어도 약 78%, 적어도 약 79%, 적어도 약 80%, 적어도 약 81%, 적어도 약 82%, 적어도 약 83%, 적어도 약 84%, 적어도 약 85%, 적어도 약 86%, 적어도 약 87%, 적어도 약 88%, 적어도 약 89%, 적어도 약 90%, 적어도 약 91%, 적어도 약 92%, 적어도 약 93%, 적어도 약 94%, 적어도 약 95%, 적어도 약 96%, 적어도 약 97%, 적어도 약 98%, 적어도 약 99%, 또는 적어도 약 100%의 파조파닙의 형태 G가 형성되는 것을 제공한다.A method of converting a crystalline form of a pazopanib (e.g. Form A) into a substance containing crystalline Form G may include up to about 50%, up to about 51%, up to about 52%, up to about 53%, up to about 54% , Less than about 55%, less than about 56%, less than about 57%, less than about 58%, less than about 59%, less than about 60%, less than about 61%, less than about 62% , About 65%, about 66%, about 67%, about 68%, about 69%, about 70% or less of the form P of the wave pan nip is formed. At least about 72%, at least about 73%, at least about 74%, at least about 75%, at least about 76%, at least about 77%, at least about 78% At least about 84%, at least about 85%, at least about 86%, at least about 87%, at least about 88%, at least about 80%, at least about 80% , At least about 89%, at least about 90%, at least about 91%, at least about 92%, at least about 93%, at least about 94%, at least about 95%, at least about 96%, at least about 97% , At least about 99%, or at least about 100%.
본 개시내용은 제제화 방법 전에 전처리되는 치료제, 예를 들어 파조파닙 모노히드로클로라이드의 결정질 형태를 제공한다. 치료제, 예를 들어 파조파닙 모노히드로클로라이드의 결정질 형태는 동결건조에 의해 전처리된다 (DMSO로부터의 동결건조 조건의 예로서 표 4 참조). DMSO (디메틸 술폭시드) 중의 약 20-60 mg/mL의 치료제, 예를 들어 파조파닙 모노히드로클로라이드 용액이 제조된다. 그 후에, 용액은 관련 기술분야의 표준 조건 하에 동결 건조된다. 본 개시내용의 건조 조건은 주위 온도보다 높은 온도이다. 예를 들어, 본 개시내용의 용액은 약 35℃ - 약 50℃ (예를 들어, 약 40℃) 하에 약 12 - 약 24시간 동안, 또한 약 50℃ - 약 65℃ (예를 들어, 약 60℃)에서 약 24 - 약 40시간 동안, 또한 약 90 - 약 110℃ (예를 들어, 약 100℃)에서 약 0.5 - 약 2시간 동안 건조된다. DMSO로부터의 동결건조는 XRPD에 의해 결정될 때, 결정질 상 형태 A를 결정질 상 형태 G로 전환시킨다.This disclosure provides a crystalline form of a therapeutic agent that is pretreated prior to the formulation method, for example pachopanip monohydrochloride. The crystalline form of the therapeutic agent, for example pachopanip monohydrochloride, is pretreated by lyophilization (see Table 4 as an example of freeze-drying conditions from DMSO). About 20-60 mg / mL of the therapeutic agent in DMSO (dimethylsulfoxide), for example Pazopanip monohydrochloride solution, is prepared. The solution is then lyophilized under standard conditions in the relevant art. The drying conditions of this disclosure are higher than the ambient temperature. For example, a solution of the present disclosure can be heated to a temperature of from about 35 ° C to about 50 ° C (eg, about 40 ° C) for a period of from about 12 to about 24 hours, and from about 50 ° C to about 65 ° C Deg.] C for about 24 to about 40 hours, and also about 90 to about 110 [deg.] C (e.g., about 100 [deg.] C) for about 0.5 to about 2 hours. Freeze drying from DMSO converts crystalline phase A to crystalline phase G when determined by XRPD.
본 개시내용의 방법에서 DMSO로부터의 동결건조 단계는 2θ로 5.6, 15.5, 16.4, 24.0 및 24.3 ± 0.2도에서 XRPD 피크를 갖는 파조파닙의 결정질 상 형태 A (추가로 2θ로 10.5, 16.8, 17.9, 26.4 및 32.9 ± 0.2도에서의 피크에 의해 특징화됨)를, 2θ로 9.6, 16.8, 19.6, 24.7 및 26.2 ± 0.2도에서의 XRPD 피크에 의해 특징화되는 형태 G (추가로 2θ로 11.8, 14.6, 15.3, 18.4, 20.3 및 23.6 ± 0.2도에서의 피크에 의해 특징화됨)로 전환시킨다.In the method of the present disclosure, the lyophilization step from DMSO is performed in the crystalline phase A (additionally 10.5, 16.8, 17.9 (in terms of 2 [theta]) of XRPD peaks having XRPD peaks at 5.6, 15.5, 16.4, 24.0 and 24.3 + , 26.4 and 32.9 + 0.2 degrees) was characterized by a form G (further characterized by an XRPD peak at 9.6, 16.8, 19.6, 24.7 and 26.2 + 0.2 degrees in 2θ of 11.8, 14.6 , 15.3, 18.4, 20.3 and 23.6 +/- 0.2 degrees).
본 개시내용은 (a) 유기 용매 (TFE 또는 DMSO) 중의 염의 용액을 제조하고; (b) 용액을 동결건조시켜 치료제의 동결건조 염을 제조하는 단계를 포함하는, 치료제의 제약상 허용되는 1가 염의 안정한 제약 제제의 제조 방법을 제공한다. 치료제의 제약상 허용되는 1가 염의 안정한 제약 제제의 제조 방법은 (c) 가용화제 및 완충제를 물에 용해시켜 용액을 제조하고; (d) 착물화제를 가용화제 및 완충제의 수용액에 용해시켜 용액을 제조하고; (e) 약 주위 온도 이상에서 (약 37℃ - 약 50℃) 동결건조 염을 용액에 첨가하고 혼합하여 용액에 용해시키고; (f) 임의로, 제제의 pH를 조정하는 단계를 추가로 포함한다. 동결건조 염은 약 37℃ 내지 약 50℃ (예를 들어, 약 37℃, 약 38℃, 약 39℃, 약 40℃, 약 41℃, 약 42℃, 약 43℃, 약 44℃, 약 45℃, 약 46℃, 약 47℃, 약 48℃, 약 49℃, 약 50℃)의 온도에서 용액에 용해된다.The present disclosure relates to a process for preparing a solution comprising: (a) preparing a solution of a salt in an organic solvent (TFE or DMSO); (b) lyophilizing the solution to produce a lyophilized salt of the therapeutic. < RTI ID = 0.0 > < / RTI > A method for preparing a stable pharmaceutical preparation of a pharmaceutically acceptable monovalent salt of a therapeutic agent comprises: (c) dissolving the solubilizing agent and the buffer in water to prepare a solution; (d) dissolving the complexing agent in an aqueous solution of a solubilizer and a buffer to prepare a solution; (e) adding a lyophilized salt at a temperature above about the ambient temperature (about 37 ° C to about 50 ° C) to the solution and mixing and dissolving in the solution; (f) optionally, adjusting the pH of the formulation. The lyophilized salt may be administered at a temperature of about 37 캜 to about 50 캜 (e.g., about 37 캜, about 38 캜, about 39 캜, about 40 캜, about 41 캜, about 42 캜, about 43 캜, About 46 DEG C, about 47 DEG C, about 48 DEG C, about 49 DEG C, about 50 DEG C).
상기 방법에 사용되는 가용화제는 PVP이다. 상기 방법에 사용되는 완충제는 히스티딘 HCl이다. 상기 방법에 사용되는 착물화제는 시클로덱스트린: 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 또는 이들의 임의의 조합(들)이다.The solubilizing agent used in the process is PVP. The buffer used in the method is histidine HCl. The complexing agent used in this method is cyclodextrin: 2-hydroxypropyl- beta -cyclodextrin, methyl- beta -cyclodextrin, random methylated-beta-cyclodextrin, ethylated- beta -cyclodextrin, triacetyl- Cyclodextrin, carboxymethyl-beta-cyclodextrin, hydroxyethyl- beta -cyclodextrin, 2-hydroxy-3- (trimethylammonio) propyl- beta -cyclodextrin, glucosyl ? -cyclodextrin, maltosyl-? -cyclodextrin, sulfobutyl ether-? -cyclodextrin, branched? -cyclodextrin, hydroxypropyl-? -cyclodextrin, random methylated-? -cyclodextrin, trimethyl- gamma -cyclodextrin, or any combination (s) thereof.
본 개시내용은 (a) 유기 용매 (TFE 또는 DMSO) 중의 염의 용액을 제조하고; (b) 용액을 동결건조시켜 파조파닙의 동결건조 염을 제조하는 단계를 포함하는, 파조파닙의 제약상 허용되는 1가 염, 예를 들어 파조파닙의 1가 클로라이드 염, 파조파닙의 1가 브로마이드 염, 파조파닙의 1가 아이오다이드 염, 또는 파조파닙의 1가 플루오라이드 염의 안정한 제약 제제의 제조 방법을 제공한다. 파조파닙의 제약상 허용되는 1가 염의 안정한 제약 제제의 제조 방법은 동결건조 후에, 파조파닙의 동결건조된 1가 할라이드 염, 예를 들어 파조파닙의 1가 클로라이드 염, 예를 들어 히드로클로라이드 염, 파조파닙의 1가 브로마이드 염, 파조파닙의 1가 아이오다이드 염, 또는 파조파닙의 1가 플루오라이드 염이 무정형인 것을 제공한다.The present disclosure relates to a process for preparing a solution comprising: (a) preparing a solution of a salt in an organic solvent (TFE or DMSO); (b) lyophilizing the solution to produce a lyophilized salt of the pachopanip. < Desc /
파조파닙의 제약상 허용되는 1가 염의 안정한 용액 제약 제제의 제조 방법은 (c) 가용화제 및 완충제를 물에 용해시켜 용액을 제조하고; (d) 착물화제를 가용화제 및 완충제의 수용액에 용해시켜 용액을 제조하고; (e) 약 주위 온도 이상에서 (예를 들어, 약 37℃ 또는 50℃) 동결건조 염을 점성 용액에 첨가하고 혼합하여 용액에 용해시키고; (f) 임의로, 제제의 pH를 조정하는 것을 추가로 포함한다. 동결건조 염은 약 37℃ 내지 약 50℃ (예를 들어, 약 37℃, 약 38℃, 약 39℃, 약 40℃, 약 41℃, 약 42℃, 약 43℃, 약 44℃, 약 45℃, 약 46℃, 약 47℃, 약 48℃, 약 49℃, 약 50℃)의 온도에서 용액에 용해된다.A stable solution of a pharmaceutically acceptable monovalent salt of pazopanib The process for preparing a pharmaceutical formulation comprises (c) dissolving a solubilizing agent and a buffer in water to prepare a solution; (d) dissolving the complexing agent in an aqueous solution of a solubilizer and a buffer to prepare a solution; (e) adding to the viscous solution a lyophilized salt at a temperature above the ambient temperature (e.g., about 37 ° C or 50 ° C) and mixing and dissolving in the solution; (f) optionally, adjusting the pH of the formulation. The lyophilized salt may be administered at a temperature of about 37 캜 to about 50 캜 (e.g., about 37 캜, about 38 캜, about 39 캜, about 40 캜, about 41 캜, about 42 캜, about 43 캜, About 46 DEG C, about 47 DEG C, about 48 DEG C, about 49 DEG C, about 50 DEG C).
1가 파조파닙 제제의 제조 방법에 사용되는 가용화제는 PVP이다. 동일한 방법에 사용되는 완충제는 히스티딘 HCl이다. 동일한 방법에 사용되는 착물화제는 시클로덱스트린: 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 또는 이들의 임의의 조합(들)이다.The solubilizing agent used in the preparation of the monodisperse parapanip formulation is PVP. The buffer used in the same method is histidine HCl. The complexing agent used in the same process is cyclodextrin: 2-hydroxypropyl- beta -cyclodextrin, methyl- beta -cyclodextrin, random methylated-beta-cyclodextrin, ethylated- beta -cyclodextrin, triacetyl- Cyclodextrin, carboxymethyl-beta-cyclodextrin, hydroxyethyl- beta -cyclodextrin, 2-hydroxy-3- (trimethylammonio) propyl- beta -cyclodextrin, glucosyl ? -cyclodextrin, maltosyl-? -cyclodextrin, sulfobutyl ether-? -cyclodextrin, branched? -cyclodextrin, hydroxypropyl-? -cyclodextrin, random methylated-? -cyclodextrin, trimethyl- gamma -cyclodextrin, or any combination (s) thereof.
파조파닙의 제약상 허용되는 1가 염의 안정한 용액 제약 제제의 제조 방법은 파조파닙의 1가 할라이드 염, 예를 들어 파조파닙의 1가 클로라이드 염, 파조파닙의 1가 브로마이드 염, 파조파닙의 1가 아이오다이드 염, 또는 파조파닙의 1가 플루오라이드 염의 안정한 제제를 제공한다. 파조파닙의 제약상 허용되는 1가 염의 안정한 제약 제제의 제조 방법은 파조파닙의 1가 클로라이드 염의 안정한 제제를 제공한다.A stable solution of a pharmaceutically acceptable monovalent salt of pazopanib The process for the preparation of a pharmaceutical formulation comprises reacting a monohydrate salt of pazopanib, for example monovalent chloride salt of pazopanib, a monovalent bromide salt of pazopanib, A stable preparation of a monovalent iodide salt of zapanib, or a monovalent fluoride salt of pazopanib. A process for the preparation of a stable pharmaceutical formulation of a pharmaceutically acceptable monovalent salt of pazopanib provides a stable formulation of monophoryl chloride salt of pazopanib.
본 개시내용은 제제의 천연 pH가 사용되는, 즉 점성 용액의 pH 조정이 필요하지 않은 방법을 제공한다. 상기 방법에서, 제약 성분의 가용화는 하나의 단계로 수행된다. PVP-10k (폴리비닐 피롤리돈, MW=10 kDa) 및 히스티딘 HCl을 칭량하여 용액을 혼합함으로써 (와동, 진탕) 적절한 양의 물에 용해시킨다. 캅티솔®을 칭량하여 첨가하고, 용액의 진탕, 와동으로 용액에 용해시킨다. 동결건조된 파조파닙을 칭량한 다음 점성 캅티솔® 용액에 첨가하고, 주위 온도 또는 승온 (약 37℃ - 약 50℃)에서 와동, 초음파 처리, 진탕에 의해 완전히 용해시킨다. 제제는 0.2 ㎛ 필터를 사용하여 여과되고, 실온에서 차광하여 저장된다.The present disclosure provides a method in which the natural pH of the formulation is used, i.e. the pH adjustment of the viscous solution is not required. In this method, the solubilization of the pharmaceutical ingredients is carried out in one step. The PVP-10k (polyvinylpyrrolidone, MW = 10 kDa) and histidine HCl are weighed and dissolved in the appropriate amount of water by mixing (vortex, shaking) the solution. Capsol® is weighed and added, and dissolved in the solution by shaking and vortexing the solution. The lyophilized wave pan nip is weighed and then added to the viscous capricol® solution and completely dissolved by ambient, sonication, shaking at ambient or elevated temperature (about 37 ° C to about 50 ° C). The preparation is filtered using a 0.2 [mu] m filter, and stored by shading at room temperature.
본 개시내용은 (a) 유기 용매 (예를 들어, 트리플루오로 에탄올, 트리플루오로 에탄올-물 혼합물, 또는 디메틸 술폭시드) 중의 염의 용액을 제조하고; (b) 용액을 동결건조시켜 치료제의 동결건조 염을 제조하고; (c) 가용화제 및 완충제를 물에 용해시켜 용액을 제조하고; 일정량의 착물화제를 용액에 용해시켜 저 점도 용액을 제조하고; 약 주위 온도 이상에서 (약 37℃ - 약 50℃) 동결건조 염을 저 점도 용액에 첨가하고 혼합하여 용액에 용해시키고; 여기서 저 점도 용액의 pH를 조정하고; 약 2x 더 많은 양의 착물화제를 저 점도 용액에 첨가하여 용해시키는 것을 포함하는, 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법을 제공하고, 여기서 염은 1가 염, 예를 들어 파조파닙 1HCl이다. 동결건조 염은 약 37℃ 내지 약 50℃ (예를 들어, 약 37℃, 약 38℃, 약 39℃, 약 40℃, 약 41℃, 약 42℃, 약 43℃, 약 44℃, 약 45℃, 약 46℃, 약 47℃, 약 48℃, 약 49℃, 약 50℃)의 온도에서 용액에 용해된다.The present disclosure provides a process for preparing a solution of a salt in an organic solvent (e.g., trifluoroethanol, trifluoroethanol-water mixture, or dimethylsulfoxide); (b) lyophilizing the solution to prepare a lyophilized salt of the therapeutic; (c) dissolving the solubilizing agent and the buffer in water to prepare a solution; Dissolving an amount of the complexing agent in a solution to prepare a low viscosity solution; The lyophilized salt is added to the low viscosity solution at about the ambient temperature (about 37 ° C to about 50 ° C) and mixed and dissolved in the solution; Wherein the pH of the low viscosity solution is adjusted; A stable solution of a pharmaceutically acceptable salt of a therapeutically active agent having a low water solubility, comprising adding about 2x greater amount of a complexing agent to a low viscosity solution to dissolve, wherein the salt is a monovalent salt , For example Parmopanib 1 HCl. The lyophilized salt may be administered at a temperature of about 37 캜 to about 50 캜 (e.g., about 37 캜, about 38 캜, about 39 캜, about 40 캜, about 41 캜, about 42 캜, about 43 캜, About 46 DEG C, about 47 DEG C, about 48 DEG C, about 49 DEG C, about 50 DEG C).
본 개시내용은 제제 제조 공정 동안에 용액의 점도를 감소시키기 위해 두 단계로 캅티솔®이 첨가되는, 치료제 제제의 제조 방법을 제공한다. 상기 방법은 예를 들어, 캅티솔® 총량의 절반을 사용하여 캅티솔® 수용액을 제조하고; 치료제 및 첨가제 (예를 들어, PVP, 히스티딘)를 용해시키고, 제제의 pH를 조정한 다음, 나머지 양의 캅티솔®을 첨가하는 것을 포함한다. 2단계 공정은 고 점도 용액보다 신속한 용해 및 pH 측정의 용이함을 제공하는 저 점도 용액을 생성한다.The present disclosure provides a method of making a therapeutic agent formulation in which capricol is added in two steps to reduce the viscosity of the solution during the formulation manufacturing process. The method includes, for example, preparing an aqueous solution of CaPtisol using half of the total amount of Captisol®; Dissolving therapeutic agents and additives (e.g., PVP, histidine), adjusting the pH of the formulation, and then adding the remaining amount of Capsytol®. The two-step process produces a low viscosity solution that provides faster dissolution and ease of pH measurement than high viscosity solutions.
제제는 완충제 (예를 들어, 유기 완충제, 예를 들어 히스티딘 또는 히스티딘 HCl) 및 pH 조정 작용제 (예를 들어, 무기 염기, 예를 들어 NaOH; 유기 염기, 예를 들어 메글루민; 또는 유기 완충제, 예를 들어 히스티딘 또는 히스티딘 HCl)를 추가로 포함한다. 일부 실시양태에서, 치료제, 예를 들어 파조파닙 제제의 pH는 무기 염기, 예를 들어 NaOH로 조정된다. 다른 실시양태에서, 치료제, 예를 들어 파조파닙 제제의 pH는 유기 염기, 예를 들어 메글루민으로 조정된다. 일부 실시양태에서, 본 개시내용은 유기 염기, 예를 들어 메글루민이 사용된, 치료제, 예를 들어 파조파닙의 비-침전 제제를 제공한다. 일부 실시양태에서, 추가 첨가제 (예를 들어, 트리아세틴, 글리세롤)가 제제의 안정성 (침전에 대한 안정성)을 증가시키기 위해 첨가된다.The formulations may also include buffers (e. G., Organic buffers such as histidine or histidine HCl) and pH adjusting agents (e. G., Inorganic bases such as NaOH; organic bases such as meglumine; For example, histidine or histidine HCl). In some embodiments, the pH of the therapeutic agent, such as pachopanim, is adjusted to an inorganic base, such as NaOH. In another embodiment, the pH of the therapeutic agent, for example pachopanim, is adjusted with an organic base, e. G. Meglumine. In some embodiments, the disclosure provides a non-precipitation formulation of a therapeutic agent, such as pachopanip, in which an organic base, e. G., Meglumine, is used. In some embodiments, additional additives (e. G., Triacetin, glycerol) are added to increase the stability of the formulation (stability to precipitation).
본 개시내용은 필요한 캅티솔®의 대략 절반을 먼저 칭량하여 적절한 양의 물을 함유하는 바이알에서 용해시킴으로써, 치료제, 예를 들어 파조파닙의 1가 염, 예를 들어 1가 히드로클로라이드 염의 안정한 용액 제제를 제조하는 방법을 제공한다. PVP-10k (폴리비닐 피롤리돈, MW=10 kDa) 및 히스티딘 HCl을 첨가하여 용액을 혼합함으로써 (와동, 초음파 처리, 진탕) 용액에 용해시킨다. 동결건조된 파조파닙을 칭량한 다음 캅티솔® 용액에 첨가한다. 필요에 따라, 소량의 염산 (HCl)을 첨가하여 용액의 pH를 약 2 이하의 pH로 조정하고 유지한다. 첨가제, 예컨대 트리아세틴 또는 글리세롤을 첨가한다. 파조파닙이 완전히 용해될 때까지, 제제를 37℃에서 또는 실온에서 교반하고 진탕시킨다. 파조파닙의 용해는 수시간이 걸릴 수 있다. 그 후에, NaOH 또는 메글루민을 첨가하여 파조파닙-캅티솔® 용액의 pH를 약 pH 6-7로 조정한다. 나머지 캅티솔®을 어어서 첨가하고 37℃에서 또는 실온에서 제제의 진탕/와동에 의해 완전히 용해시킨다. pH를 확인하고, 제제를 0.2 ㎛ 필터를 사용하여 여과하기 전에, 필요에 따라 조정한다. 제제는 실온에서 차광하여 저장된다. 제제의 함량 및 순도는 HPLC 및 UV에 의해 시험한다.This disclosure is based on the need to first weigh approximately one-half of the required capricol, and dissolve in vials containing the appropriate amount of water to form a stable solution of a therapeutic agent, for example a monovalent salt of pazopanib, for example a monohydric hydrochloride salt To provide a method of making the formulation. The solution is dissolved in the solution (vortex, sonication, shaking) by adding PVP-10k (polyvinylpyrrolidone, MW = 10 kDa) and histidine HCl. The lyophilized wave pan nip is weighed and then added to the capricol solution. If necessary, a small amount of hydrochloric acid (HCl) is added to adjust and maintain the pH of the solution to a pH of about 2 or less. Additives such as triacetin or glycerol are added. The formulation is stirred and shaken at < RTI ID = 0.0 > 37 C < / RTI > or at room temperature until the rippa nip is completely dissolved. Dissolution of pachopanip may take several hours. Thereafter, the pH of the Pazopanib-capricosol solution is adjusted to about pH 6-7 by adding NaOH or meglumine. Add the remaining Capsytol ® completely and dissolve completely at 37 ° C or at room temperature by shaking / vortexing the formulation. The pH is checked and the formulation is adjusted as necessary before filtration using a 0.2 [mu] m filter. The formulation is stored at room temperature for shading. The content and purity of the preparation are tested by HPLC and UV.
본 개시내용은 모든 고체 부형제 (캅티솔®, PVP 및 히스티딘-HCl) 및 치료제 (예를 들어, 동결건조된 파조파닙 1HCl)가 측정되고 먼저 바이알에서 함께 혼합되는 제제화 방법을 제공한다. 연속적 혼합을 사용하여, 필요한 물의 점진적인 첨가가 수행된다. 형성된 분산물의 가용화는 주위 온도에서 또는 승온에서 (예를 들어, 약 37℃ 또는 약 50℃) 수행될 수 있고; 승온의 사용은 균질 용액을 달성하는데 필요한 시간을 단축시킨다 (예를 들어, 약 24시간 → 약 4시간). 그 후에, 제제는 천연 pH 약 3 - 약 4에서 사용되거나, 또는 NaOH 용액에 의한 pH 조정 (pH 약 6 - 약 7) 후에 사용된다.The present disclosure provides a method of formulation in which all solid excipients (Capsytol®, PVP and histidine-HCl) and therapeutic agents (eg, lyophilized Pazopanib® HCl) are measured and first mixed together in vials. Using continuous mixing, gradual addition of the required water is carried out. Solubilization of the formed dispersion can be performed at ambient temperature or at elevated temperature (e.g., about 37 ° C or about 50 ° C); The use of elevated temperatures shortens the time required to achieve a homogeneous solution (e. G., About 24 hours to about 4 hours). Thereafter, the formulation is used at a natural pH of about 3 to about 4, or after pH adjustment by a NaOH solution (pH of about 6 to about 7).
본 개시내용은 적어도 가용화제, 완충제, 착물화제, 및 동결건조 염이 약 주위 온도 이상에서 물을 첨가하면서 연속적으로 혼합되는 방법을 제공한다. 제제의 pH는 염기를 사용하여 약 6-7로 조정된다.The present disclosure provides a method wherein at least the solubilizing agent, the buffering agent, the complexing agent, and the lyophilized salt are continuously mixed while adding water at or above about the ambient temperature. The pH of the formulation is adjusted to about 6-7 using a base.
본 개시내용은 적어도 가용화제, 완충제, 착물화제, 및 파조파닙의 동결건조된 모노히드로클로라이드 염이 약 주위 온도 이상에서 물을 첨가하면서 연속적으로 혼합되는 방법을 제공한다. 제제의 pH는 염기를 사용하여 약 6-7로 조정된다. 상기 방법에서 착물화제는 시클로덱스트린: 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 또는 이들의 임의의 조합(들)이고; 가용화제는 폴리(비닐 피롤리돈) (PVP)이며; 완충제는 히스티딘 HCl이다.The present disclosure provides a method wherein at least the solubilizing agent, the buffering agent, the complexing agent, and the lyophilized monohydrochloride salt of pachopanip are continuously mixed while adding water at a temperature above the ambient temperature. The pH of the formulation is adjusted to about 6-7 using a base. In this method, the complexing agent is selected from the group consisting of cyclodextrin: 2-hydroxypropyl-beta-cyclodextrin, methyl-beta-cyclodextrin, random methylated- beta -cyclodextrin, ethyl- beta -cyclodextrin, triacetyl- beta -cyclodextrin , Peracetylated-beta-cyclodextrin, carboxymethyl-beta-cyclodextrin, hydroxyethyl-beta-cyclodextrin, 2-hydroxy- 3- (trimethylammonio) propyl- beta -cyclodextrin, glucosyl-beta - cyclodextrin, maltosyl-beta-cyclodextrin, sulfobutyl ether-beta-cyclodextrin, branched-beta-cyclodextrin, hydroxypropyl- gamma -cyclodextrin, random methylated- gamma- cyclodextrin, trimethyl- Cyclodextrin, or any combination (s) thereof; The solubilizing agent is poly (vinylpyrrolidone) (PVP); The buffer is histidine HCl.
본 개시내용은 적어도 가용화제, 완충제, 착물화제, 및 파조파닙의 동결건조된 모노히드로클로라이드 염이 약 주위 온도 이상에서 물을 첨가하면서 연속적으로 혼합되는 방법을 제공한다. 상기 방법에서, 제제의 pH는 조정되지 않는다. 상기 방법에서 착물화제는 시클로덱스트린: 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 또는 이들의 임의의 조합(들)이고; 가용화제는 폴리(비닐 피롤리돈) (PVP)이며; 완충제는 히스티딘 HCl이다.The present disclosure provides a method wherein at least the solubilizing agent, the buffering agent, the complexing agent, and the lyophilized monohydrochloride salt of pachopanip are continuously mixed while adding water at a temperature above the ambient temperature. In this method, the pH of the formulation is not adjusted. In this method, the complexing agent is selected from the group consisting of cyclodextrin: 2-hydroxypropyl-beta-cyclodextrin, methyl-beta-cyclodextrin, random methylated- beta -cyclodextrin, ethyl- beta -cyclodextrin, triacetyl- beta -cyclodextrin , Peracetylated-beta-cyclodextrin, carboxymethyl-beta-cyclodextrin, hydroxyethyl-beta-cyclodextrin, 2-hydroxy- 3- (trimethylammonio) propyl- beta -cyclodextrin, glucosyl-beta - cyclodextrin, maltosyl-beta-cyclodextrin, sulfobutyl ether-beta-cyclodextrin, branched-beta-cyclodextrin, hydroxypropyl- gamma -cyclodextrin, random methylated- gamma- cyclodextrin, trimethyl- Cyclodextrin, or any combination (s) thereof; The solubilizing agent is poly (vinylpyrrolidone) (PVP); The buffer is histidine HCl.
치료제의 방출 속도Release rate of therapeutic agent
본 개시내용은 37℃에서 PDS에 의해 수용 유체 (PBS 완충액)로 방출된 치료제의 양에 의해 측정되는 치료제 방출 속도를 제공한다. 치료제 방출 시험은 인큐베이터에서 37℃로 유지된, 유리체를 대표하는 유체로 PDS에 의해 방출된 치료제의 양을 측정함으로써 수행된다. PDS는 포스페이트 완충 식염수를 함유하는 용기에 부유되어 있다. 주기적으로, PDS는 새로운 용기로 옮겨지고 치료제의 농도가 이전 용기의 유체에서 측정된다. 속도는 방출된 치료제의 양을 샘플 수집 기간으로 나누어 계산한다. 누적 방출 %는 치료 장치 (PDS)에 초기에 충전된 치료제의 양으로 나눈 치료제의 누적 양으로부터 계산한다. 반감기는 4주째의 누적 방출 %로부터 계산한다. 본 개시내용은 소량의 제제가 다량의 완충 용액으로 방출될 때의 조건을 제공한다. 약물이 희석 (방출) 시에 침전된다면, 이는 전달 장치의 폐색 및/또는 수용 유체 중의 고체 약물은 측정가능하지 않을 것이므로 약물의 손실을 초래할 수 있다. 게다가, 침전은 생체내에서 약물을 사용하기 어렵게 하여 활성제에 의한 효과적인 치료를 방해한다.The present disclosure provides a therapeutic agent release rate as measured by the amount of therapeutic agent released to the receiving fluid (PBS buffer) by PDS at 37 占 폚. The therapeutic agent release test is performed by measuring the amount of therapeutic agent released by the PDS as a fluid representative of the vitreous, maintained at 37 占 폚 in an incubator. The PDS is suspended in a container containing phosphate buffered saline. Periodically, the PDS is transferred to a new container and the concentration of the therapeutic agent is measured in the fluid of the previous vessel. The rate is calculated by dividing the amount of therapeutic released by the sample collection period. The cumulative release rate is calculated from the cumulative amount of therapeutic agent divided by the amount of therapeutic agent initially charged to the treatment device (PDS). The half-life is calculated from the cumulative% of release at 4 weeks. The present disclosure provides conditions under which a small amount of the formulation is released into a large amount of buffer solution. If the drug is precipitated at the time of dilution (release), it can lead to loss of drug because the obstruction of the delivery device and / or the solid drug in the receiving fluid will not be measurable. In addition, sedimentation makes the drug difficult to use in vivo, thereby hindering effective treatment with active agents.
본 개시내용은 약물 침전 시험을 위한 조건을 제공하고, 여기서 제제는 예를 들어, 포스페이트 완충 식염수 용액 (예를 들어, 0.1% 아지드화나트륨)으로 330배 희석되는데, 예를 들어 약 3 μL의 제제가 1 mL의 PBS 완충액에 첨가된다. 용액은 온도조절기에서 유지되고 (예를 들어, 37℃의 온도), 결정 성장/침전의 출현에 대하여 주기적으로 확인된다.본 개시내용은 상이한 약물 샘플 (파조파닙 HCl의 샘플-1 또는 2)로 제조된 제제가 희석 시에 침전에 대하여 상이한 안정성을 나타낸다는 것을 제공한다.The present disclosure provides conditions for drug sedimentation testing wherein the formulation is diluted 330 times with, for example, a phosphate buffered saline solution (e.g., 0.1% sodium azide), for example, about 3 μL of The formulation is added to 1 mL of PBS buffer. The solution is maintained in a thermostat (for example at a temperature of 37 [deg.] C) and is periodically checked for the appearance of crystal growth / precipitation. The present disclosure provides that formulations made with different drug samples (Sample 1 or 2 of Pazopapin HCI) exhibit different stability with respect to precipitation upon dilution.
<표 5><Table 5>

*약물 농도는 파조파닙 염 (mg/mL)에 대한 것임* Drug concentration is for prazopanib salt (mg / mL).
다양한 충전 농도 하에 약 1 mg/ml 내지 약 100 mg/mL의 제제 중의 본 개시내용의 치료제의 방출 속도는 1일째의 약 100 ㎍/mL 내지 140일째의 약 0.01 ㎍/mL에서 달라진다. 본 개시내용은 방출 속도가 파조파닙의 제약상 허용되는 염의 HCl 함량 및/또는 결정질 형태의 함수라는 것을 제공한다. 파조파닙 1HCl의 방출 속도와 비교하여, 파조파닙 2HCl의 방출 속도가 더 빠르고 100일 넘게 지속될 수 있다.The release rate of the present disclosure in a formulation of from about 1 mg / ml to about 100 mg / ml under various fill concentrations varies from about 100 占 퐂 / mL on day one to about 0.01 占 퐂 / mL on
동결건조는 고 결정질 약물을, 보다 유리한 용해도 특성을 갖는 거의 무정형 고체로 전환시키는 것으로 생각된다. 도 1에 도시된 바와 같이, 동결건조된 1HCl 염 파조파닙 활성 작용제로부터 제조된 제제는 고 결정질 1HCl 형태와 비교하여 상당히 개선된 안정성 (침전/결정화에 대한 안정성)을 가지며, 또한 2HCl 형태와 유사한 약물 방출 특징을 초래하였다.It is believed that lyophilization converts the high crystalline drug into an almost amorphous solid with more favorable solubility characteristics. As shown in Figure 1, formulations made from lyophilized < RTI ID = 0.0 > 1HCl < / RTI > Drug release characteristics.
본 개시내용은 제제의 pH가 약 6.5로 조정된, 캅티솔®, 가용화제, 예를 들어 PVP, 및 완충제, 예를 들어 히스티딘 HCl의 제제로부터 비-동결건조된 파조파닙 1HCl (샘플-2)의 1일째의 약 12 ㎍/일 내지 대략 20일경 또는 약 20일을 지났을 때의 약 1 - 약 3 ㎍/일의 방출 속도를 제공한다. 약물 방출의 반감기는 약 99일이다. 게다가, 비-동결건조된 파조파닙 1HCl은 약물 방출 중에 용액으로부터 침전된다. 제제 중의 비-동결건조된 파조파닙 1HCl (샘플-2)은 약 40 mg/mL, 약 41 mg/mL, 약 42 mg/mL, 약 43 mg/mL, 약 44 mg/mL, 약 45 mg/mL, 약 46 mg/mL, 약 47 mg/mL, 약 48 mg/mL, 약 49 mg/mL, 약 50 mg/mL, 약 51 mg/mL, 약 52 mg/mL, 약 53 mg/mL, 약 53 mg/mL, 약 54 mg/mL, 약 55 mg/mL, 약 56 mg/mL, 약 57 mg/mL, 약 58 mg/mL, 약 59 mg/mL, 약 60.0 mg/mL, 약 61 mg/mL, 약 62 mg/mL, 약 63 mg/mL, 약 64 mg/mL, 약 64 mg/mL, 또는 약 65 mg/mL이다. 제제 중의 캅티솔®:파조파닙 1HCl의 비는 약 9:1, 약 8:1, 약 7:1, 약 6:1, 약 5:1, 약 4:1, 약 3:1, 또는 약 2:1이다. 예를 들어, CD:파조파닙 1HCl의 비는 약 2.2:1; 약 2.5:1; 약 3.7:1; 약 5:1; 약 8:1; 또는 약 9:1이다. 예를 들어, CD:파조파닙 1HCl의 비는 약 2.5:1 또는 2.2:1이다.The present disclosure discloses the use of a non-lyophilized wave pan nipHCl (sample-2) from a formulation of CaPHTHOSOL, a solubilizer, such as PVP, and a buffer, such as histidine HCl, Day to about 20 days or about 20 days to about 1 to about 3 micrograms per day. The half-life of drug release is about 99 days. In addition, the non-lyophilized rapamycin N HCI precipitates from solution during drug release. The non-lyophilized rapamycin IHCl (Sample-2) in the formulation is about 40 mg / mL, about 41 mg / mL, about 42 mg / mL, about 43 mg / mL, about 44 mg / about 47 mg / mL, about 48 mg / mL, about 49 mg / mL, about 50 mg / mL, about 51 mg / mL, about 52 mg / mL, about 53 mg / mL , About 53 mg / mL, about 54 mg / mL, about 55 mg / mL, about 56 mg / mL, about 57 mg / mL, about 58 mg / mL, about 59 mg / mL, about 60.0 mg / 61 mg / mL, about 62 mg / mL, about 63 mg / mL, about 64 mg / mL, about 64 mg / mL, or about 65 mg / mL. The ratio of Capsytol ®: Pazopanib HCl in the formulation is about 9: 1, about 8: 1, about 7: 1, about 6: 1, about 5: 1, about 4: 1, about 3: 2: 1. For example, the ratio of CD: ripple nip I HCl is about 2.2: 1; About 2.5: 1; About 3.7: 1; About 5: 1; About 8: 1; Or about 9: 1. For example, the ratio of CD: ripple nip N HCI is about 2.5: 1 or 2.2: 1.
본 개시내용은 약 60 mg/mL의 파조파닙 2HCl, 약 660 mg/mL의 캅티솔®, 약 1%의 PVP-10kD, 약 6 mg/mL의 히스티딘 HCl을 갖는 약 pH 6의 안정한 제제를 제공하여, 파조파닙 2HCl이 주위 조건에서의 저장 동안 최대 1년간 침전되지 않으며, 희석되었을 때 적어도 40-120일 이내에는 침전되지 않거나 또는 단지 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 약 30℃ - 약 50℃ (예를 들어, 약 37℃)에서 최대 약 350배까지 (예를 들어, 약 50-100배, 약 100-150배, 약 150-200배, 약 200-250배, 약 250-300배, 약 300-310배, 약 310-320배, 약 320-330배, 약 330-340배, 또는 약 340-350배) 희석되었을 때 적어도 40-120일간 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 본 개시내용의 약물 전달 장치로부터 신체 부위, 예를 들어 눈의 유리체로 방출되는 동안에 또한/또는 그러한 방출 후에 적어도 40-120일 이내에는 침전되지 않거나 또는 최소한으로 침전된다.This disclosure describes a stable formulation at about pH 6 with about 60 mg / mL PazopapinH 2 Cl, about 660 mg / mL Capsysol®, about 1% PVP-10 kD, about 6 mg / mL histidine HCl So that the ripope nip 2HCI is not precipitated for up to one year during storage at ambient conditions and is not precipitated or only minimally precipitated within at least 40-120 days when diluted. The wave pan nip 1 HCl in the formulations of the present invention can range from about 30 ° C to about 50 ° C (eg, about 37 ° C) up to about 350 times (eg, about 50-100 times, about 100-150 times, About 200-350 times, about 300-310 times, about 310-320 times, about 320-330 times, about 330-340 times, or about 340-350 times) Gt; at least < / RTI > 40-120 days. The pachipapenib 1HCl in the formulations of the present invention does not precipitate or at least dissolve within at least 40-120 days after release from the drug delivery device of the present disclosure to the body site, e.g., into the vitreous of the eye, and / Precipitated.
본 개시내용은 제제의 pH가 약 6.5로 조정된, 캅티솔®, 가용화제, 예를 들어 PVP, 및 완충제, 예를 들어 히스티딘 HCl의 제제로부터 파조파닙 2HCl (샘플-1)의 1일째의 약 20 ㎍/일 내지 약 140일경 또는 약 140일을 지났을 때의 약 2 - 약 4 ㎍/일의 방출 속도를 제공한다. 약물 방출의 반감기는 약 53일이다. 제제 중의 파조파닙 2HCl (샘플-1)은 약 40 mg/mL, 약 41 mg/mL, 약 42 mg/mL, 약 43 mg/mL, 약 44 mg/mL, 약 45 mg/mL, 약 46 mg/mL, 약 47 mg/mL, 약 48 mg/mL, 약 49 mg/mL, 약 50 mg/mL, 약 51 mg/mL, 약 52 mg/mL, 약 53 mg/mL, 약 53 mg/mL, 약 54 mg/mL, 약 55 mg/mL, 약 56 mg/mL, 약 57 mg/mL, 약 58 mg/mL, 약 59 mg/mL, 약 60.0 mg/mL, 약 61 mg/mL, 약 62 mg/mL, 약 63 mg/mL, 약 64 mg/mL, 약 64 mg/mL, 또는 약 65 mg/mL이다. 제제 중의 캅티솔®:파조파닙 2HCl의 비는 약 9:1, 약 8:1, 약 7:1, 약 6:1, 약 5:1, 약 4:1, 약 3:1, 또는 약 2:1이다. 예를 들어, CD:파조파닙 2HCl의 비는 약 2.2:1; 약 2.5:1; 약 3.7:1; 약 5:1; 약 8:1; 또는 약 9:1이다. 예를 들어, CD:파조파닙 2HCl의 비는 약 2.5:1 또는 2.2:1이다.The present disclosure relates to a method for the preparation of a pharmaceutical composition for the first day of Pachopapenib 2HCl (Sample-1) from a formulation of Capsicol®, a solubilizing agent such as PVP, and a buffer, such as histidine HCl, About 20 μg / day to about 140 days, or about 2 to about 4 μg / day over about 140 days. The half-life of drug release is about 53 days. About 45 mg / mL, about 46 mg / mL, about 42 mg / mL, about 43 mg / mL, about 44 mg / mL, about 45 mg / about 53 mg / mL, about 47 mg / mL, about 48 mg / mL, about 49 mg / mL, about 50 mg / mL, about 51 mg / mL, about 52 mg / mL, about 55 mg / mL, about 56 mg / mL, about 57 mg / mL, about 58 mg / mL, about 59 mg / mL, about 60.0 mg / mL, about 61 mg / About 62 mg / mL, about 63 mg / mL, about 64 mg / mL, about 64 mg / mL, or about 65 mg / mL. The ratio of Capsytol®: PazopapinH 2 Cl 2 in the formulation is about 9: 1, about 8: 1, about 7: 1, about 6: 1, about 5: 1, about 4: 1, about 3: 2: 1. For example, the ratio of CD: ripple nip 2HCI is about 2.2: 1; About 2.5: 1; About 3.7: 1; About 5: 1; About 8: 1; Or about 9: 1. For example, the ratio of CD: ripple nip 2HCI is about 2.5: 1 or 2.2: 1.
본 개시내용은 약 62 mg/mL의 파조파닙 1HCl, 약 660 mg/mL의 캅티솔®, 약 1%의 PVP-10kD, 약 6 mg/mL의 히스티딘 HCl을 갖는 약 pH 6의 안정한 제제를 제공하여, 파조파닙 1HCl이 주위 조건에서의 저장 동안 최대 9개월간 침전되지 않으며, 희석되었을 때 5-10일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 약 30℃ - 약 50℃ (예를 들어, 약 37℃)에서 최대 약 350배까지 (예를 들어, 약 50-100배, 약 100-150배, 약 150-200배, 약 200-250배, 약 250-300배, 약 300-310배, 약 310-320배, 약 320-330배, 약 330-340배, 또는 약 340-350배) 희석되었을 때 5-10일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 본 개시내용의 약물 전달 장치로부터 신체 부위, 예를 들어 눈의 유리체로 방출되는 동안에 또한/또는 그러한 방출 후에 5-10일 이내에는 침전되지 않거나 또는 최소한으로 침전된다.This disclosure describes a stable formulation at about pH 6 with about 62 mg / mL Pazopapenip HCl, about 660 mg / mL Capsysol®, about 1% PVP-10 kD, about 6 mg / mL histidine HCl , So that rapamycin N HCl is not precipitated for up to 9 months during storage at ambient conditions and is not precipitated or minimally precipitated within 5-10 days when diluted. The wave pan nip 1 HCl in the formulations of the present invention can range from about 30 ° C to about 50 ° C (eg, about 37 ° C) up to about 350 times (eg, about 50-100 times, about 100-150 times, About 200-350 times, about 300-310 times, about 310-320 times, about 320-330 times, about 330-340 times, or about 340-350 times) It is not precipitated or at least precipitated within 5-10 days. The rapamycin nip1HCl in the formulation of the present invention does not precipitate within the 5-10 days after release from the drug delivery device of the present disclosure, such as into the vitreous of the eye, e.g., the eye, and / do.
본 개시내용은 약 40 mg/mL의 파조파닙 1HCl, 약 660 mg/mL의 캅티솔®, 약 1%의 PVP-10kD, 약 6 mg/mL의 히스티딘 HCl을 갖는 약 pH 6의 안정한 제제를 제공하여, 파조파닙 1HCl이 주위 조건에서의 저장 동안 적어도 9개월까지는 침전되지 않으며, 희석되었을 때 적어도 13-20일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 약 30℃ - 약 50℃ (예를 들어, 약 37℃)에서 최대 약 350배까지 (예를 들어, 약 50-100배, 약 100-150배, 약 150-200배, 약 200-250배, 약 250-300배, 약 300-310배, 약 310-320배, 약 320-330배, 약 330-340배, 또는 약 340-350배) 희석되었을 때 적어도 13-20일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 본 개시내용의 약물 전달 장치로부터 신체 부위, 예를 들어 눈의 유리체로 방출되는 동안에 또한/또는 그러한 방출 후에 적어도 13-20일 이내에는 침전되지 않거나 또는 최소한으로 침전된다.This disclosure describes a stable formulation at about pH 6 with about 40 mg / mL Pazopapenip HCl, about 660 mg / mL Capsysol®, about 1% PVP-10 kD, about 6 mg / mL histidine HCl So that the ripple nip HCI is not precipitated for at least 9 months during storage at ambient conditions and is not precipitated or minimally precipitated within at least 13-20 days when diluted. The wave pan nip 1 HCl in the formulations of the present invention can range from about 30 ° C to about 50 ° C (eg, about 37 ° C) up to about 350 times (eg, about 50-100 times, about 100-150 times, About 200-350 times, about 300-310 times, about 310-320 times, about 320-330 times, about 330-340 times, or about 340-350 times) It is not precipitated or precipitated to a minimum within at least 13-20 days. The pachipapenib 1HCl in the formulations of the present invention does not precipitate or at least dissolve within at least 13-20 days after release from the drug delivery device of the present disclosure to the body site, e.g., into the vitreous of the eye, and / Precipitated.
본 개시내용은 제제의 pH가 약 6.5로 조정된, 캅티솔®, 가용화제, 예를 들어 PVP, 및 완충제, 예를 들어 히스티딘 HCl의 제제로부터 TFE 동결건조된 파조파닙 1HCl (샘플-2)의 1일째의 약 12 ㎍/일 내지 약 140일경 또는 약 140일을 지났을 때의 약 1 - 약 2 ㎍/일의 방출 속도를 제공한다. 약물 방출의 반감기는 약 45일이다. 제제 중의 파조파닙 2HCl (샘플-1)은 약 30 mg/mL, 약 31 mg/mL, 약 32 mg/mL, 약 33 mg/mL, 약 34 mg/mL, 약 35 mg/mL, 약 36 mg/mL, 약 37 mg/mL, 약 38 mg/mL, 약 39 mg/mL, 40 mg/mL, 약 41 mg/mL, 약 42 mg/mL, 약 43 mg/mL, 약 44 mg/mL, 약 45 mg/mL, 약 46 mg/mL, 약 47 mg/mL, 약 48 mg/mL, 약 49 mg/mL, 약 50 mg/mL, 약 51 mg/mL, 약 52 mg/mL, 약 53 mg/mL, 약 53 mg/mL, 약 54 mg/mL, 약 55 mg/mL, 약 56 mg/mL, 약 57 mg/mL, 약 58 mg/mL, 약 59 mg/mL, 약 60.0 mg/mL, 약 61 mg/mL, 약 62 mg/mL, 약 63 mg/mL, 약 64 mg/mL, 약 64 mg/mL, 또는 약 65 mg/mL이다. 제제 중의 캅티솔®:파조파닙 2HCl의 비는 약 9:1, 약 8:1, 약 7:1, 약 6:1, 약 5:1, 약 4:1, 약 3:1, 또는 약 2:1이다. 예를 들어, CD:파조파닙 2HCl의 비는 약 2.2:1; 약 2.5:1; 약 3.7:1; 약 5:1; 약 8:1; 또는 약 9:1이다. 예를 들어, CD:파조파닙 2HCl의 비는 약 4:1이다. 제제 중의 PVP는 약 1%이고, 제제 중의 히스티딘 HCl은 약 25 mg/mL이다.The present disclosure discloses the use of TFE lyophilized Pafopapin® HCl (Sample-2) from a formulation of Capsytol®, a solubilizing agent, such as PVP, and a buffer, eg, histidine HCl, Day to about 140 days or about 1 day to about 2 days per day for about 140 days. The half-life of drug release is about 45 days. ML), about 35 mg / mL, about 36 mg / mL, about 32 mg / mL, about 33 mg / mL, about 34 mg / about 41 mg / mL, about 42 mg / mL, about 43 mg / mL, about 38 mg / mL, about 39 mg / mL, about 40 mg / About 45 mg / mL, about 46 mg / mL, about 47 mg / mL, about 48 mg / mL, about 49 mg / mL, about 50 mg / mL, about 51 mg / mL, about 52 mg / About 53 mg / mL, about 54 mg / mL, about 55 mg / mL, about 56 mg / mL, about 57 mg / mL, about 58 mg / mL, about 59 mg / mL, about 60.0 mg / mL, about 61 mg / mL, about 62 mg / mL, about 63 mg / mL, about 64 mg / mL, about 64 mg / mL, or about 65 mg / mL. The ratio of Capsytol®: PazopapinH 2 Cl 2 in the formulation is about 9: 1, about 8: 1, about 7: 1, about 6: 1, about 5: 1, about 4: 1, about 3: 2: 1. For example, the ratio of CD: ripple nip 2HCI is about 2.2: 1; About 2.5: 1; About 3.7: 1; About 5: 1; About 8: 1; Or about 9: 1. For example, the ratio of CD: ripple nip 2HCI is about 4: 1. The PVP in the formulation is about 1% and the histidine HCl in the formulation is about 25 mg / mL.
본 개시내용은 가용화제, 예를 들어 약 1%의PVP, 및 완충제, 예를 들어 약 25 mg/ml의 히스티딘 HCl의 존재 하에, 캅티솔®이 약 4:1의 캅티솔®:파조파닙의 비로 존재하고, 제제의 pH가 약 6.5로 조정된 제제로부터 약 36.0 mg/mL의 TFE 동결건조된 파조파닙 1HCl (샘플-2)의 1일째의 약 12 ㎍/일 내지 약 140일경 또는 약 140일을 지났을 때의 약 1 - 약 2 ㎍/일의 제제로부터의 방출 속도를 제공한다. 약물 방출의 반감기는 약 45일이다.This disclosure is directed to solubilizing agents, for example, about 1% PVP, and a buffer, e.g., about 25 mg / ml of histidine HCl, is present in the ratio of CaPTISOL® to Pachopanip about 4: 1 and the pH of the formulation is adjusted to about 6.5 From about 12 μg / day to about 140 days or about 1 to about 2 μg / day of about 36.0 mg / ml of the lyophilized TFE lyophilized Pafopapin® HCl (Sample-2) Lt; / RTI > release rate of the formulation. The half-life of drug release is about 45 days.
본 개시내용은 약 41 mg/mL의 TFE 동결건조된 파조파닙 1HCl, 약 660 mg/mL의 캅티솔®, 약 1%의 PVP-10kD, 약 6 mg/mL의 히스티딘 HCl을 갖는 약 pH 7의 안정한 제제를 제공하여, 파조파닙 1HCl이 주위 조건에서의 저장 동안 최대 6개월 이내에는 침전되지 않으며, 희석되었을 때 적어도 41-120일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 약 30℃ - 약 50℃ (예를 들어, 약 37℃)에서 최대 약 350배까지 (예를 들어, 약 50-100배, 약 100-150배, 약 150-200배, 약 200-250배, 약 250-300배, 약 300-310배, 약 310-320배, 약 320-330배, 약 330-340배, 또는 약 340-350배) 희석되었을 때 적어도 41-120일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 본 개시내용의 약물 전달 장치로부터 신체 부위, 예를 들어 눈의 유리체로 방출되는 동안에 또한/또는 그러한 방출 후에 적어도 41-120일 이내에는 침전되지 않거나 또는 최소한으로 침전된다.The present disclosure provides a pharmaceutical composition comprising about 41 mg / mL of TFE lyophilized pefafenib HCI, about 660 mg / mL of capricol, about 1% PVP-10 kD, about pH 7 with about 6 mg / mL histidine HCl So that Pazopapanib 1HCl is not precipitated within a maximum of 6 months during storage at ambient conditions and is not precipitated or minimally precipitated within at least 41-120 days when diluted. The wave pan nip 1 HCl in the formulations of the present invention can range from about 30 ° C to about 50 ° C (eg, about 37 ° C) up to about 350 times (eg, about 50-100 times, about 100-150 times, About 200-350 times, about 300-310 times, about 310-320 times, about 320-330 times, about 330-340 times, or about 340-350 times) Lt; RTI ID = 0.0 > 41-120 < / RTI > days. The breakfar nip1HCl in the formulation of the present invention does not precipitate or at least does not precipitate within the period of at least 41-120 days after release from the drug delivery device of this disclosure to the body site, e.g., into the vitreous of the eye, and / Precipitated.
본 개시내용은 제제의 pH가 약 3.4로 조정된, 캅티솔®, 가용화제, 예를 들어 PVP, 및 완충제, 예를 들어 히스티딘 HCl의 제제로부터 DMSO 동결건조된 파조파닙 1HCl (샘플-2)의 1일째의 약 16 ㎍/일 내지 약 140일경 또는 약 140일을 지났을 때의 약 1 - 약 3 ㎍/일의 방출 속도를 제공한다. 약물 방출의 반감기는 약 45일이다. 제제 중의 파조파닙 2HCl (샘플-1)은 약 30 mg/mL, 약 31 mg/mL, 약 32 mg/mL, 약 33 mg/mL, 약 34 mg/mL, 약 35 mg/mL, 약 36 mg/mL, 약 37 mg/mL, 약 38 mg/mL, 약 39 mg/mL, 40 mg/mL, 약 41 mg/mL, 약 42 mg/mL, 약 43 mg/mL, 약 44 mg/mL, 약 45 mg/mL, 약 46 mg/mL, 약 47 mg/mL, 약 48 mg/mL, 약 49 mg/mL, 약 50 mg/mL, 약 51 mg/mL, 약 52 mg/mL, 약 53 mg/mL, 약 53 mg/mL, 약 54 mg/mL, 약 55 mg/mL, 약 56 mg/mL, 약 57 mg/mL, 약 58 mg/mL, 약 59 mg/mL, 약 60.0 mg/mL, 약 61 mg/mL, 약 62 mg/mL, 약 63 mg/mL, 약 64 mg/mL, 약 64 mg/mL, 또는 약 65 mg/mL이다. 제제 중의 캅티솔®:파조파닙 2HCl의 비는 약 9:1, 약 8:1, 약 7:1, 약 6:1, 약 5:1, 약 4:1, 약 3:1, 또는 약 2:1이다. 예를 들어, CD:파조파닙 2HCl의 비는 약 2.2:1; 약 2.5:1; 약 3.7:1; 약 5:1; 약 8:1; 또는 약 9:1이다. 예를 들어, CD:파조파닙 2HCl의 비는 약 3:1이다. 제제 중의 PVP는 약 1%이고, 제제 중의 히스티딘 HCl은 약 6 mg/mL이다.The present disclosure discloses a DMSO lyophilized wave pan nip 1 HCl (sample-2) from a formulation of Capsytol®, a solubilizing agent such as PVP, and a buffer, such as histidine HCl, Day to about 140 days, or about 1 to about 3 micrograms per day over about 140 days. The half-life of drug release is about 45 days. ML), about 35 mg / mL, about 36 mg / mL, about 32 mg / mL, about 33 mg / mL, about 34 mg / about 41 mg / mL, about 42 mg / mL, about 43 mg / mL, about 38 mg / mL, about 39 mg / mL, about 40 mg / About 45 mg / mL, about 46 mg / mL, about 47 mg / mL, about 48 mg / mL, about 49 mg / mL, about 50 mg / mL, about 51 mg / mL, about 52 mg / About 53 mg / mL, about 54 mg / mL, about 55 mg / mL, about 56 mg / mL, about 57 mg / mL, about 58 mg / mL, about 59 mg / mL, about 60.0 mg / mL, about 61 mg / mL, about 62 mg / mL, about 63 mg / mL, about 64 mg / mL, about 64 mg / mL, or about 65 mg / mL. The ratio of Capsytol®: PazopapinH 2 Cl 2 in the formulation is about 9: 1, about 8: 1, about 7: 1, about 6: 1, about 5: 1, about 4: 1, about 3: 2: 1. For example, the ratio of CD: ripple nip 2HCI is about 2.2: 1; About 2.5: 1; About 3.7: 1; About 5: 1; About 8: 1; Or about 9: 1. For example, the ratio of CD: ripple nip 2HCI is about 3: 1. The PVP in the formulation is about 1% and the histidine HCl in the formulation is about 6 mg / mL.
본 개시내용은 약 60 mg/mL의 DMSO 동결건조된 파조파닙 1HCl, 약 660 mg/mL의 캅티솔®, 약 1%의 PVP-10kD, 임의로 약 6 mg/mL의 히스티딘 HCl을 갖는 약 pH 6의 안정한 제제를 제공하여, 파조파닙 1HCl이 주위 조건에서의 저장 동안 최대 40일 이내에는 침전되지 않으며, 희석되었을 때 적어도 10-14일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 약 30℃ - 약 50℃ (예를 들어, 약 37℃)에서 최대 약 350배까지 (예를 들어, 약 50-100배, 약 100-150배, 약 150-200배, 약 200-250배, 약 250-300배, 약 300-310배, 약 310-320배, 약 320-330배, 약 330-340배, 또는 약 340-350배) 희석되었을 때 적어도 10-14일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 본 개시내용의 약물 전달 장치로부터 신체 부위, 예를 들어 눈의 유리체로 방출되는 동안에 또한/또는 그러한 방출 후에 적어도 10-14일 이내에는 침전되지 않거나 또는 최소한으로 침전된다.The present disclosure relates to a pharmaceutical composition having a pH of about pH 6.0 with about 60 mg / mL of DMSO lyophilized pachopyrimid HC1, about 660 mg / mL capricol, about 1% PVP-10 kD, optionally about 6 mg / mL histidine HCl 6, so that Pazopapanib 1HCl is not precipitated within a maximum of 40 days during storage at ambient conditions, and is not precipitated or minimally precipitated within at least 10-14 days when diluted. The wave pan nip 1 HCl in the formulations of the present invention can range from about 30 ° C to about 50 ° C (eg, about 37 ° C) up to about 350 times (eg, about 50-100 times, about 100-150 times, About 200-350 times, about 300-310 times, about 310-320 times, about 320-330 times, about 330-340 times, or about 340-350 times) Or at least within 10-14 days. The pachipapenib 1HCl in the formulation of the present invention is not precipitated or released to the body part, such as the eye, during the release to the vitreous of the body and / or within at least 10-14 days after such release Precipitated.
본 개시내용은 약 45 mg/mL의 DMSO 동결건조된 파조파닙 1HCl, 약 660 mg/mL의 캅티솔®, 약 1%의 PVP-10kD, 약 6 mg/mL의 히스티딘 HCl을 갖는 약 pH 3.5의 안정한 제제를 제공하여, 파조파닙 1HCl이 주위 온도에서의 저장 동안에 침전되지 않으며, 희석되었을 때 적어도 50일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 약 30℃ - 약 50℃ (예를 들어, 약 37℃)에서 최대 약 350배까지 (예를 들어, 약 50-100배, 약 100-150배, 약 150-200배, 약 200-250배, 약 250-300배, 약 300-310배, 약 310-320배, 약 320-330배, 약 330-340배, 또는 약 340-350배) 희석되었을 때 적어도 50일 이내에는 침전되지 않거나 또는 최소한으로 침전된다. 본 발명의 제제 중의 파조파닙 1HCl은 본 개시내용의 약물 전달 장치로부터 신체 부위, 예를 들어 눈의 유리체로 방출되는 동안에 또한/또는 그러한 방출 후에 적어도 50일 이내에는 침전되지 않거나 또는 최소한으로 침전된다.This disclosure describes a pharmaceutical composition comprising about 45 mg / mL of DMSO lyophilized Pachopanip HCl, about 660 mg / mL capricol, about 1% PVP-10 kD, about pH 3.5 with about 6 mg / mL histidine HCl So that the rapamycin N HCl is not precipitated during storage at ambient temperature and is not precipitated or minimally precipitated within at least 50 days when diluted. The wave pan nip 1 HCl in the formulations of the present invention can range from about 30 ° C to about 50 ° C (eg, about 37 ° C) up to about 350 times (eg, about 50-100 times, about 100-150 times, About 200-350 times, about 300-310 times, about 310-320 times, about 320-330 times, about 330-340 times, or about 340-350 times) Or at least within 50 days. The pachipapenib 1HCl in the formulations of the present invention is not precipitated or minimally precipitated during, and / or within, at least 50 days after release from the drug delivery device of the present disclosure to the body site, e.g., the vitreous of the eye .
본 개시내용은 가용화제, 예를 들어 약 1%의PVP, 및 완충제, 예를 들어 약 6 mg/ml의 히스티딘 HCl의 존재 하에, 캅티솔®이 약 3:1의 캅티솔®:파조파닙의 비로 존재하고, 제제의 pH가 약 3.4로 조정된 제제로부터의 약 50.0 mg/mL의 DMSO 동결건조된 파조파닙 1HCl (샘플-2)의 방출 속도가 1일째의 약 16 ㎍/일 내지 약 140일경 또는 약 140일을 지났을 때의 약 1 - 약 3 ㎍/일인 것을 제공한다. 약물 방출의 반감기는 약 45일이다.This disclosure is directed to solubilizing agents, for example, about 1% PVP, and a buffering agent, such as about 6 mg / ml of histidine HCl, is present in the ratio of Capsytol® to Pazopanib of about 3: 1 and the pH of the formulation is adjusted to about 3.4 The release rate of about 50.0 mg / mL of DMSO lyophilized rapamycin HCI (Sample-2) from the formulation is about 1 to about 140 days or about 1 day to about 140 days 3 [mu] g / day. The half-life of drug release is about 45 days.
본 개시내용은 전달 장치로부터의 파조파닙 제제의 방출 속도를 제공하고, 여기서 제제는 완충제 없이 제조되고, 파조파닙 1HCl (약 30 mg/ml 내지 약 40 mg/mL), 약 660 - 약 700 mg/mL (예를 들어, 약 660 mg/mL)의 캅티솔®, 중합체 (예를 들어, PVP)를 포함하며, 천연 pH를 갖는다. 충전 농도는 약 30 mg/mL - 약 35 mg/mL (대형 동결건조 제제) 또는 약 35 mg/mL - 약 45 mg/mL (소형 동결건조 제제)이다. 표 6은 본 개시내용의 제제의 방출 속도의 비제한적 예를 제공한다.The present disclosure provides a release rate of the ripodip nip formulation from a delivery device wherein the formulation is prepared without buffer and comprises a ripapan iHCl (about 30 mg / ml to about 40 mg / ml), about 660 to about 700 (e.g., about 660 mg / mL) of Capsytol®, a polymer (e.g., PVP), and has a natural pH. The charge concentration is about 30 mg / mL to about 35 mg / mL (large lyophilized formulation) or about 35 mg / mL to about 45 mg / mL (small lyophilized formulation). Table 6 provides a non-limiting example of the release rate of the formulation of the present disclosure.
<표 6><Table 6>

키트Kit
본 개시내용은 본 개시내용의 안정한 제제 중 어느 하나가 치료 장치의 저장소 챔버에 함유되어 있으며, 여기서 저장소 챔버는 눈의 유리체에서의 치료제의 제어 방출을 위해 다공성 구조에 커플링된 것인 키트를 제공한다.The present disclosure provides a kit in which any one of the stable formulations of the present disclosure is contained in a reservoir chamber of a treatment device wherein the reservoir chamber is coupled to a porous structure for controlled release of a therapeutic agent in the vitreous of the eye do.
치료 장치Treatment device
치료 장치는 다수의 구성 및 물리적 속성을 포함하는데, 예를 들어 치료 장치의 물리적 특징은 시력이 손상되지 않도록 하는 봉합, 위치결정 및 사이징(sizing)을 갖는 치료제 전달 장치 (포트 전달 시스템 (PDS)), 및 생체적합성 물질 중 적어도 하나를 포함한다. 예를 들어, 장치는 약 0.005 cc 내지 약 0.2 cc, 예를 들어 약 0.01 cc 내지 약 0.1 cc의 저장소 용량, 및 약 2 cc 이하의 장치 부피를 포함한다. 0.1 cc를 초과하는 장치 부피에 대해서는 유리체 절제술이 수행된다. 치료 장치의 길이는 환자의 시력을 저해하지 않으며, 장치의 형상 뿐만 아니라, 이식 장치의 눈에 대한 위치에 따라 좌우된다. 장치의 길이는 또한 장치의 삽입 각도에 따라 좌우된다. 예를 들어, 장치의 길이는 약 4 내지 6 mm를 포함한다. 눈의 직경이 약 24 mm이므로, 공막으로부터 유리체로 약 6 mm 이하로 연장되는 장치는 환자의 시력에 대하여 최소한의 영향을 미친다.Therapeutic devices include a number of configurations and physical attributes, for example, the physical characteristics of the treatment device include a therapeutic delivery device (Port Delivery System (PDS)) with suture, positioning and sizing to prevent visual impairment, , And a biocompatible material. For example, the device comprises a storage volume of from about 0.005 cc to about 0.2 cc, such as from about 0.01 cc to about 0.1 cc, and a device volume of about 2 cc or less. For device volumes in excess of 0.1 cc, vitrectomy is performed. The length of the treatment device does not interfere with the vision of the patient and depends on the shape of the device as well as the position of the implant on the eye. The length of the device also depends on the insertion angle of the device. For example, the length of the device includes about 4 to 6 mm. Since the diameter of the eye is about 24 mm, devices that extend from the sclera to less than about 6 mm of vitreous will have minimal effect on the patient's visual acuity.
변형예는 이식 치료제 전달 장치 (포트 전달 시스템 (PDS))의 다수의 조합을 포함한다. 치료 장치는 치료제 및 결합제를 포함한다. 장치는 또한 막, 개구부, 확산 장벽, 치료량의 치료제를 연장된 시간 동안 방출시키기 위한 확산 메카니즘 중 적어도 하나를 포함한다. 장치의 여러 변형예가 WO 2012/065006, WO2012/019047, WO2013/003620, WO 2012/019136, WO 2012/019176, 및 미국 특허 8,277,830에서 개시되었으며, 이들은 각각 그 전문이 본원에 참조로 포함된다.Variations include multiple combinations of implant treatment delivery devices (Port Delivery Systems (PDS)). Therapeutic devices include therapeutic agents and binding agents. The apparatus also includes at least one of a membrane, an opening, a diffusion barrier, and a diffusion mechanism for releasing a therapeutic amount of the therapeutic agent for an extended period of time. Various modifications of the apparatus have been disclosed in WO 2012/065006, WO2012 / 019047, WO2013 / 003620, WO 2012/019136, WO 2012/019176, and U.S. Patent 8,277,830, each of which is incorporated herein by reference in its entirety.
도 2a는 눈의 망막을 치료하기 위해 눈의 유리액으로 치료제를 방출하는, 결막(16) 아래에 이식되어 공막(24)을 통해 연장되는 치료 장치(100)를 도시한다. 치료 장치(100)는 결막이 치료 장치를 덮어서 치료 장치(100)를 보호하도록, 결막 아래에서 공막을 따라 위치하도록 구성된 활면 돌출부와 같은 보유 구조(120)를 포함한다. 치료제(110)가 장치(100) 내로 삽입될 때는, 결막의 리프팅, 절개, 또는 바늘 천자에 의해 치료 장치에 접근한다. 눈은 눈의 공막을 상직근에 커플링하는 상직근의 힘줄 착생부(27)를 포함한다. 실시양태에서, 장치(100)는, 예를 들어 힘줄에서 떨어진 편평부 영역의 여러 위치 및 힘줄 아래의 하나 이상의 힘줄 후방부에 위치하거나, 또는 치료 장치의 비측 또는 이측 위치도 가능하다.Figure 2a shows a
이식물이 다수의 방식으로 눈에 위치할 수 있지만, 변형예와 관련된 작업은 편평부 영역에서의 위치가 치료제를 유리체로 방출하여 망막을 치료함을 시사하고, 예를 들어 치료제는 거대 분자로 구성된 활성 성분을 포함한다.Although the implant may be located in the eye in a number of ways, the work associated with the variant suggests that the location in the flattened area releases the therapeutic to the vitreous to treat the retina, for example, Active ingredients.
장치(100)와 함께 사용하기에 적합한 치료제(110)는 다수의 치료제, 예를 들어 표 1에 나열된 것을 포함한다. 장치(100)의 치료제(110)는 치료제의 활성 성분, 치료제의 제제, 치료제 제제의 성분, 의사가 조제한 치료제 제제, 또는 약사가 조제한 치료제 제제 중 1종 이상을 포함한다.
치료 장치(100)는 환자에게 도움이 되고 유익한 동안에는 눈에 이식되어 눈을 치료할 수 있다. 예를 들어, 장치는 적어도 약 5년 동안, 예컨대 환자의 평생 동안 영구적으로 이식된다. 선택적으로 또는 조합하여, 장치는 환자의 치료에 더이상 도움이 되지 않거나 또는 유익하지 않을 때 제거된다.
도 2b는 도 2a에서와 같이 눈에 위치하도록 구성된 치료 장치(100)의 구조를 도시한다. 장치는 장치(100)를 공막에 커플링하기 위한 보유 구조(120), 예를 들어 장치의 근위 단부에 배치된 돌출부를 포함한다. 장치(100)는 보유 구조(120)에 부착된 용기(130)를 포함한다. 활성 성분, 예를 들어 치료제(110)는 저장소(140), 예를 들어 장치의 용기(130)에 의해 한정된 챔버(132) 내에 함유된다. 용기(130)는 다공성 물질(152), 예를 들어 다공성 유리 프리트(frit)(154)를 포함하는 다공성 구조(150), 및 치료제의 방출을 억제하는 장벽(160), 예를 들어 불투과성 막(162)을 포함한다. 불투과성 막(162)은 실질적으로 불투과성인 물질(164)을 포함한다. 불투과성 막(162)은 치료량의 치료제(110)를 연장된 시간 동안 방출하도록 사이징된 개구부(166)를 포함한다. 다공성 구조(150)는 치료량의 치료제를 연장된 시간 동안 방출하도록 개구부(166)와 함께 구성된 두께(150T) 및 세공 크기를 포함한다. 용기(130)는 연장된 시간 동안의 방출을 위해 치료량의 치료제(110)를 함유하도록 사이징된 부피(142)의 챔버를 갖는 저장소(140)를 포함한다. 장치는 바늘 정지부(170)를 포함한다. 유리액 중의 단백질이 장치에 들어가서 다공성 구조 상의 흡착 부위에 대하여 경쟁함으로써, 치료제의 방출에 기여한다. 저장소(140)에 함유된 치료제(110)는 유리액 중의 단백질과 평형을 달성하므로, 시스템이 평형 쪽으로 유도되고 치료제(110)가 치료량으로 방출된다.FIG. 2B shows the structure of a
불투과성 물질, 예컨대 불투과성 막(162), 다공성 물질(152), 저장소(140), 및 보유 구조(120)는 치료제(110)를 전달하는 다수의 구성을 포함한다. 불투과성 막(162)은 치료제를 방출하기 위해 디스크 상에 형성된 적어도 하나의 개구부를 갖는 디스크에 의해 연결된 환상 튜브를 포함한다. 다공성 물질(152)은 환상 다공성 유리 프리트(154) 및 그 위에 배치된 원형 단부를 포함한다. 저장소(140)는 삽입의 용이함을 위해 형상-변화형인데; 즉, 공막을 통한 삽입 동안에는 얇게 신장된 형상을 취하고, 그 후에 치료제가 충전되면, 연장된 부푼 형상을 취한다.Impermeable materials such as
다공성 구조(150)는 의도된 방출 프로파일에 따라 치료제를 방출하도록 다수의 방식으로 구성될 수 있다. 다공성 구조는 장벽 물질, 예컨대 경질 플라스틱 또는 금속을 통해 연장되는 단일 구멍 또는 복수 개의 구멍을 포함한다. 선택적으로 또는 조합하여, 다공성 구조는 저장소와 대향하는 제1 측면 상의 복수 개의 개구부 및 유리액과 대향하는 제2 측면 상의 복수 개의 개구부를 가지고, 복수 개의 상호연결 채널이 그 사이에 배치되어 제1 측면의 개구부를 제2 측면의 개구부와 커플링하는 다공성 구조, 예를 들어 소결 경질 물질을 포함한다. 다공성 구조(150)는 투과성 막, 반투과성 막, 그 안에 배치된 적어도 하나의 구멍을 갖는 물질, 나노-채널, 경질 물질에 에칭된 나노-채널, 레이저 에칭된 나노-채널, 모세관 채널, 복수 개의 모세관 채널, 하나 이상의 굴곡이 있는 채널, 굴곡이 있는 마이크로채널, 소결 나노-입자, 개방 셀 발포체 또는 히드로겔, 예컨대 개방 셀 히드로겔 중 하나 이상을 포함한다.The
도 2c는 삽입 기구(200)의 삽입 캐뉼라(210)에 로딩된 치료 장치(100)를 도시하고, 여기서 장치(100)는 공막으로의 삽입을 위해 신장된 협폭 형상을 포함하고, 또한 장치는 적어도 부분적으로 공막에서의 보유를 위해 제2의 신장된 광폭 형상으로 팽창되도록 구성된다.2C illustrates a
도 2d는 캐뉼라에 로딩하기에 적합한 저장소(140)를 포함하는 치료 장치(100)를 도시하고, 여기서 저장소(140)는 눈에 위치할 때 팽창된 구성을 포함한다.2D illustrates a
도 2e는 도 2a에서와 같이 눈에 위치하는 치료 장치(100)를 도시한다. 장치는 공막에 커플링하기 위한 보유 구조(120), 예를 들어 공막과의 민면(flush)을 포함하고, 장벽(160)은 튜브(168)를 포함한다. 치료제(110)를 포함하는 활성 성분(112)은 불투과성 물질(164)을 포함하는 튜브(168) 내에 함유된다. 다공성 물질(152)을 포함하는 다공성 구조(150)가 튜브(168)의 원위 단부에 배치되어, 연장된 기간 동안 치료 농도의 치료제의 서방성 방출을 제공한다. 불투과성 물질(164)은 다공성 물질(152) 둘레에서 말단으로 연장되어, 장치가 눈에 삽입될 때 다공성 물질(152)을 유리액에 커플링하기 위한 개구부를 한정한다.FIG. 2E shows an
도 2f는 치료 장치(100)와의 통합을 위해 적합한 접속 포트(180)를 도시한다. 접속 포트(180)는 본원에 기재된 치료 장치와 조합된다. 접속 포트는 장치의 근위 단부에 배치된다. 접속 포트(180)는 그 위에 배치된 격막(186)을 포함하는 관통성 장벽(184)을 갖는 보유 구조(120)에 형성된 개구부를 포함한다. 관통성 장벽은 본원에 기재된 제제(190)가 통과하도록 사이징된 바늘(189)을 수용한다. 접속 포트(180)는 환자의 결막(16) 아래에서 공막(24) 위에 위치하도록 구성된다.FIG. 2F illustrates an
치료제의 장치로부터의 전달Delivery of the therapeutic agent from the device
본 개시내용의 약물 전달 제제는 눈의 유리체에서의 치료제의 제어 방출을 위해 치료제 전달 시스템 내의 다공성 구조에 커플링된 저장소 챔버에 함유되고; 여기서 제제의 다공성 구조로부터의 제어 방출은 저장소 챔버에서의 치료제 농도보다 적어도 두 자릿수가 더 낮은 유리체에서의 치료제 농도를 발생시킨다. 저장소 챔버는 재충전가능하고 장치가 눈에 삽입된 후에 제제가 재충전된다.The drug delivery formulations of the present disclosure are contained in a storage chamber coupled to a porous structure in a therapeutic delivery system for controlled release of a therapeutic agent in the vitreous of the eye; Wherein the controlled release from the porous structure of the formulation results in a therapeutic agent concentration in the vitreous which is at least two orders of magnitude lower than the therapeutic agent concentration in the reservoir chamber. The reservoir chamber is rechargeable and the formulation is recharged after the device is inserted into the eye.
저장소 챔버는 장치가 눈에 30 - 90일 동안, 또는 최대 6개월 동안 위치한 후에 제제가 재충전된다. 본 개시내용의 제제 중 어느 하나를 전달하는데 사용되는 전달 장치는 WO 2010/088548에 개시되어 있으며, 단지 전달 장치에 관한 '548 공보의 개시내용은 본원에 참조로 포함된다.The reservoir chamber is recharged after the device has been placed in the eye for 30-90 days, or for up to 6 months. A delivery device used for delivering any of the presently disclosed formulations is disclosed in WO 2010/088548, the disclosure of which is incorporated herein by reference in its entirety, the disclosure of which is incorporated herein by reference.
본 발명의 실시양태의 PDS 이식물로부터의 서방성 방출을 위한 치료제 전달 제제의 디자인은 여러 고려사항에 기반한다. 예를 들어, PDS로부터의 치료제 용출은 불규칙적인 형상의 채널로 이루어진, 방출 제어 요소 (RCE)를 통한 분자 확산에 기반한다. 불규칙적인 형상의 채널은 WO 2012/065006에 기재되어 있고, RCE에 관한 그의 내용은 그 전문이 본원에 포함된다.The design of therapeutic delivery formulations for sustained release from PDS implants of embodiments of the present invention is based on several considerations. For example, therapeutic agent elution from PDS is based on molecular diffusion through the emission control element (RCE), which is composed of irregularly shaped channels. Channels of irregular shape are described in WO < RTI ID = 0.0 > 2012/06006, < / RTI >
게다가, 확산은 양방향으로, 즉 충전된 PDS로부터 유리체로, 또한 유리체로부터 PDS로의 치료제 확산으로부터 발생한다. 이러한 가역적인 확산은 시간이 지날수록 제제 함량이 유리체와 평형을 달성하도록 한다. PDS와 유리체 사이의 양방향 확산으로 인해, 디자인된 제제는 유리체 성분 및 유리체 pH와 상용성이어야 한다. 제제는 또한 PDS 저장소로부터의 방출 시에 유리체로의 고 희석과 상용성이어야 한다.In addition, diffusion occurs in both directions, i.e. from the filled PDS to the vitreous body and also from the diffusion of the therapeutic agent from the vitreous to the PDS. This reversible diffusion allows the formulation to achieve equilibrium with the vitreous over time. Due to the bidirectional diffusion between the PDS and the vitreous, the designed formulation should be compatible with the vitreous component and the vitreous pH. The formulation should also be compatible with high dilution to the vitreous at the time of release from the PDS reservoir.
본 개시내용의 제제는 유리체 성분 및 유리체 pH와 상용성이다. 본 발명의 실시양태에 기재된 제제는 PDS 저장소로부터의 방출 시에 유리체로의 고 희석과 상용성이다.The formulations of this disclosure are compatible with the vitreous component and the vitreous pH. The formulations described in the embodiments of the present invention are compatible with high dilution to the vitreous in release from the PDS reservoir.
본 개시내용은 목적하는 서방성 방출 프로파일 및 목적하는 조직 수준을 달성하기 위해 PDS 이식물 저장소로부터의 치료제 전달 속도를 조절하는 것을 제공한다. 본 개시내용에 따라서, 조절은 치료제 방출을 제어하기 위해 다공성 구조를 포함하는, PDS 이식물의 디자인에 의해 달성된다. 다공성 구조는 다공성 및 기하학적 차원을 추가로 갖는 만곡성을 가지며, 티타늄, 중합체, 및/또는 코팅물과 같은 물질이고, 표면의 기능을 갖는다. 전달 속도의 조절은 또한 저장소 부피를 달리함으로써 달성된다.The present disclosure provides for controlling the rate of therapeutic delivery from the PDS implant repository to achieve the desired sustained release profile and the desired tissue level. According to the present disclosure, modulation is achieved by the design of a PDS implant, including a porous structure to control release of the therapeutic agent. The porous structure has curvature with additional porosity and geometric dimensions and is a material such as titanium, polymer, and / or coating and has the function of a surface. Adjustment of the delivery speed is also achieved by varying the storage volume.
치료제 전달 속도의 조절은 제제 조성, 제제화 작용제, pH, 착물화제의 특성, 착물화제의 농도, 제제 점도, 및/또는 저장소에서의 치료제 농도에 따라 좌우된다.Control of therapeutic delivery rates will depend on the formulation, the formulation agent, the pH, the nature of the complexing agent, the concentration of the complexing agent, the viscosity of the formulation, and / or the concentration of the therapeutic agent in the reservoir.
본 개시내용의 제제는 확실하고 고도로 예측가능한 치료제 전달 특징 및 프로파일을 발생시키도록 디자인된다. 일부 실시양태에서, 선택된 착물화제의 사용은 상기 선택된 착물화제로 제제화된 다양한 화합물에 대하여 매우 유사한 치료제 전달 특징 (예컨대, PDS 저장소로부터의 치료제 전달의 반감기)을 달성한다. 본 개시내용은 제제 중의 일정 범위의 착물화제 농도 내에서 상이한 치료제의 반감기가 유사한 것을 제공한다. 이러한 제제에 대하여 PDS 이식물을 통한 치료제 전달 성능 및 확산은 비-착화 단일 분자 개체의 것과 유사하다.The preparations of this disclosure are designed to generate reliable and highly predictable therapeutic delivery characteristics and profiles. In some embodiments, the use of the selected complexing agent achieves very similar therapeutic delivery characteristics (e.g., half-life of therapeutic delivery from the PDS reservoir) for the various compounds formulated with the selected complexing agent. The present disclosure provides that the half life of different therapeutic agents is similar within a range of complexing agent concentrations in the formulation. For this formulation, the therapeutic delivery performance and diffusion through the PDS implant is similar to that of non-complex single molecule entities.
본 개시내용의 전달 장치는 저장소 및 다공성 구조를 포함한다. 예를 들어, 장치는 WO 2012/019176에 기재된 것이며, 저장소에 관한 그의 내용은 그 전문이 본원에 포함된다. 본 발명의 실시양태의 것과 유사한 다공성 구조가 WO 2012/065006에 기재되어 있으며, 다공성 구조에 관한 그의 내용은 그 전문이 본원에 포함된다.The delivery device of the present disclosure includes a reservoir and a porous structure. For example, the device is described in WO 2012/019176, the contents of which are incorporated herein by reference. Porous structures similar to those of embodiments of the present invention are described in WO < RTI ID = 0.0 > 2012/065006 < / RTI >
일부 실시양태에서, 다공성 구조는 저장소에 커플링된 제1 측면 및 유리체에 커플링된 제2 측면을 포함한다. 제1 측면은 제1 면적을 포함하고 제2 측면은 제2 면적을 포함할 수 있다.In some embodiments, the porous structure comprises a first side coupled to the reservoir and a second side coupled to the vitreous. The first aspect may comprise a first area and the second aspect may comprise a second area.
저장소의 부피는 약 5 μL 내지 약 50 μL의 치료제, 또는 예를 들어 약 10 μL 내지 약 25 μL, 예를 들어 23 μL의 치료제를 포함한다.The volume of the reservoir may comprise from about 5 [mu] L to about 50 [mu] L of the therapeutic agent or, for example, from about 10 [mu] L to about 25 [mu] L, for example, 23 [
용기의 저장소에 저장된 치료제는 치료제를 포함하는 고체, 치료제를 포함하는 용액, 치료제를 포함하는 현탁물, 그에 흡착된 치료제를 포함하는 입자, 또는 치료제와 가역적으로 결합된 입자 중 적어도 1종을 포함한다. 저장소는 완충제 및 약 1 mg/ml 내지 약 100 mg/mL, 예컨대 약 1 mg/ml 내지 약 40 mg/mL 범위 내의 용해도를 포함하는 치료제의 현탁물을 포함한다.The therapeutic agent stored in the reservoir of the container includes at least one of a solid including the therapeutic agent, a solution containing the therapeutic agent, a suspension containing the therapeutic agent, particles comprising the therapeutic agent adsorbed thereon, or particles reversibly bound to the therapeutic agent . The reservoir contains a buffer and a suspension of the therapeutic agent comprising a solubility in the range of about 1 mg / ml to about 100 mg / ml, such as about 1 mg / ml to about 40 mg / ml.
실시양태에서, 제제 중의 치료제의 농도는 착물화제, pH 조정 작용제, 가용화제/안정화제, 양쪽 친매성 작용제, 완충제, 비수성 용매, 또는 이들의 임의의 조합 중 임의의 1종 이상을 사용하여 물 또는 수용액 중에서의 작용제의 용해도를 증가시키는 것에 따라 좌우된다. 이러한 실시양태의 치료제는 본질적으로 물 또는 수용액 중에서 조금 녹는 것 (용질 1부를 위해 필요한 용매 30 내지 100부), 녹기 어려운 것 (용질 1부를 위해 필요한 용매 100 내지 1000부), 매우 녹기 어려운 것 (용질 1부를 위해 필요한 용매 1000 내지 10,000부), 또는 거의 녹지 않는 것 또는 불용성인 것 (용질 1부를 위해 필요한 용매 ≥10,000부)이다.In an embodiment, the concentration of the therapeutic agent in the formulation is at least about 10% by weight, more preferably at least about 10%, more preferably at least about 10%, and most preferably at least about 10% Or increasing the solubility of the agent in an aqueous solution. The therapeutic agents of this embodiment are essentially those which dissolve slightly in water or aqueous solution (30-100 parts solvent required for one solute), those that are difficult to dissolve (100-1000 parts solvent required for one solute) 1000 to 10,000 parts of solvent required for one part), or substantially insoluble or insoluble (solvent required for one part of solubility ≥ 10,000 parts).
방출 속도 지수는 다양한 값을 포함하며, 예를 들어 다수의 실시양태에서 현탁물의 방출 속도 지수가 용액에 대한 것보다 어느 정도 높다.The release rate index includes various values, such as, in many embodiments, the release rate index of the suspension is somewhat higher than for the solution.
다공성 구조는 바늘의 관통을 제한하는 바늘 정지부를 포함한다. 다공성 구조는 치료제의 연장된 방출을 위해 구성된 복수 개의 채널을 포함한다. 다공성 구조는 물질의 서방성 방출을 위해 적합한 특징을 갖는 경질 소결 물질을 포함한다.The porous structure includes a needle stop which limits the penetration of the needle. The porous structure comprises a plurality of channels configured for prolonged release of the therapeutic agent. The porous structure includes a hard sintered material having suitable characteristics for sustained release of the material.
저장소 및 다공성 구조는 치료량의 치료제를 다수의 방식으로 방출하도록 구성된다. 저장소 및 다공성 구조는 적어도 약 3개월의 연장된 기간 동안 유리액 1 ml 당 적어도 약 0.1 ㎍ 또는 0.1-25 ㎍/일의 농도에 상응하는 치료량의 치료제를 방출하도록 구성된다. 저장소 및 다공성 구조는 적어도 약 3개월의 연장된 기간 동안 유리액 1 ml 당 적어도 약 0.1 ㎍ 내지 유리액 1 ml 당 약 10 ㎍ 이하의 농도에 상응하는 치료량의 치료제를 방출하도록 구성된다. 일부 실시양태에서, 치료제는 서방성 방출에 적합한 소분자 치료제이다.The reservoir and the porous structure are configured to release a therapeutic amount of the therapeutic agent in a number of ways. The reservoir and the porous structure are configured to release a therapeutic amount of therapeutic agent corresponding to a concentration of at least about 0.1 [mu] g or 0.1-25 [mu] g / day per ml of the vitreous fluid for an extended period of at least about 3 months. The reservoir and the porous structure are configured to release a therapeutic amount of therapeutic agent corresponding to a concentration of at least about 0.1 占 퐂 per ml of the vitreous liquid to about 10 占 퐂 per ml of the vitreous fluid for an extended period of at least about 3 months. In some embodiments, the therapeutic agent is a small molecule therapeutic agent suitable for sustained release.
저장소 및 다공성 구조는 적어도 약 3개월 또는 적어도 약 6개월의 연장된 기간 동안 유리액 1 ml 당 적어도 약 0.1 ㎍ 내지 유리액 1 ml 당 약 10 ㎍ 이하의 농도에 상응하는 치료량의 치료제를 방출하도록 구성된다. 예를 들어, 저장소 및 다공성 구조는 적어도 약 12개월 또는 적어도 약 2년 또는 적어도 약 3년의 연장된 기간 동안 유리액 1 ml 당 적어도 약 0.1 ㎍ 내지 유리액 1 ml 당 약 10 ㎍ 이하의 농도에 상응하는 치료량의 치료제를 방출하도록 구성된다. 예를 들어, 저장소 및 다공성 구조는 적어도 약 3개월 또는 6개월 또는 12개월 또는 24개월의 연장된 기간 동안 유리액 1 ml 당 적어도 약 0.01 ㎍ 내지 유리액 1 ml 당 약 300 ㎍ 이하의 농도에 상응하는 치료량의 치료제를 방출하도록 구성된다.The reservoir and the porous structure are configured to release a therapeutic amount of therapeutic agent corresponding to a concentration of at least about 0.1 [mu] g per ml of the vitreous fluid to about 10 [mu] g per ml of the lyophilized solution for an extended period of at least about 3 months or at least about 6 months do. For example, the reservoir and porous structure may be present at a concentration of at least about 0.1 [mu] g per ml of the vial to about 10 [mu] g per ml of the vial for at least about 12 months, or at least about 2 years, or at least about 3 years And to release a corresponding therapeutic amount of the therapeutic agent. For example, the reservoir and the porous structure correspond to a concentration of at least about 0.01 [mu] g per ml of the vial to about 300 [mu] g per ml of the vial for at least about 3 months or 6 months, or 12 months or 24 months, To release a therapeutic amount of the therapeutic agent.
치료제의 용해도를 증가시키기 위해 첨가된 제제화 성분은 표적 조직에서의 효능이 적어도 일부 경우에 이상적이지 않을 정도로 매우 강력하게 치료제와 결합한다. 예를 들어, 착물화제, 예컨대 시클로덱스트린은 낮은 수용해도의 치료제를 고 농도로 함유하는 제제를 가능하게 한다. 그러나, 예를 들어 문헌 [Stella et al.,Advanced Drug Delivery Reviews, 36: 3-16 (1999)]; 및 [Brewster and Loftsson,Advanced Drug Delivery Reviews, 59: 645-666 (2007)]에서 논의된 바와 같이, 치료제를 방출시키기 위해서는 다량의 희석이 필요하다. 적어도 10의 희석 배율, 때로는 적어도 100 또는 1000 또는 심지어 10,000의 희석 배율이 일반적으로 시클로덱스트린과의 착물로부터 높은 분율의 치료제를 방출시키기 위해 필요하다.The formulation components added to increase the solubility of the therapeutic agent are very strongly associated with the therapeutic agent so that the efficacy in the target tissue is not ideal, at least in some cases. For example, complexing agents, such as cyclodextrins, make preparations containing high therapeutic concentrations of low water solubility agents. However, for example, Stella et al.,Advanced Drug Delivery Reviews , 36: 3-16 (1999); And Brewster and Loftsson,Advanced Drug Delivery Reviews , 59: 645-666 (2007), a large amount of dilution is required to release the therapeutic. A dilution factor of at least 10, sometimes at least 100 or 1000 or even a dilution factor of 10,000 is generally required to release a higher fraction of the therapeutic agent from the complex with the cyclodextrin.
착물화제, 예컨대 시클로덱스트린을 함유하는 제제와 조합된 치료제 전달 장치 (PDS)는 시클로덱스트린의 이전의 모든 적용과 비교하여 특이한 장점을 제공한다. PDS의 저장소 및 다공성 구조는 연장된 기간 동안 시클로덱스트린 착물로부터 치료제를 방출시키기 위해 필요한 희석을 달성하도록 구성된다. 예를 들어, 인간의 눈에 이식된 23 μL의 부피 및 RRI = 0.007 mm의 PDS는 연장된 기간 동안, 예를 들어 수개월 동안 10,000을 초과하는 희석 배율을 달성한다. 지속적인 고 희석은, 시클로덱스트린 제제가 국소 점안제로서 눈에 적용될 때 발생하는 최소 희석과 매우 상이하다. 추가적으로, 수개월의 기간 동안 고 희석과 함께 발생하는 PDS로부터의 서방성 전달은 시클로덱스트린 제제의 정맥내 주사에 상응하는 단기간 (예를 들어, 수시간)과 비교하여 특이하다.Therapeutic delivery devices (PDS) in combination with complexing agents, such as those containing cyclodextrin, offer unique advantages over all previous applications of cyclodextrins. The reservoir and porous structure of the PDS are configured to achieve the dilution required to release the therapeutic agent from the cyclodextrin complex for extended periods of time. For example, a volume of 23 μL implanted in the human eye and a PDS with RRI = 0.007 mm achieve a dilution magnification of over 10,000 for extended periods of time, eg, over several months. Continuous high dilution is very different from the minimum dilution that occurs when the cyclodextrin formulation is applied to the eye as a topical eye drop. Additionally, sustained release from PDS that occurs with high dilution over a period of months is unusual compared to a short term (e.g., hours) corresponding to intravenous injection of a cyclodextrin preparation.
실시양태에서, 다공성 구조는 연장된 기간 동안 치료량을 방출하도록 구성된 다공성, 두께, 채널 파라미터 및 표면적을 포함한다. 예를 들어, 다공성 물질은 물질 내에서 연장되는 채널의 공극 공간의 분율에 상응하는 다공성을 포함한다. 예를 들어, 다공성은 약 3% 내지 약 70% 범위 내의 값을 포함한다. 다른 실시양태에서, 다공성은 약 5% 내지 약 10%, 또는 약 10% 내지 약 25%, 또는 예를 들어 약 15% 내지 약 20% 범위 내의 값을 포함한다. 다공성은 중량 및 거시적 부피로부터 결정되거나 또는 질소 기체 흡착을 통해 측정된다.In an embodiment, the porous structure includes porosity, thickness, channel parameters and surface area configured to release a therapeutic dose over an extended period of time. For example, the porous material includes porosity corresponding to a fraction of the void space of the channels extending in the material. For example, the porosity includes values within the range of about 3% to about 70%. In another embodiment, the porosity comprises a value within the range of from about 5% to about 10%, or from about 10% to about 25%, or for example, from about 15% to about 20%. The porosity is determined from the weight and macroscopic volume or is measured through nitrogen gas adsorption.
다공성 구조는 복수 개의 다공성 구조를 포함하고, 계산을 위해 방정식에 사용되는 면적은 복수 개의 다공성 구조의 합친 면적을 포함한다.The porous structure includes a plurality of porous structures, and the area used in the equation for calculation includes the combined area of the plurality of porous structures.
징후 및 치료 방법Signs and treatment methods
눈의 질환 또는 병태, 특히 망막병증 및 안구 신생혈관화를 치료 및/또는 완화시키는 방법이 개시되어 있다. 이러한 질환 또는 병태의 비제한적 예는 당뇨병성 황반 부종, AMD, CNV, NV, DR, 안구 허혈, 망막 정맥 폐쇄 (중심 또는 분지), 안구 외상, 수술 유래 부종, 수술 유래 신생혈관화, 낭포황반 부종, 포도막염 등을 포함한다. 이러한 질환 또는 병태는 진행성인지 또는 비진행성인지에 상관없이, 급성 질환 또는 병태의 결과인지 또는 만성 질환 또는 병태의 결과인지에 상관없이, 안구 혈관구조의 변화를 특징으로 한다.Methods for treating and / or ameliorating diseases or conditions of the eye, particularly retinopathy and ocular neovascularization, are disclosed. Non-limiting examples of such diseases or conditions include diabetic macular edema, AMD, CNV, NV, DR, ocular ischemia, retinal vein occlusion (central or branchial), ocular trauma, surgery-induced edema, , Uveitis, and the like. Regardless of whether the disease or condition is progressive or non-progressive, it is characterized by a change in the ocular vascular structure, whether it is the result of an acute disease or condition or a chronic disease or condition.
본 개시내용은 위축성 AMD의 치료 및/또는 완화에 있어서의 본원에 기재된 제제의 용도를 제공한다. 제제는 신생혈관 (삼출성 또는 습성) AMD의 치료에 사용된다. 본 개시내용의 제제는 망막에서의 혈관 누수 및/또는 신생혈관화를 치료하거나, 그의 진행을 예방하거나, 또는 그의 증상을 완화시킨다.The present disclosure provides for the use of the agents described herein in the treatment and / or alleviation of atrophic AMD. The agent is used to treat neovascular (exudative or habitual) AMD. The preparations of the present disclosure treat vascular leakage and / or neovascularization in the retina, prevent its progression, or alleviate its symptoms.
개시된 방법은 개시된 치료제 및 그의 제제 중 1종 이상을 대상체에게 투여함으로써, 병리학적 신생혈관화 (NV)의 진행을 예방 또는 제어하거나, 또는 NV의 발병과 관련있는 질환 또는 병태를 치료하는 것에 관한 것이다. 개시된 방법은 착물화제, 가용화제/안정화제, pH 조정 작용제, 완충제, 양쪽 친매성 작용제, 비수성 용매, 장성 작용제, 또는 이들의 조합을 포함하는 1종 이상의 제제화 작용제가 포함된 제제 중의 파조파닙의 제약상 허용되는 염의 유효량을 대상체에게 투여함으로써, NV를 치료하거나 또는 그의 진행을 예방하는 것에 관한 것이다. NV를 치료하거나 또는 예방하기 위한 제제에 사용되는 착물화제는 시클로덱스트린, 예를 들어 캅티솔®이다.The disclosed methods relate to the prevention or control of the progression of pathological neovascularization (NV), or the treatment of a disease or condition associated with the onset of NV, by administering one or more of the disclosed therapeutic agents and their agents to a subject . The disclosed methods can also be used to formulate pajopap nips in formulations containing one or more formulatory agents, including complexing agents, solubilizers / stabilizers, pH adjusting agents, buffers, amphiphilic agents, non-aqueous solvents, , ≪ / RTI > by administering to a subject an effective amount of a pharmaceutically acceptable salt of a compound of formula (I). The complexing agent used in the formulation for treating or preventing NV is cyclodextrin, for example, Captisol.
개시된 방법은 본 개시내용의 제제의 유리체내 전달을 통해, 안구 신생혈관화를 예방 또는 제어하거나 또는 안구 신생혈관화의 발병과 관련있는 질환 또는 병태를 치료하는 것에 관한 것이다.The disclosed methods relate to the prevention or control of ocular neovascularization through the delivery of a formulation of the present disclosure through a vitreous body, or to the treatment of a disease or condition associated with the onset of ocular neovascularization.
또 다른 개시된 방법은 티로신 키나제 억제제 및 착물화제, 예를 들어 시클로덱스트린을 포함하는 제제의 유리체내 전달을 통해, 망막 부종 또는 망막 신생혈관화를 예방 또는 제어하거나 또는 망막 부종 또는 망막 신생혈관화의 발병과 관련있는 질환 또는 병태를 치료하는 것에 관한 것이다.Another disclosed method is to prevent or control retinal edema or retinal neovascularization through delivery of a formulation comprising a tyrosine kinase inhibitor and a complexing agent, such as a cyclodextrin, into the vitreous, or to prevent the onset of retinal edema or retinal neovascularization ≪ RTI ID = 0.0 > and / or < / RTI >
본 개시내용은 티로신 키나제 억제제 및 착물화제, 예를 들어 시클로덱스트린을 포함하는 제제의 유리체내 전달을 통해, 비증식성 망막병증의 증식성 망막병증으로의 진행을 지연시키거나 또는 예방하는 방법에 관한 것이다.This disclosure relates to a method of delaying or preventing the progression of non-proliferative retinopathy to proliferative retinopathy through the in vivo delivery of a formulation comprising a tyrosine kinase inhibitor and a complexing agent, for example a cyclodextrin .
추가의 개시된 방법은 티로신 키나제 억제제, 파조파닙 1HCl 또는 파조파닙 2HCl, 및 착물화제, 예를 들어 시클로덱스트린을 포함하는 제제의 유리체내 전달을 통해, 당뇨병성 망막병증을 치료, 그의 진행을 예방 및/또는 제어하거나, 또는 당뇨병성 망막병증의 발병과 관련있거나 또는 그에 의해 야기되는 질환 또는 병태를 치료하는 것에 관한 것이다.A further disclosed method is to treat diabetic retinopathy, prevent its progression, through the delivery of a tyrosine kinase inhibitor, Pazopanip HCI or Pazopanib 2HCI, and a complexing agent, such as cyclodextrin, And / or to treat or ameliorate a disease or condition associated with or caused by the onset of diabetic retinopathy.
당뇨병성 증식성 망막병증은 신생혈관화를 특징으로 한다. 새로운 혈관은 취약하고 출혈을 일으키기 쉽다. 그 결과는 망막 반흔 뿐만 아니라, 새로운 혈관의 과형성으로 인해 눈을 통과하는 광 경로의 폐쇄 또는 완전한 차단이다. 전형적으로 당뇨병성 황반 부종을 갖는 대상체는 비증식성 단계의 당뇨병성 망막병증을 앓고 있지만; 종종 대상체가 증식성 단계의 발병 시에 황반 부종 증후를 나타내기 시작한다.Diabetic proliferative retinopathy is characterized by neovascularization. New blood vessels are vulnerable and prone to bleeding. The result is blockage or complete blockage of the optical pathway through the eye due to hyperplasia of the new blood vessels as well as retinal scarring. Subjects with diabetic macular edema typically suffer from diabetic retinopathy at the non-proliferative stage; Often the subject begins to exhibit macular edema symptoms at the onset of the proliferative stage.
또 다른 추가의 개시된 방법은 티로신 키나제 억제제 및 착물화제, 예를 들어, 시클로덱스트린을 포함하는 제제의 유리체내 전달을 통해, 당뇨병성 황반 부종을 예방 또는 제어하거나, 또는 당뇨병성 황반 부종의 발병과 관련있는 질환 또는 병태를 치료하는 것에 관한 것이다.Another additional disclosed method is to prevent or control diabetic macular edema through the delivery of a tyrosine kinase inhibitor and complexing agent, for example, a formulation comprising a cyclodextrin, into the vitreous, or to the onset of diabetic macular edema Lt; RTI ID = 0.0 > disease or condition. ≪ / RTI >
일반 정의General Definition
본 명세서 및 하기 청구범위에서, 하기의 의미를 갖는 것으로 정의될, 다수의 용어가 언급된다: 본원에서 모든 백분율, 비 및 비율은 달리 특정되지 않는 한 중량 기준이다. 모든 온도는 달리 특정되지 않는 한 섭씨 온도 (℃)이다.In this specification and in the claims that follow, a number of terms are defined that have the following meanings: All percentages, ratios and ratios herein are by weight unless otherwise specified. All temperatures are in degrees Celsius (° C) unless otherwise specified.
"제약상 허용되는"이란 생물학적으로 또는 달리 바람직한 물질을 의미하는데, 즉 물질이 임상학적으로 허용되지 않는 생물학적 효과를 초래하지 않으면서 또는 그것이 함유된 제약 조성물의 임의의 다른 성분과 유해한 방식으로 상호작용하지 않으면서, 관련된 활성 화합물과 함께 개체에게 투여될 수 있다."Pharmaceutically acceptable" means a biologically or otherwise preferred substance, that is, a substance that does not result in a clinically unacceptable biological effect or that interacts in a deleterious manner with any other ingredients of the pharmaceutical composition in which it is contained , But may be administered to an individual together with the active compound concerned.
성분의 중량 퍼센트는, 구체적으로 그 반대로 명시되지 않는 한, 성분이 포함된 제제 또는 조성물의 총 중량을 기준으로 한다.The weight percent of the ingredient is based on the total weight of the formulation or composition containing the ingredient, unless specifically stated otherwise.
본원에 사용된 "유효량"이란 "목적하는 또는 치료학적 결과를 달성하는데 필요한 기간 동안 투여 시에 효과적인, 개시된 화합물 중 1종 이상의 양"을 의미한다. 유효량은 관련 기술분야에서 공지된 인자, 예컨대 치료받는 인간 또는 동물의 질환 상태, 연령, 성별 및 체중에 따라 달라질 수 있다. 특정 투여 요법이 본원의 실시예에 기재되어 있을 수 있지만, 관련 기술분야의 통상의 기술자라면 투여 요법이 최적의 치료 반응을 제공하도록 변형될 수 있음을 알 것이다. 예를 들어, 여러번 분할된 용량이 매일 투여될 수 있거나 또는 용량이 치료 환경의 위급 상황에 의한 지시에 비례해서 감소할 수 있다. 또한, 본 개시내용의 조성물은 치료량을 달성하기 위해 필요한 만큼 자주 투여될 수 있다.As used herein, "effective amount" means "amount of one or more of the disclosed compounds that is effective upon administration for the period required to achieve the desired or therapeutic result. The effective amount may vary depending on factors known in the art, such as the disease state, age, sex, and body weight of the human or animal being treated. Although specific dosage regimens may be described in the examples herein, those skilled in the art will appreciate that the dosage regimen may be modified to provide the optimal therapeutic response. For example, multiple divided doses may be administered daily, or the dose may decrease in proportion to an indication by an emergency in the therapeutic environment. In addition, the compositions of the present disclosure may be administered as often as necessary to achieve therapeutic dosages.
"작용제"는 본원에서 치료상 또는 생물학적 활성 화합물이 아닌, 개시된 억제제 중 1종 이상에 함유되거나 또는 그와 조합될 수 있는 임의의 다른 화합물을 포함시키기 위해 사용된다. 그러므로, 작용제는 제약상 또는 생물학적으로 허용되거나 또는 적절해야 한다 (예를 들어, 작용제는 일반적으로 대상체에 대하여 비독성이어야 함). "작용제"는 단독의 그러한 화합물을 포함하고, 또한 복수의 작용제도 포함시키고자 한다. 본 개시내용의 목적상, 용어 "작용제" 및 "담체"는 본 개시내용의 상세한 설명에서 상호교환하여 사용되고, 상기 용어들은 본원에서 "안전하고 효과적인 제약 조성물의 제제화를 실시할 때 사용되는 성분"으로서 정의된다."Agonist" is used herein to include any other compound that may be contained in, or be combined with, one or more of the disclosed inhibitors, but not a therapeutically or biologically active compound. Therefore, the agonist should be pharmaceutically or biologically acceptable or appropriate (e.g., the agonist should generally be non-toxic to the subject). "Agents" encompasses such compounds alone and is intended to include a plurality of functional mechanisms. For purposes of this disclosure, the terms " agonist "and" carrier "are used interchangeably in the description of the present disclosure and the terms are used herein as" components used in the formulation of safe and effective pharmaceutical compositions " Is defined.
어구 "제약상 허용되는 담체"는 관련 기술분야에 알려져 있으며, 예를 들어 임의의 보충제 또는 조성물, 또는 이들의 성분을 하나의 기관 또는 신체 부위에서 또 다른 기관 또는 신체 부위로 운반 또는 수송하거나, 또는 작용제를 눈의 표면으로 전달하는데 관여하는, 제약상 허용되는 물질, 조성물 또는 비히클, 예컨대 액체 또는 고체 충전제, 희석제, 부형제, 용매 또는 캡슐화 물질을 말한다. 각각의 담체는 조성물의 다른 성분과 상용성이라는 점에서 "허용되고" 환자에게 해롭지 않아야 한다. 특정 실시양태에서, 제약상 허용되는 담체는 비발열성이다. 제약상 허용되는 담체로서 사용될 수 있는 물질의 일부 예는 (1) 당, 예컨대 락토스, 글루코스 및 수크로스; (2) 전분, 예컨대 옥수수 전분 및 감자 전분; (3) 셀룰로스 및 그의 유도체, 예컨대 소듐 카르복시메틸 셀룰로스, 히드록시프로필메틸 셀룰로스, 에틸 셀룰로스 및 셀룰로스 아세테이트; (4) 분말형 트래거캔트(tragacanth); (5) 맥아; (6) 젤라틴; (7) 활석; (8) 부형제, 예컨대 코코아 버터 및 좌제 왁스; (9) 오일, 예컨대 낙화생유, 면실유, 해바라기유, 참깨유, 올리브유, 옥수수유 및 대두유; (10) 글리콜, 예컨대 프로필렌 글리콜; (11) 폴리올, 예컨대 글리세린, 소르비톨, 만니톨 및 폴리에틸렌 글리콜; (12) 에스테르, 예컨대 에틸 올리에이트 및 에틸 라우레이트; (13) 한천; (14) 완충제, 예컨대 마그네슘 히드록시드 및 알루미늄 히드록시드; (15) 알긴산; (16) 발열성 물질 제거수; (17) 등장성 식염수; (18) 링거액(Ringer's solution); (19) 에틸 알콜; (20) 포스페이트 완충 용액; (21) 검(gum), 예컨대 HP-구아(HP-guar); (22) 중합체; 및 (23) 제약 제제에 사용되는 다른 비독성 상용성 물질을 포함한다.The phrase "pharmaceutically acceptable carrier" is known in the art, for example, to transport or transport any supplement or composition, or component thereof, from one organs or body region to another organ or body region, or Refers to a pharmaceutically acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, which is involved in delivering the agent to the surface of the eye. Each carrier should be "permitted" in terms of compatibility with the other ingredients of the composition and not deleterious to the patient. In certain embodiments, the pharmaceutically acceptable carrier is non-heat-degradable. Some examples of materials that can be used as pharmaceutically acceptable carriers include (1) sugars such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) Cellulose and derivatives thereof such as sodium carboxymethyl cellulose, hydroxypropylmethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients such as cocoa butter and suppository wax; (9) oils such as peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols such as propylene glycol; (11) polyols such as glycerin, sorbitol, mannitol, and polyethylene glycol; (12) esters such as ethyl oleate and ethyl laurate; (13) agar; (14) Buffering agents such as magnesium hydroxide and aluminum hydroxide; (15) alginic acid; (16) the number of exothermic substances removed; (17) isotonic saline solution; (18) Ringer's solution; (19) ethyl alcohol; (20) phosphate buffer solution; (21) gums such as HP-guar; (22) a polymer; And (23) other non-toxic compatible materials used in pharmaceutical formulations.
용어 "제약상 허용되는"은 담체, 희석제 또는 작용제가 제제의 다른 성분과 상용성이고 수용자에게 유해하지 않아야 한다는 사실을 나타낸다.The term " pharmaceutically acceptable "indicates that the carrier, diluent or agent is compatible with the other ingredients of the formulation and not deleterious to the recipient.
본원에 사용된 "대상체"란 개체를 의미한다. 따라서, "대상체"는 사육 동물 (예를 들어, 고양이, 개 등), 가축 (예를 들어, 소, 말, 돼지, 양, 염소 등), 실험실 동물 (예를 들어, 마우스, 토끼, 래트, 기니 피그(guinea pig) 등), 및 조류를 포함할 수 있다. "대상체"는 또한 포유동물, 예컨대 영장류 또는 인간을 포함할 수 있다."Object" as used herein means an individual. Thus, the term "subject" refers to any animal, including a rabbit, a rabbit, a rabbit, Guinea pigs, etc.), and algae. The "subject" may also include mammals, such as primates or humans.
"감소시키다" 또는 상기 용어의 다른 형태, 예컨대 "감소시키는" 또는 "감소"는 사건 또는 특징 (예를 들어, 혈관 누수)을 줄이는 것을 의미한다. 이는 전형적으로 일부 표준값 또는 기대값과 비교되어, 달리 말하면 상대적이나, 항상 표준값 또는 상대값이 언급될 필요는 없음이 이해된다."Reducing" or other forms of the term, such as "reducing" or "decreasing", means reducing an event or characteristic (eg, a blood vessel leak). It is understood that this is typically compared to some standard or expected value, in other words, relative, but not always the standard or relative value.
용어 "치료하다" 또는 상기 용어의 다른 형태, 예컨대 "치료하는" 또는 "치료"는 본원에서 본 발명의 치료제의 투여가 수용자에게서 질환 또는 장애를 경감시키고/거나 장애와 연관된 특정한 특징 또는 사건 (예를 들어, 혈관 누수)을 감소, 억제, 또는 제거한다는 것을 의미하기 위해 사용된다.The term " treating "or other form of the term, such as" treating "or" treating ", as used herein refers to administering a therapeutic agent of the present invention to a recipient to relieve a disease or disorder and / For example, a blood vessel leak, for example).
본 발명의 방법이 장애의 예방에 관한 것이라면, 용어 "예방하다"는 질환 상태가 완전히 저지되어야 하는 것은 아닌 것으로 이해된다. 오히려, 본원에 사용된 예방이라는 용어는 본 발명의 화합물의 투여가 질환의 발병 전에 발생할 수 있도록, 장애에 민감한 집단을 동정하는 통상의 기술자의 능력을 말한다. 상기 용어는 질환 상태가 완전히 회피되는 것을 내포하지 않는다.If the method of the present invention is directed to prevention of a disorder, the term "preventing" is understood to mean that the disease state is not to be completely inhibited. Rather, the term prophylactic used herein refers to the ability of a conventional technician to identify a population that is susceptible to a disorder so that administration of a compound of the present invention occurs prior to the onset of the disease. The term does not imply that the disease state is completely avoided.
용어 "증상을 완화시키는" 또는 상기 용어의 다른 형태, 예컨대 "증상을 완화시키다"는 본원에서 본 발명의 치료제의 투여가 수용자에게서 질환 또는 장애의 하나 이상의 증상을 경감시키고/거나 치료제의 투여 전 및/또는 후에 질환 또는 장애와 연관된 특정 증상을 감소, 억제, 또는 제거한다는 것을 의미하기 위해 사용된다.The term "alleviating the symptoms" or other forms of the term, e. G., "Alleviating the symptoms ", as used herein means that administration of the therapeutic agent of the invention alleviates one or more symptoms of the disease or disorder in the recipient and / / &Quot; is used to mean reducing, inhibiting, or eliminating certain symptoms associated with a disease or disorder.
개시된 화합물은 수용체 티로신 키나제를 억제함으로써 혈관 누수에 영향을 준다.The disclosed compounds affect vascular leakage by inhibiting receptor tyrosine kinases.
본 명세서의 상세한 설명 및 청구범위 전체에서, 용어 "포함하다" 및 상기 용어의 다른 형태, 예컨대 "포함하는" 및 "포함한다"는 비제한적으로 포함하는 것을 의미하고, 예를 들어 다른 첨가제, 성분, 정수, 또는 단계를 제외시키려는 것이 아니다.Throughout the description and claims of this specification, the term " comprises "and other forms of the term, such as" comprising "and" , Integers, or steps.
상세한 설명 및 첨부된 청구범위에서 사용된 단수 형태 "하나의", "한" 및 "그"는, 문맥에서 명백하게 달리 나타내지 않는 한, 복수의 지시대상을 포함한다.The singular forms "a," " an "and" the ", as used in the description and the appended claims, include a plurality of referents unless the context clearly dictates otherwise.
"임의적" 또는 "임의적으로"는 그 다음에 기재된 사건 또는 상황이 발생할 수 있거나 또는 발생하지 않을 수 있고, 또한 그의 기재는 사건 또는 상황이 발생한 경우 및 발생하지 않은 경우를 포함한다는 것을 의미한다."Optional" or "optionally" means that the subsequently described event or circumstance may or may not occur, and that the description includes instances where the event or circumstance occurs and instances in which it does not.
본원에서 "약"으로 수식된 하나의 특정 값부터의 범위, 및/또는 "약"으로 수식된 또 다른 특정 값까지의 범위가 표시될 수 있다. 이러한 범위가 표시될 때, 또 다른 측면은 하나의 특정 값부터, 또한/또는 다른 특정 값까지를 포함한다. 마찬가지로, 값이 선행되는 "약"을 사용하여 근사값으로서 표시되면, 특정 값이 또 다른 측면을 구성하는 것으로 이해된다. 추가로, 각각의 범위의 종점이 다른 종점과 관련하여, 또한 다른 종점과 상관없이 유의한 것으로 이해된다. 추가로, 본원에 개시된 다수의 값이 존재하고, 또한 각각의 값은 본원에서 그 값 자체와 함께 "약"으로 수식된 그 특정 값으로서 개시된 것으로 이해된다. 예를 들어, "10"이라는 값이 개시되면, "약 10"이 또한 개시된다. 또한, 어떤 값이 개시되면, 통상의 기술자에 의해 적절하게 이해되는 바와 같이, "그 값 이하", "그 값 이상" 및 이들 값 사이의 가능한 범위가 또한 개시된 것으로 이해된다. 예를 들어, "10"이라는 값이 개시되면, "10 이하" 및 "10 이상"이 또한 개시된다. 또한, 본 출원 전체에서 데이터가 다수의 상이한 포맷으로 제공되고, 또한 이러한 데이터는 종점과 시작점 및 데이터 포인트가 임의로 조합된 범위를 나타내는 것으로 이해된다. 예를 들어, 특정 데이터 포인트 "10" 및 특정 데이터 포인트 "15"가 개시되면, 10 및 15 초과, 이상, 미만, 이하의 값, 및 그와 동일한 값 뿐만 아니라, 10 내지 15가 개시된 것으로 고려됨이 이해된다. 또한, 2개의 특정 단위 사이의 각각의 단위가 또한 개시된 것으로 이해된다. 예를 들어, 10 내지 15의 범위가 개시되면, 11, 12, 13, 및 14가 또한 개시된다.Ranges may range from one particular value modified by the term " about " and / or to another specific value modified by "about " When such a range is indicated, another aspect includes from one particular value, and / or to another specific value. Likewise, if a value is indicated as an approximation using the preceding "about ", it is understood that the particular value constitutes another aspect. Additionally, it is understood that the endpoints of each range are significant with respect to the other endpoints, and also with respect to the other endpoints. In addition, it is understood that there are a plurality of values disclosed herein, and each value is also disclosed as its specific value herein modified with "about " with its value itself. For example, when a value of "10" is initiated, "about 10" is also initiated. It is also understood that, when a value is disclosed, it is understood that "below that value", "above that value", and the possible range between these values are also disclosed, as is well understood by those skilled in the art. For example, when a value of "10" is initiated, "10 or less" and "10 or more" It is also understood that throughout the present application the data is provided in a number of different formats, and that such data represents an endpoint and a starting point and any combination of data points. For example, when a particular data point "10" and a particular data point "15" are initiated, 10 to 15 and more, less than, less than, less than, and the same value, . It is also understood that each unit between two specific units is also disclosed. For example, if a range of 10 to 15 is initiated, 11, 12, 13, and 14 are also disclosed.
본원에 사용된 어구 "제약상 허용되는 염(들)"은, 달리 지시되지 않는 한, 산성 또는 염기성 기의 염을 포함한다.As used herein, the phrase " pharmaceutically acceptable salt (s) " includes salts of acidic or basic groups, unless otherwise indicated.
용어 "키나제"는 포스페이트 기의 단백질 잔기에의 첨가를 촉매하는 임의의 효소를 말하고; 예를 들어 세린 및 트레오닌 키나제는 포스페이트 기의 세린 및 트레오닌 잔기에의 첨가를 촉매한다.The term "kinase" refers to any enzyme that catalyzes the addition of a phosphate group to a protein residue; For example, serine and threonine kinases catalyze the addition of a phosphate group to the serine and threonine residues.
용어 "VEGFR 키나제", "VEGFR"은 임의의 혈관 내피 성장 인자 수용체를 말한다.The term "VEGFR kinase "," VEGFR "refers to any vascular endothelial growth factor receptor.
용어 "VEGF 신호전달" 및 "VEGF 캐스케이드"는 VEGF 신호전달 캐스케이드의 상류 및 하류 성분을 둘다 말한다.The terms "VEGF signaling" and "VEGF cascade" refer to both upstream and downstream components of the VEGF signaling cascade.
용어 "화합물의 투여" 또는 "화합물을 투여하는 것"은 본 발명의 화합물 또는 제약 조성물을, 치료를 필요로 하는 대상체에게 제공하는 행위를 말한다.The term " administration of a compound "or" administering a compound "refers to the act of providing a compound of the invention or a pharmaceutical composition to a subject in need of treatment.
본 개시내용에서 "조성물" 및 "제제"는 상호교환하여 사용되고, 조성물 또는 제제에 관한 관련 기술분야에서 공지된 통상적인 의미를 나타낸다. 본원에 개시된 "제제"는 치료제 및/또는 제제 부형제 또는 제제화 작용제의 용액, 현탁물, 반고체, 또는 반액체 혼합물을 포함할 수 있다.In the present disclosure, the terms "composition" and "preparation" are used interchangeably and refer to conventional meanings known in the related art relating to a composition or preparation. The "formulations" disclosed herein may include solutions, suspensions, semisolids, or semi-liquid mixtures of therapeutic and / or pharmaceutical excipients or formulation agents.
본 개시내용에 따른 "용액"은 용매 또는 상호 혼화성인 용매의 혼합물에 용해된 1종 이상의 화학 물질을 함유하는 투명한 균질 액체 형태이다. 용액은 적합한 용매 또는 상호 혼화성인 용매의 혼합물 중에 1종 이상의 용해 화학 물질을 함유하는 액체 제제이다. 용액 중의 치료제 물질 분자가 균일하게 분산되어 있으므로, 투여 형태로서의 용액의 사용은 일반적으로 투여 시에 균일한 투여의 보장 및 용액이 희석되거나 또는 달리 혼합될 때 양호한 정확성을 제공한다. 본원에 개시된 "용액"은 통용되는 최신 기술에 기반한 임의의 변형예 또는 관련 기술분야의 통상의 기술자에 의해 달성되는 변형예가 고려된다.A "solution" in accordance with the present disclosure is in the form of a transparent homogeneous liquid containing one or more chemicals dissolved in a solvent or a mixture of solvents that are compatible with each other. A solution is a liquid formulation containing one or more dissolution chemicals in a suitable solvent or a mixture of solvents that are compatible with each other. Since the therapeutic agent molecules in solution are uniformly dispersed, the use of the solution as the dosage form generally provides for uniform delivery at the time of administration and good accuracy when the solution is diluted or otherwise mixed. It is contemplated that the "solution" disclosed herein may be any variation based on the state of the art or a variation achieved by one of ordinary skill in the relevant art.
본 개시내용에 따른 "현탁물"은 액체 비히클에 분산된 고체 입자를 함유하는 액체 형태이다. 본원에 개시된 "현탁물"은 통용되는 최신 기술에 기반한 임의의 변형예 또는 관련 기술분야의 통상의 기술자에 의해 달성되는 변형예가 고려된다."Suspension" in accordance with the present disclosure is in the form of a liquid containing solid particles dispersed in a liquid vehicle. "Suspensions" disclosed herein may take any variant based on the state-of-the-art techniques or variations achieved by one of ordinary skill in the relevant art.
"치료제 전달 장치" 및 "포트 전달 시스템" ("PDS")은 본 명세서에서 상호교환하여 사용된다. 본원에 개시된 "치료제 전달 장치" 또는 "포트 전달 시스템" ("PDS")은 치료제를 대상체에게 특별히 전달하는 유사한 목적을 달성하도록 디자인된 개시 장치의 임의의 변형예가 고려된다. 예를 들어, "치료제 전달 장치" 또는 "포트 전달 시스템" ("PDS")은 막, 개구부, 확산 장벽, 연장된 기간, 예를 들어 30일, 60일, 90일, 120일 또는 그보다 긴 기간 동안 치료량의 치료제를 방출시키기 위한 확산 메카니즘을 포함하는 디자인을 가질 수 있다. 장치의 다수의 변형예가 WO 2012/065006, WO2012/019047, WO2013/003620, WO 2012/019136, WO 2012/019176, 및 미국 특허 8,277,830에서 개시되었고, 이들은 각각 그 전문이 본원에 참조로 포함된다."Therapeutic delivery device" and "Port delivery system" ("PDS") are used interchangeably herein. A "therapeutic delivery device" or "port delivery system" ("PDS") as disclosed herein contemplates any variation of the initiating device designed to accomplish a similar purpose of delivering a therapeutic agent specifically to a subject. For example, a "therapeutic delivery device" or "port delivery system" ("PDS") may be a membrane, an opening, a diffusion barrier, Lt; RTI ID = 0.0 > a < / RTI > dose of therapeutic agent for a period of time. Many variations of the apparatus are disclosed in WO < RTI ID = 0.0 > 2012/065006, < / RTI > WO2012 / 019047, WO2013 / 003620, WO 2012/019136, WO 2012/019176, and U.S. Patent 8,277,830, each of which is incorporated herein by reference in its entirety.
본원에 사용된 용어 "급성"은 급속 발병, 및 단기간이긴 하지만 중증의 증상을 갖는 병태를 나타낸다.As used herein, the term "acute" refers to a condition that has a rapid onset and a short-term but severe symptoms.
본원에 사용된 용어 "진통제"는 장기간의 사용에 적합한, 간헐적 및/또는 만성적 신체 불편감을 관리하기 위한 화합물/제제를 나타낸다.The term "analgesic " as used herein refers to a compound / formulation for managing intermittent and / or chronic physical discomfort suitable for long term use.
본원에 사용된 용어 "마취제" 또는 "마취"는 단기간의, 일시적인 사용에 적합한, 급성 신체 통증을 관리하기 위한 화합물/제제를 나타내고, 이는 화합물/제제가 투여되는 신체 부위/기관에서 둔감화 또는 감소한 감수성 (예를 들어, 눈의 감소한 각막 감수성)을 유발하는 효과를 갖는다.The term " anesthetic "or" anesthesia " as used herein refers to a compound / agent for managing acute bodily pain suitable for short-term, transient use, which is either dulled or reduced in body parts / (For example, reduced corneal sensitivity of the eyes).
용어 "수성"은 전형적으로 담체가 >50 중량%, 보다 바람직하게는 >75 중량%, 특히 90 중량% 정도까지 물인 수성 조성물을 나타낸다.The term "aqueous" typically refers to an aqueous composition wherein the carrier is water up to> 50 wt%, more preferably> 75 wt%, especially up to 90 wt%.
본원에 정의된 용어 "만성"은 지속적으로 계속되는 병태, 또는 빈번한 재발을 특징으로 하는 병태, 바람직하게는 3개월 넘게, 보다 바람직하게는 6개월 넘게, 보다 바람직하게는 12개월 넘게, 보다 더욱 바람직하게는 24개월 넘게 계속되고/재발되는 병태를 의미한다.The term "chronic ", as defined herein, refers to a condition characterized by a persistent condition, or a condition characterized by frequent recurrence, preferably over three months, more preferably over six months, more preferably over twelve months, Means a condition that lasts for more than 24 months / relapses.
본원에 사용된 용어 "편안감"은 통증, 작열감, 자통, 가려움, 과민, 또는 신체 불편감과 연관된 다른 증상의 신체 감각과 대조되는, 신체적 건강 또는 안도감의 감각을 나타낸다.As used herein, the term "comfort" refers to a sense of physical health or relief, contrasted with body sensations of pain, burning sensation, dizziness, irritability, or other symptoms associated with physical discomfort.
본원에 사용된 용어 "증상"은 질환, 질병, 부상, 또는 신체에서 잘못된 어떤 것의 징후로서 정의된다. 증상은 증상을 겪고 있는 개체가 느끼거나 또는 감지하지만, 타인에 의해서는 용이하게 감지되지 않을 수 있다. 타인은 비-의료 전문가로서 정의된다.The term "symptom" as used herein is defined as a disease, a disease, an injury, or a symptom of something erroneous in the body. Symptoms may be felt or detected by the individual experiencing the symptoms, but may not be easily detected by others. Others are defined as non-medical professionals.
본원에 사용된 용어 "증후" 또한 신체에서 잘못된 어떤 것의 징후로서 정의된다. 그러나, 증후는 의사, 간호사, 또는 다른 의료 전문가에 의해 확인될 수 있는 것으로서 정의된다.The term " symptom ", as used herein, is also defined as a sign of something erroneous in the body. However, the symptoms are defined as being identifiable by a physician, nurse, or other health care professional.
본 개시내용에 사용된 용어 "보다 많은"은 무한수의 가능성을 포함하지 않는다. 본 개시내용에 사용된 용어 "보다 많은"은 그것이 사용된 문맥에서 관련 기술분야의 통상의 기술자가 이해하는 바와 같이 사용된다.The term "more than" as used in this disclosure does not include the possibility of an infinite number. The term "more" as used in this disclosure is used as it will be understood by one of ordinary skill in the art in the context in which it is used.
본 개시내용에 사용된 바와 같이, 청구항의 연결 어구에 위치하는지 또는 중심 어구에 위치하는지에 상관없이, 용어 "포함하다(포함한다)" 및 "포함하는"은 개방적 의미를 갖는 것으로 해석되어야 한다. 즉, 상기 용어들은 어구 "적어도 갖는" 또는 "적어도 포함하는"과 동의어로 해석되어야 한다. 방법의 문맥에 사용될 때, 용어 "포함하는"은 방법이 나열된 단계를 적어도 포함하나, 추가 단계를 포함할 수 있음을 의미한다. 분자, 화합물, 또는 조성물의 문맥에 사용될 때, 용어 "포함하는"은 화합물 또는 조성물이 나열된 특징 또는 성분을 적어도 포함하나, 또한 추가 특징 또는 성분을 포함할 수 있음을 의미한다.As used in this disclosure, the terms "comprises" and "comprising" are to be interpreted as having open meaning, regardless of whether they are located in the connection or the center phrase of the claim. That is, the terms should be construed as synonymous with the phrase "having at least" or "including at least ". When used in the context of a method, the term "comprising " means that the method includes at least the listed steps, but may include additional steps. The term "comprising " when used in the context of a molecule, compound, or composition, means that the compound or composition includes at least the listed features or components, but may also include additional features or components.
본원에 기재된 실시양태의 이해를 용이하게 하기 위해, 바람직한 실시양태를 참조하고 그것을 기재하기 위해 특정 언어가 사용된다. 본원에 사용된 용어들은 단지 특정 실시양태를 기재하기 위한 것이며, 본 발명의 범주를 제한하려는 것이 아니다. 본 개시내용 전체에서 사용된 단수 형태 "하나의", "한" 및 "그"는, 문맥에서 명백하게 달리 나타내지 않는 한, 복수의 지시대상을 포함한다. 따라서, 예를 들어, "조성물"의 언급은 복수의 그러한 조성물 뿐만 아니라, 단일 조성물을 포함하고, "치료제"의 언급은 1종 이상의 치료 및/또는 제약 작용제 및 관련 기술분야의 통상의 기술자에게 공지된 이들의 등가물 등을 나타내는 것이다. 본원에 사용된 모든 백분율 및 비는, 달리 지시되지 않는 한, 중량 기준이다.In order to facilitate an understanding of the embodiments described herein, certain languages are used to refer to and describe the preferred embodiments. The terminology used herein is for the purpose of describing particular embodiments only and is not intended to limit the scope of the invention. The singular forms "a," " an "and" the ", as used throughout this disclosure, include a plurality of referents unless the context clearly dictates otherwise. Thus, for example, reference to a "composition" includes a plurality of such compositions, as well as a single composition, and reference to "a therapeutic agent" refers to one or more therapeutic and / or pharmaceutical agents and to those skilled in the art And equivalents thereof. All percentages and ratios used herein are by weight unless otherwise indicated.
하기 실시예는 본 발명의 방법 및 조성물을 비제한적으로 예시하는 것이다. 본 개시내용의 화합물의 합성 및 사용 시에 보통 직면하게 되고 관련 기술분야의 통상의 기술자에게 명백한 다양한 조건 및 파라미터의 다른 적합한 수정 및 적합화는 본 개시내용의 취지 및 범주 내에 포함된다.The following examples illustrate without limitation the methods and compositions of the present invention. Other suitable modifications and adaptations of the various conditions and parameters that are normally encountered in the synthesis and use of the compounds of this disclosure and which are obvious to one of ordinary skill in the art are included within the spirit and scope of this disclosure.
추가 실시양태Additional embodiments
1. 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염, 및 1종 이상의 제제화 작용제의 안정한 제약 제제이며, 여기서 제약상 허용되는 염은 1가 또는 2가 염이고, 1종 이상의 제제화 작용제는 착물화제, 가용화제, 및 임의로 완충제를 포함하고; 치료제의 염은 제제에 용해되어 있는 것인 안정한 제약 제제.1. A stable pharmaceutical formulation of a therapeutic agent having a low water solubility and at least one formulation agent, wherein the pharmaceutically acceptable salt is a monovalent or divalent salt, wherein at least one formulation agent is a complexing agent , A solubilizing agent, and optionally a buffering agent; Wherein the salt of the therapeutic agent is dissolved in the formulation.
2. 제1항에 있어서, 치료제가 파조파닙인 제제.2. The preparation according to claim 1, wherein the therapeutic agent is parapipap.
3. 제2항에 있어서, 제약상 허용되는 염이 1가 또는 2가 할라이드 염인 제제.3. The formulation of claim 2, wherein the pharmaceutically acceptable salt is a monovalent or divalent halide salt.
4. 제3항에 있어서, 염이 클로라이드 염인 제제.4. The formulation of claim 3, wherein the salt is a chloride salt.
5. 제4항에 있어서, 1가 염이 제제에서 약 60 mg/mL의 농도까지 안정한 것인 제제.5. The formulation of
6. 제4항에 있어서, 2가 염이 제제에서 약 70 mg/ml의 농도까지 안정한 것인 제제.6. The formulation of
7. 제4항에 있어서, 제제화 전의 2가 염 결정 구조가 XRPD에 의해 결정된 형태 XIV인 제제.7. The formulation of
8. 제4항에 있어서, 제제에서의 1가 염의 안정성이 제제화 작용제가 포함된 용액에서의 가용화 전에 유기 용매로부터의 치료제의 동결건조를 수행함으로써 증가하는 것인 제제.8. The formulation of
9. 제8항에 있어서, 유기 용매가 디메틸 술폭시드 (DMSO) 또는 트리플루오로 에탄올 (TFE)인 제제.9. The preparation according to claim 8, wherein the organic solvent is dimethylsulfoxide (DMSO) or trifluoroethanol (TFE).
10. 제9항에 있어서, DMSO로부터의 동결건조가 치료제의 하나의 결정질 상 형태를 또 다른 형태로 전환시키는 것인 제제.10. The formulation of claim 9 wherein lyophilization from DMSO converts one of the crystalline forms of the therapeutic agent to another form.
11. 제10항에 있어서, DMSO로부터의 동결건조가 XRPD에 의해 결정될 때, 결정질 상 형태 A를 적어도 70%의 결정질 상 형태 G를 함유하는 물질로 전환시키는 것인 제제.11. The formulation of claim 10, wherein when lyophilization from DMSO is determined by XRPD, the crystalline phase A is converted to a material containing at least 70% crystalline phase G.
12. 제9항에 있어서, TFE로부터의 동결건조가 결정질 상 형태 A를 부분적으로 또는 완전히 무정형 상으로 전환시키는 것인 제제.12. The formulation of claim 9, wherein lyophilization from TFE converts the crystalline phase A partially or completely into an amorphous phase.
13. 제8항에 있어서, pH가 치료제의 제제화 동안에 조정되는 것인 제제.13. The formulation of claim 8, wherein the pH is adjusted during the formulation of the therapeutic.
14. 제8항에 있어서, pH가 치료제의 제제화 동안에 조정되지 않는 것인 제제.14. The formulation of claim 8, wherein the pH is not adjusted during the formulation of the therapeutic.
15. 제4항에 있어서, 착물화제가 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 및 이들의 조합으로 이루어진 군으로부터 선택된 시클로덱스트린인 제제.15. The composition of
16. 제4항에 있어서, 가용화제가 폴리(비닐 피롤리돈) (PVP)인 제제.16. The preparation according to
17. 제4항에 있어서, 완충제가 히스티딘 HCl인 제제.17. The formulation of
18. 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법이며, 여기서 염이 2가 염이고, 방법은 (a) 염을, 착물화제, 가용화제, 및 임의로 완충제를 포함하는 1종 이상의 제제화 작용제의 용액에 용해시키고, (b) 염을 제제화 작용제에 용해시킨 후에 pH를 최적의 값으로 조정하는 것을 포함하는 것인, 안정한 용액 제약 제제의 제조 방법.18. A stable solution of a pharmaceutical acceptable salt of a therapeutic agent having a low water solubility, wherein the salt is a divalent salt, the method comprising: (a) contacting the salt with a complexing agent, a solubilizing agent, and optionally a buffering agent. (B) dissolving the salt in the formulation agent, and then adjusting the pH to an optimal value. ≪ Desc / Clms Page number 14 >
19. 제18항에 있어서, 치료제가 파조파닙인 방법.19. The method of claim 18, wherein the therapeutic agent is psoriopanib.
20. 제18항에 있어서, 제약상 허용되는 염이 할라이드 염인 방법.20. The method of claim 18, wherein the pharmaceutically acceptable salt is a halide salt.
21. 제20항에 있어서, 염이 클로라이드 염인 방법.21. The method of
22. 제21항에 있어서, 착물화제가 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 및 이들의 조합으로 이루어진 군으로부터 선택된 시클로덱스트린인 방법.22. The composition of claim 21, wherein the complexing agent is selected from the group consisting of 2-hydroxypropyl- beta -cyclodextrin, methyl- beta -cyclodextrin, random methylated- beta -cyclodextrin, ethyl- beta -cyclodextrin, triacetyl- Cyclodextrin, carboxymethyl-beta-cyclodextrin, hydroxyethyl- beta -cyclodextrin, 2-hydroxy-3- (trimethylammonio) propyl- beta -cyclodextrin, glucosyl ? -cyclodextrin, maltosyl-? -cyclodextrin, sulfobutyl ether-? -cyclodextrin, branched? -cyclodextrin, hydroxypropyl-? -cyclodextrin, random methylated-? -cyclodextrin, trimethyl- gamma -cyclodextrin, gamma -cyclodextrin, and combinations thereof.
23. 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법이며, 여기서 염은 1가 염이고, 방법은 (a) 염을 염기로 처리하고; (b) 염기 처리된 염을, 착물화제, 가용화제, 및 임의로 완충제를 포함하는 1종 이상의 제제화 작용제의 용액에 용해시키고, (c) pH를 산을 사용하여 약 2 이하의 pH로 조정하는 것을 포함하고, 여기서 염기 처리는 제제 중의 총 염 함량을 증가시키고 산에 의한 pH 조정은 제제에서의 염의 용해도를 증가시키는 것인, 안정한 용액 제약 제제의 제조 방법.23. A stable solution of a pharmaceutical acceptable salt of a therapeutic agent having a low water solubility, wherein the salt is a monovalent salt, the method comprising: (a) treating the salt with a base; (b) dissolving the base treated salt in a solution of one or more formulating agents, including complexing agents, solubilizing agents, and optionally buffering agents, and (c) adjusting the pH to a pH of about 2 or less Wherein the base treatment increases the total salt content in the formulation and the pH adjustment by the acid increases the solubility of the salt in the formulation.
24. 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염의 안정한 용액 제약 제제의 제조 방법이며, 여기서 염은 1가 염이고, 방법은 (a) 유기 용매 중의 염의 용액을 제조하고; (b) 용액을 동결건조시켜 치료제의 동결건조 염을 제조하는 것을 포함하는 것인, 안정한 용액 제약 제제의 제조 방법.24. A stable solution of a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility, wherein the salt is a monovalent salt, the method comprising: (a) preparing a solution of a salt in an organic solvent; (b) lyophilizing the solution to prepare a lyophilized salt of the therapeutic agent.
25. 제24항에 있어서, 가용화제 및 완충제를 물에 용해시켜 용액을 제조하고; 착물화제를 용액에 용해시키고; 약 주위 온도 이상에서 동결건조 염을 용액에 첨가하고 혼합하여 염을 용액에 용해시키는 것을 추가로 포함하고; 여기서 제제의 pH가 임의로 조정되는 것인 방법.25. The method of
26. 제24항에 있어서, 가용화제 및 완충제를 물에 용해시켜 용액을 제조하고; 일정량의 착물화제를 용액에 용해시켜 저 점도 용액을 제조하고; 약 주위 온도 이상에서 동결건조 염을 저 점도 용액에 첨가하고 혼합하여 용액에 용해시키고; 여기서 저 점도 용액의 pH가 조정되고; 약 2x 더 많은 양의 착물화제를 저 점도 용액에 첨가하여 용해시키는 것을 추가로 포함하는 방법.26. The method of
27. 제25항 또는 제26항에 있어서, 가용화제가 폴리(비닐 피롤리돈) (PVP)인 방법.27. The method of
28. 제25항 또는 제26항에 있어서, 완충제가 히스티딘 HCl인 방법.28. The method of
29. 제25항 또는 제26항에 있어서, 착물화제가 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 및 이들의 조합으로 이루어진 군으로부터 선택된 시클로덱스트린인 방법.29. The method of
30. 제25항 또는 제26항에 있어서, 치료제가 파조파닙인 방법.30. The method according to claim 25 or 26, wherein the therapeutic agent is Pachopanip.
31. 제30항에 있어서, 1가 염이 할라이드인 방법.31. The method of claim 30, wherein the monovalent salt is a halide.
32. 제31항에 있어서, 할라이드가 클로라이드인 방법.32. The method of claim 31, wherein the halide is chloride.
33. 제32항에 있어서, 동결건조 염이 무정형인 방법.33. The method of claim 32, wherein the freeze-dried salt is amorphous.
34. 제32항에 있어서, 동결건조 단계가 XRPD에 의해 결정될 때, 결정질 상 형태 A를 적어도 약 70%의 파조파닙의 형태 G를 함유하는 물질로 전환시키는 것인 방법.34. The method of claim 32, wherein when the lyophilization step is determined by XRPD, the crystalline phase A is converted to a material containing at least about 70% of the ripopanib form G.
35. 제25항 또는 제26항에 있어서, 동결건조 염이 약 37℃ 내지 약 50℃의 온도에서 용액에 용해되는 것인 방법.35. The method of
36. 제24항에 있어서, 약 주위 온도 이상에서 물을 첨가하면서, 적어도 가용화제, 완충제, 착물화제, 및 동결건조 염을 연속적으로 혼합하는 것을 추가로 포함하는 방법.36. The method of
37. 제36항에 있어서, 제제의 pH가 염기를 사용하여 약 6-7로 조정되는 것인 방법.37. The method of claim 36, wherein the pH of the formulation is adjusted to about 6-7 using a base.
38. 제36항에 있어서, pH가 조정되지 않는 것인 방법.38. The method of claim 36, wherein the pH is not adjusted.
39. 제36항에 있어서, 치료제가 파조파닙인 방법.39. The method of claim 36, wherein the therapeutic agent is psapopanib.
40. 제39항에 있어서, 제약상 허용되는 염이 할라이드 염인 방법.40. The method of claim 39, wherein the pharmaceutically acceptable salt is a halide salt.
41. 제40항에 있어서, 염이 1가 클로라이드 염인 방법.41. The method of
42. 제41항에 있어서, 착물화제가 2-히드록시프로필-β-시클로덱스트린, 메틸-β-시클로덱스트린, 랜덤 메틸화-β-시클로덱스트린, 에틸화-β-시클로덱스트린, 트리아세틸-β-시클로덱스트린, 퍼아세틸화-β-시클로덱스트린, 카르복시메틸-β-시클로덱스트린, 히드록시에틸-β-시클로덱스트린, 2-히드록시-3-(트리메틸암모니오)프로필-β-시클로덱스트린, 글루코실-β-시클로덱스트린, 말토실-β-시클로덱스트린, 술포부틸 에테르-β-시클로덱스트린, 분지형-β-시클로덱스트린, 히드록시프로필-γ-시클로덱스트린, 랜덤 메틸화-γ-시클로덱스트린, 트리메틸-γ-시클로덱스트린, 및 이들의 조합으로 이루어진 군으로부터 선택된 시클로덱스트린인 방법.42. The method of claim 41, wherein the complexing agent is selected from the group consisting of 2-hydroxypropyl- beta -cyclodextrin, methyl- beta -cyclodextrin, random methylated- beta -cyclodextrin, ethyl- beta -cyclodextrin, triacetyl- Cyclodextrin, carboxymethyl-beta-cyclodextrin, hydroxyethyl- beta -cyclodextrin, 2-hydroxy-3- (trimethylammonio) propyl- beta -cyclodextrin, glucosyl ? -cyclodextrin, maltosyl-? -cyclodextrin, sulfobutyl ether-? -cyclodextrin, branched? -cyclodextrin, hydroxypropyl-? -cyclodextrin, random methylated-? -cyclodextrin, trimethyl- gamma -cyclodextrin, gamma -cyclodextrin, and combinations thereof.
43. 제41항에 있어서, 가용화제가 폴리(비닐 피롤리돈) (PVP)인 방법.43. The method of Claim 41, wherein the solubilizing agent is poly (vinylpyrrolidone) (PVP).
44. 제41항에 있어서, 완충제가 히스티딘 HCl인 방법.44. The method of claim 41, wherein the buffer is histidine HCl.
45. 제8항에 있어서, 유기 용매가 트리플루오로 에탄올, 트리플루오로 에탄올-물 혼합물, 또는 디메틸 술폭시드인 제제.45. The formulation of claim 8, wherein the organic solvent is trifluoroethanol, a trifluoroethanol-water mixture, or dimethylsulfoxide.
46. 제24항에 있어서, 유기 용매가 트리플루오로 에탄올, 트리플루오로 에탄올-물 혼합물, 또는 디메틸 술폭시드인 방법.46. The method of
47. 제45항에 있어서, 유기 용매가 디메틸 술폭시드인 제제.47. The preparation of claim 45, wherein the organic solvent is dimethylsulfoxide.
48. 제46항에 있어서, 유기 용매가 디메틸 술폭시드인 방법.48. The method of Claim 46, wherein the organic solvent is dimethylsulfoxide.
49. 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하는, 파조파닙의 결정 형태 A를 적어도 약 70%의 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법.49. A method of converting crystalline Form A of pazopapanib into a substance containing crystalline Form G of at least about 70% pazopanib, comprising dissolving Form A in DMSO and lyophilizing the resulting solution.
50. 대상체 눈의 망막에서의 혈관 누수 또는 신생혈관화 (NV)를 특징으로 하는 장애를 치료하거나, 그의 진행을 예방하거나, 또는 그의 증상을 완화시키는 방법에서의, 제1항, 제18항, 제23항, 및 제24항 중 어느 한 항의 제제의 용도.50. A method of treating a disorder characterized by vascular leakage or neovascularization (NV) in the retina of a subject's eye, preventing the progression thereof, or alleviating its symptoms, 24. Use of an agent according to any one of claims 23 to 24.
51. 대상체 눈의 망막에서의 혈관 누수 또는 신생혈관화 (NV)를 특징으로 하는 장애를 치료하거나, 그의 진행을 예방하거나, 또는 그의 증상을 완화시키는 방법에 사용하기 위한 의약의 제조에 있어서의, 제1항, 제18항, 제23항, 및 제24항 중 어느 한 항의 제제의 용도.51. A method for the manufacture of a medicament for use in a method of treating a disorder characterized by vascular leakage or neovascularization (NV) in the retina of a subject's eye, preventing the progression thereof, or alleviating its symptoms, Use of an agent according to any one of
52. 치료 장치의 저장소 챔버에 함유된 제1항 내지 제48항 중 어느 한 항의 안정한 제제를 포함하고, 여기서 저장소 챔버는 눈의 유리체에서의 치료제의 제어 방출을 위해 다공성 구조에 커플링된 것인 키트.52. A stable formulation of any one of claims 1 to 48 contained in a reservoir chamber of a therapeutic device wherein the reservoir chamber is coupled to a porous structure for controlled release of a therapeutic agent in the vitreous of the eye, Kits.
53. 제1항 내지 제48항 중 어느 한 항에 있어서, 눈의 유리체에서의 치료제의 제어 방출을 위해 치료제 전달 시스템 내의 다공성 구조에 커플링된 저장소 챔버에 함유되고; 여기서 제제의 다공성 구조로부터의 제어 방출이 저장소 챔버에서의 치료제 농도보다 적어도 두 자릿수가 더 낮은 유리체에서의 치료제 농도를 발생시키는 것인 약물 전달 제제.53. A pharmaceutical composition according to any one of claims 1 to 48, which is contained in a storage chamber coupled to a porous structure in a therapeutic delivery system for controlled release of a therapeutic agent in the vitreous of the eye; Wherein the controlled release from the porous structure of the formulation results in a therapeutic agent concentration in the vitreous which is at least two orders of magnitude lower than the therapeutic agent concentration in the reservoir chamber.
54. 제1항 내지 제48항 중 어느 한 항에 있어서, 안구 약물 전달 방법에 사용되는 제제.54. The formulation according to any one of claims 1 to 48 for use in an ocular drug delivery method.
55. 제54항에 있어서, 유리체내 전달 제제인 제제.55. The formulation according to claim 54, which is a delivery system in the vitreous body.
56. 제54항에 있어서, 점안제가 아닌 제제.56. The formulation of claim 54, which is not an eye drops.
57. 제54항에 있어서, 국소 전달 제제가 아닌 제제.57. The formulation of claim 54, which is not a topical delivery formulation.
58. 제54항에 있어서, 경구 전달 제제 또는 비경구 전달 제제가 아닌 제제.58. The formulation of claim 54, which is not an oral delivery formulation or a parenteral delivery formulation.
59. 제54항에 있어서, 눈주위 전달 제제가 아닌 제제.59. The formulation of claim 54, which is not an ocular delivery agent.
60. 낮은 수용해도를 갖는 치료제의 제약상 허용되는 염, 및 1종 이상의 제제화 작용제의 안정한 제약 제제를, 다공성 구조에 커플링된 저장소 챔버를 포함하는 유리체내 전달 장치로부터 전달하는 것을 포함하고, 여기서 제제는 장치의 저장소에 함유되어 있으며, 제제의 다공성 구조를 통한 저장소로부터의 제어 방출이 유리체에서의 치료제의 반감기를 증가시키고;60. A pharmaceutical composition comprising a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility and a stable pharmaceutical formulation of one or more formulating agents from a delivery system comprising a storage chamber coupled to the porous structure, The formulation is contained in a reservoir of the device and controlled release from the reservoir through the porous structure of the formulation increases the half-life of the therapeutic agent in the vitreous;
여기서 제약상 허용되는 염은 1가 또는 2가 염이고, 1종 이상의 제제화 작용제는 착물화제, 가용화제 및 완충제를 포함하고; 치료제의 염이 제제에 용해되어 있는 것인, 후안부의 안과 질환 또는 장애를 치료 및/또는 완화시키는 방법.Wherein the pharmaceutically acceptable salt is a monovalent or divalent salt, wherein the at least one formulation agent comprises a complexing agent, a solubilizing agent and a buffering agent; Wherein the salt of the therapeutic agent is dissolved in the formulation.
61. 제60항에 있어서, 질환 또는 장애가 당뇨병성 망막병증, 연령-관련 황반 변성 (AMD), 병리학적 맥락막 신생혈관화 (CNV), 병리학적 망막 신생혈관화, 포도막염, 망막 정맥 폐쇄, 안구 외상, 수술 유래 부종, 수술 유래 신생혈관화, 낭포양 황반 부종, 안구 허혈, 미숙아 망막병증, 코츠병, 겸상 적혈구 망막병증, 및 신생혈관 녹내장으로부터 선택된 것인 방법.61. The method of
62. 제60항에 있어서, 저장소 챔버가 재충전가능하고, 장치가 눈에 삽입된 후에 제제가 재충전되는 것인 방법.62. The method of
63. 제62항에 있어서, 장치가 눈에 30 - 90일 동안, 또는 최대 6개월 동안 위치한 후에, 제제가 저장소 챔버에 재충전되는 것인 방법.63. The method of claim 62, wherein the formulation is refilled into the reservoir chamber after the device is placed in the eye for 30-90 days, or for up to 6 months.
64. 결정 형태를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 적어도 약 70%의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법.64. The method of claim 1, wherein the crystalline form is dissolved in DMSO and the resulting solution is lyophilized; Wherein a form G of the at least about 70% of the wave pan nips is formed, is converted to a material containing the crystalline form G of the wave pan nip.
65. 제64항에 있어서, 약 100%의 파조파닙의 형태 G가 형성되는 방법.65. The method of claim 64, wherein form G of about 100% of the wave pan nip is formed.
66. 제64항에 있어서, 적어도 약 70% - 약 95%의 파조파닙의 형태 G가 형성되는 방법.66. The method of claim 64, wherein at least about 70% to about 95% of the form G of the wave pan nip is formed.
67. 형태 A를 DMSO에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 적어도 약 70%의 파조파닙의 형태 G가 형성되는 것인, 파조파닙의 결정 형태 A를 파조파닙의 결정 형태 G를 함유하는 물질로 전환시키는 방법.67. The method of claim 1, wherein Form A is dissolved in DMSO and the resulting solution is lyophilized; Wherein a form G of the at least about 70% of the wave pan nip is formed.
68. 제67항에 있어서, 약 100%의 파조파닙의 형태 G가 형성되는 방법.68. The method of claim 67, wherein form G of about 100% of the wave pan nip is formed.
69. 제67항에 있어서, 적어도 약 70% - 약 95%의 파조파닙의 형태 G가 형성되는 방법.69. The method of claim 67, wherein at least about 70% to about 95% of the form G of the wave pan nip is formed.
70. 결정 형태를 TFE에 용해시키고 생성 용액을 동결건조시키는 것을 포함하고; 여기서 약 96% 이하의 또는 적어도 약 96%의 무정형 파조파닙이 형성되는 것인, 파조파닙의 결정 형태를 파조파닙의 무정형 형태로 전환시키는 방법.70. The method of claim 1, wherein the crystalline form is dissolved in TFE and the resulting solution is lyophilized; Wherein at least about 96% or at least about 96% of the amorphous ripshin nip is formed, wherein the crystalline form of the ripopanip is converted to the amorphous form of the ripshnop nip.
71. 제70항에 있어서, 약 96% 이하의 무정형 파조파닙이 형성되는 방법.71. The method of claim 70, wherein up to about 96% amorphous ripple nip is formed.
72. 제70항에 있어서, 적어도 약 96%의 무정형 파조파닙이 형성되는 방법.72. The method of claim 70, wherein at least about 96% of the amorphous ripple nip is formed.
73. 전달 장치로부터의 유리체내 전달을 위한, 착물화제, 가용화제, 및 임의로 완충제를 포함하는 파조파닙 1HCl의 안정한 제약 제제이며, 여기서 디메틸 술폭시드 (DMSO)로부터의 동결건조에 의해 적어도 약 70%의 파조파닙 1HCl의 결정질 상 형태 A가 결정질 상 형태 G로 전환됨으로써 제제 중의 파조파닙 1HCl의 안정성이 증가하고; 희석될 때 및/또는 유리체 내로의 전달 중에 또는 전달 시에 적어도 50일 동안 침전되지 않는 안정한 제약 제제.73. A stable pharmaceutical formulation of PazopapanipHCl, comprising complexing agent, solubilizer, and optionally buffer, for delivery into the vitreous body from a delivery device, wherein it is freeze-dried from lyophilization from dimethylsulfoxide (DMSO) The conversion of the crystalline phase A of the% ripafanib 1HCI to the crystalline phase G increases the stability of the ripple nip HCl in the formulation; And is not precipitated during dilution and / or during delivery into the vitreous or during delivery for at least 50 days.
74. 전달 장치로부터의 유리체내 전달을 위한, 착물화제, 가용화제, 및 임의로 완충제를 포함하는 파조파닙 1HCl의 안정한 제약 제제이며, 여기서 트리플루오로 에탄올 (TFE)로부터의 동결건조에 의해 파조파닙 1HCl의 결정질 상 형태 A가 부분적으로 또는 완전히 무정형 및/또는 미세결정질 상으로 전환됨으로써 제제 중의 파조파닙 1HCl의 안정성이 증가하고; 희석될 때 및/또는 유리체 내로의 전달 중에 또는 전달 시에 적어도 50일 동안 침전되지 않는 안정한 제약 제제.74. A stable pharmaceutical formulation of Pazopapanib 1HCl, comprising complexing agent, solubilizer, and optionally buffer, for delivery into the vitreous body from a delivery device, wherein freeze-drying from lyophilization with trifluoroethanol (TFE) The stability of the ripopanib 1HCl in the formulation is increased by converting the crystalline phase A of the nip HCl to a partially or fully amorphous and / or microcrystalline phase; And is not precipitated during dilution and / or during delivery into the vitreous or during delivery for at least 50 days.
75. 제73항 또는 제74항에 있어서, 약 30 내지 약 70 mg/mL의 파조파닙 1HCl이 전달 장치 내의 제제에서 안정한 것인 안정한 제약 제제.75. The stable pharmaceutical formulation of claim 73 or 74, wherein about 30 to about 70 mg / mL of PachopapnipHCl is stable in the formulation in the delivery device.
76. 제75항에 있어서, 착물화제가 시클로덱스트린이고, 가용화제가 제약상 허용되는 담체인 안정한 제약 제제.76. The stable pharmaceutical formulation of claim 75, wherein the complexing agent is cyclodextrin and the solubilizing agent is a pharmaceutically acceptable carrier.
77. 제76항에 있어서, 제약상 허용되는 담체가 폴리(비닐 피롤리돈) (PVP)인 안정한 제약 제제.77. The stable pharmaceutical formulation of claim 76, wherein the pharmaceutically acceptable carrier is poly (vinylpyrrolidone) (PVP).
78. 제73항 또는 제74항에 있어서, 완충제가 히스티딘 HCl인 안정한 제약 제제.78. The stable pharmaceutical formulation of claim 73 or 74, wherein the buffer is histidine HCl.
79. 제73항 또는 제74항의 안정한 제약 제제의 제조 방법.79. The method of producing a stable pharmaceutical preparation of paragraph 73 or 74.
실시예Example
하기 실시예는 본 개시내용의 제제의 제조 방법 및 유리체내 전달 시에 유리체에서의 그의 특징을 평가하는 방법을 제공한다.The following examples provide methods of making the preparations of this disclosure and methods of evaluating their properties in the vitreous body upon delivery into the body.
실시예Example 1 One
파조파닙Pazopanip2HCl2HCl 제제의 제조 방법 Manufacturing method of preparation
각각의 파조파닙 분자 당 2개의 HCl을 함유하는 API (화순 바이오테크놀로지 코포레이션(Hwasun Biotechnology Corp.) 제조; 마누스-액테바(Manus-Aktteva) 유통)를 60 mg/ml 이하의 약물 농도로 제제화하였다. 파조파닙을 캅티솔® 용액에 용해시키고 필요한 모든 부형제를 첨가한 후에, pH를 목적하는 값으로 조정하였다.An API (manufactured by Hwasun Biotechnology Corp., Manus-Aktteva circulation) containing two HCl per each ripple nip molecule was formulated at a drug concentration of 60 mg / ml or less . After the Pazopanib was dissolved in the Captisol solution and all the required excipients were added, the pH was adjusted to the desired value.
필요한 양의 캅티솔®, 산 및 작용제를 물에 용해시켜 제제를 제조하였다. 파조파닙 2HCl을 첨가하고 용해될 때까지 혼합하였다. 그 후에, 최종 pH에 도달할 때까지 수산화나트륨을 첨가하였다. 제제를 여과한 다음, PDS 이식물에 주입하여, 치료제 방출 시험을 수행하였다.The formulation was prepared by dissolving the required amount of Capsytol®, acid and agonist in water. Pazopapenib 2HCl was added and mixed until dissolved. Then, sodium hydroxide was added until the final pH was reached. The formulation was filtered and then injected into the PDS implant to perform a therapeutic release test.
실시예Example 2 2
파조파닙Pazopanip1HCl1HCl 제제의 제조 방법 - 동결건조 생략 Preparation method of preparation - Freeze-drying omission
필요한 캅티솔®의 대략 절반을 바이알에서 칭량하여 적절한 양의 물에 용해시켰다. PVP-10k (폴리비닐 피롤리돈, MW=10 kDa) 및 히스티딘 HCl을 첨가하고 용액을 혼합함으로써 (와동, 초음파 처리, 진탕) 용해시켰다. 파조파닙 API (헤테로 랩스 리미티드(Hetero Labs Limited) 제조)를 칭량한 다음, 캅티솔® 용액에 첨가하였다. 필요에 따라, 소량의 염산 (HCl)을 첨가하여 용액의 pH를 2 이하의 pH로 조정하고 유지하였다. 첨가제, 예컨대 트리아세틴 또는 글리세롤을 첨가하였다. 파조파닙이 완전히 용해될 때까지 37℃ 또는 실온에서 제제를 교반하고 진탕시켰다. 파조파닙의 용해는 수시간이 걸릴 수 있다. 그 후에, NaOH 또는 메글루민을 첨가하여 파조파닙-캅티솔® 용액의 pH를 pH 6-7로 조정하였다. 나머지 캅티솔®을 이어서 첨가하고, 37℃ 또는 실온에서의 제제의 진탕/와동에 의해 완전히 용해시켰다. pH를 확인하고, 0.2 ㎛ 필터를 사용하여 제제를 여과하기 전에, 필요에 따라 조정하였다. 제제를 실온에서 차광하여 저장하였다. 제제의 함량 및 순도를 HPLC 및 UV에 의해 시험하였다.Approximately half of the required CaPTISOL® was weighed in a vial and dissolved in the appropriate amount of water. PVP-10k (polyvinylpyrrolidone, MW = 10 kDa) and histidine HCl were added and the solution was mixed (vortex, sonication, shaking). Pazopap Nip API (manufactured by Hetero Labs Limited) was weighed, and then added to the capricol solution. If necessary, a small amount of hydrochloric acid (HCl) was added to adjust the pH of the solution to a pH of 2 or less. Additives such as triacetin or glycerol were added. The formulation was stirred and shaken at 37 [deg.] C or room temperature until the rippa nip was completely dissolved. Dissolution of pachopanip may take several hours. The pH of the Pazopanib-capricosol solution was then adjusted to pH 6-7 by addition of NaOH or meglumine. The remaining Captisol® was then added and completely dissolved by shaking / vortexing the formulation at 37 ° C or room temperature. The pH was checked and adjusted as necessary prior to filtration of the formulation using a 0.2 [mu] m filter. The formulation was shaded at room temperature and stored. The content and purity of the preparation were tested by HPLC and UV.
실시예Example 3 3
염기, 예를 들어 NaOH가 포함된 분산물 중에서 활성 제약 성분을 실온에서 약 30분 동안 교반 및/또는 진탕시켜, 치료제, 예를 들어 파조파닙 1HCl의 고 농도 제제를 제조하였다. 분산물의 조성은 예를 들어, 1 N NaOH 중 약 275 mg/mL였다.The active pharmaceutical ingredient is stirred and / or shaken at room temperature for about 30 minutes in a dispersion containing a base, for example NaOH, to prepare a high concentration preparation of a therapeutic agent, for example PazopapenHCI. The composition of the dispersion was, for example, about 275 mg / mL in 1 N NaOH.
상기 방법에서, 필요한 양의 시클로덱스트린, 산 및 작용제를 물에 용해시켜 제제를 제조하였다. NaOH 처리된 파조파닙 1HCl을 첨가하여 용해될 때까지 혼합하였다. 그 후에, pH 6-7에 도달할 때까지 수산화나트륨을 첨가하였다. 제제를 여과한 다음, PDS 이식물에 주입하여 치료제 방출 시험을 수행하였다.In this method, formulations were prepared by dissolving the required amount of cyclodextrin, acid and agonist in water. The NaOH-treated Pazopapanib HCI was added and mixed until dissolved. Then, sodium hydroxide was added until a pH of 6-7 was reached. The formulation was filtered and then injected into a PDS implant to perform a therapeutic release test.
2HCl 및 1HCl 파조파닙 형태 둘 모두에서 유사한 약물 고 농도가 달성되었지만, 1HCl 파조파닙 제제는 매우 불안정하였다. 1HCl 파조파닙은 그대로 보존한 제제 (즉, 저장 중에) 및 희석된 제제 (즉, 약물 방출 중에)의 두 경우 모두에서 용이하게 결정화되었다.Similar drug high concentrations were achieved in both the 2HCl and 1HCl wave pan nip types, but the 1HCl wave pan nip formulations were very unstable. The 1HCl wave pan nip was easily crystallized in both the preserved formulation (i. E. During storage) and the diluted formulation (i. E. During drug release).
1HCl 파조파닙 제제의 안정성은 약물 농도를 40 mg/mL 미만으로 낮춤으로써 개선되었다. 표 5를 참조한다.The stability of 1HCl wave-nip preparations was improved by lowering the drug concentration to less than 40 mg / mL. See Table 5.
실시예Example 4 4
안정성의 개선 뿐만 아니라, 보다 용이한 제제화 방법을 위해, 1종 이상의 제제화 작용제에서의 가용화 전에, 파조파닙 1HCl의 동결건조를 수행하였다. 동결건조는 트리플루오로 에탄올 (TFE), 트리플루오로 에탄올-물 (90-10) 혼합물 또는 디메틸 술폭시드 (DMSO)로부터 수행되었다. 동결건조는 고 결정질 약물을, 보다 유리한 용해도 특성을 갖는 거의 무정형 고체로 전환시키는 것으로 생각된다. XRPD 분석을 수행하여 2HCl, 1HCl 및 1HCl 동결건조 약물 생성물의 결정질 구조를 비교하였고; 그 결과가 표 3에 나타나 있다. 동결건조 방법은 표 4에 요약되어 있다.For ease of formulation as well as improvement of stability, lyophilization of Pazopanib 1HCI was performed prior to solubilization in one or more formulation agents. Lyophilization was carried out from trifluoroethanol (TFE), a mixture of trifluoroethanol-water (90-10) or dimethylsulfoxide (DMSO). It is believed that lyophilization converts the high crystalline drug into an almost amorphous solid with more favorable solubility characteristics. XRPD analysis was performed to compare the crystalline structures of the 2HCl, < 1 > HCl and lHCl lyophilized drug products; The results are shown in Table 3. The freeze-drying method is summarized in Table 4.
파조파닙Pazopanip1HCl1HCl 제제의 제조 방법 - Method of preparing the preparation -DMSODMSO 동결건조 Freeze-dried
DMSO로부터의 동결건조: DMSO (디메틸 술폭시드) 중의 약 20-60 mg/mL의 파조파닙 1HCl 용액을 제조하였다. 그 후에, 용액을 관련 기술분야에 널리 공지된 조건 하에 동결-건조시켰다. 용액을 35℃ - 50℃ (예를 들어, 약 40℃) 하에 약 12시간 내지 약 24시간 동안, 또한 약 50℃ - 65℃ (예를 들어, 약 60℃)에서 약 24시간 내지 약 40시간 동안, 또한 약 90℃ - 110℃ (예를 들어, 약 100℃)에서 약 0.5시간 내지 약 2시간 동안 건조시켰다.Lyophilization from DMSO: Approximately 20-60 mg / mL Pazopapenip HCl solution in DMSO (dimethylsulfoxide) was prepared. The solution was then freeze-dried under conditions well known in the relevant art. The solution is heated at a temperature of from 35 ° C to 50 ° C (eg, about 40 ° C) for about 12 hours to about 24 hours, and from about 50 ° C to about 65 ° C (for example, about 60 ° C) , And also from about 90 ° C to 110 ° C (for example, about 100 ° C) for about 0.5 hours to about 2 hours.
파조파닙Pazopanip1HCl1HCl 제제의 제조 방법 - Method of preparing the preparation -TFETFE 동결건조 Freeze-dried
트리플루오로 에탄올 중의 약 60 mg/mL의 파조파닙 1HCl 결정질 형태를 제조하였다. 약 1% 내지 약 30%의 물 (예를 들어, 약 20%의 물)을 또한 트리플루오로 에탄올 중의 치료제 용액에 첨가하였다. 그 후에, 용액을 관련 기술분야의 표준 조건 하에 동결 건조시켰다 (물의 첨가와 함께 또는 물의 첨가 없이). 용액을 35℃ - 50℃ (예를 들어, 약 40℃) 하에 약 12시간 내지 약 24시간 동안 또는 약 50℃ - 65℃ (예를 들어, 약 60℃)에서 약 4 내지 약 8시간 동안 건조시켰다.Approximately 60 mg / mL of Pazopapanil 1HCl crystalline form in trifluoroethanol was prepared. About 1% to about 30% water (e.g., about 20% water) was also added to the therapeutic solution in trifluoroethanol. The solution was then lyophilized (with or without the addition of water) under standard conditions in the relevant art. The solution is dried for from about 12 hours to about 24 hours or from about 50 ° C to about 65 ° C (eg, about 60 ° C) for about 4 hours to about 8 hours at 35 ° C to 50 ° C .
DMSO 및 TFE 동결건조된 파조파닙 1HCl 둘 모두에 대하여 1-단계 또는 2-단계 제제화 방법을 사용하여 제제를 제조하였다.The formulation was prepared using either a one-step or two-step formulation method for both DMSO and TFE lyophilized rapamycin N HCI.
1-단계: 천연 pH가 사용될 때, 즉 점성 용액의 pH 조정이 필요하지 않을 때, 제약 성분의 가용화를 하나의 단계로 수행하였다. PVP-10k (폴리비닐 피롤리돈, MW=10 kDa) 및 히스티딘 HCl을 칭량하여 용액을 혼합함으로써 (와동, 진탕) 적절한 양의 물에 용해시켰다. 캅티솔®을 칭량하여 첨가하고, 용액의 진탕, 와동으로 용액에 용해시켰다. 동결건조된 파조파닙을 칭량한 다음 캅티솔® 용액에 첨가하고, 주위 온도 또는 승온 (예를 들어, 약 37℃ - 50℃)에서 와동, 초음파 처리, 진탕에 의해 완전히 용해시킨다. 제제를 0.2 ㎛ 필터를 사용하여 여과하고, 실온에서 차광하여 저장하였다.Step 1: Solubilization of the pharmaceutical ingredients was carried out in one step when natural pH was used, i.e. when pH adjustment of the viscous solution was not necessary. PVP-10k (polyvinylpyrrolidone, MW = 10 kDa) and histidine HCl were weighed and dissolved in the appropriate amount of water by mixing the solution (vortex, shaking). Capsol® was weighed and added, and dissolved in the solution by shaking and vortexing the solution. The lyophilized wave pan nip is weighed and then added to the Captisol® solution and completely dissolved by ambient, sonication, shaking at ambient or elevated temperature (eg, about 37 ° C. to 50 ° C.). The formulation was filtered using a 0.2 [mu] m filter, shaded at room temperature and stored.
또 다른 방법에서, 모든 고체 부형제 (캅티솔®, PVP 및 히스티딘-HCl) 및 치료제 (파조파닙 1HCl)를 측정하여 먼저 바이알에서 함께 혼합하였다. 연속적 혼합을 사용하여, 필요한 물의 점진적인 첨가를 수행하였다. 형성된 분산물의 가용화는 주위 온도에서 또는 승온에서 (예를 들어, 약 37℃ - 50℃) 수행될 수 있고; 승온의 사용은 균질 용액을 달성하는데 필요한 시간을 단축시킬 수 있다 (예를 들어, 약 24시간 - 4시간). 제제는 그대로 (천연 pH 3 - 4) 또는 NaOH 용액에 의한 pH 조정 (pH 6 - 7) 후에 사용될 수 있다.In another method, all of the solid excipients (Capsytol®, PVP and histidine-HCl) and the therapeutic agent (Pazopanib® HCl) were measured and mixed together first in the vial. Using continuous mixing, gradual addition of the required water was carried out. Solubilization of the formed dispersion can be performed at ambient temperature or at elevated temperature (e.g., from about 37 ° C to 50 ° C); The use of elevated temperatures can shorten the time required to achieve a homogenous solution (e.g., about 24 hours to 4 hours). The preparation can be used as is (natural pH 3-4) or after pH adjustment (pH 6-7) with NaOH solution.
동결건조된 치료제를 사용하는 2-단계 제제화 방법: 필요한 캅티솔®의 대략 절반을 바이알에서 칭량하여 적절한 양의 물에 용해시켰다. PVP-10k (폴리비닐 피롤리돈, MW=10 kDa) 및 히스티딘 HCl을 첨가하여 용액을 혼합함으로써 (와동, 초음파 처리, 진탕) 용해시켰다. 동결건조된 파조파닙 1HCl (TFE 또는 DMSO로부터의 동결건조)을 칭량한 다음 캅티솔® 용액에 첨가하였다. 필요에 따라, 소량의 염산 (HCl)을 첨가하여 용액의 pH를 2 이하의 pH로 조정하고 유지하였다. 첨가제, 예컨대 트리아세틴 또는 글리세롤을 첨가하였다. 파조파닙이 완전히 용해될 때까지, 제제를 37℃ 또는 실온에서 교반하고 진탕시켰다. 파조파닙의 용해는 수시간이 걸릴 수 있다. 그 후에, NaOH를 첨가하여 파조파닙-캅티솔® 용액의 pH를 pH 6-7로 조정하였다. 나머지 캅티솔®을 이어서 첨가하고, 37℃ 또는 실온에서의 제제의 진탕/와동에 의해 완전히 용해시켰다. pH를 확인하고, 0.2 ㎛ 필터를 사용하여 제제를 여과하기 전에, 필요에 따라 조정하였다. 제제를 실온에서 차광하여 저장하였다. 제제의 함량 및 순도를 HPLC 및 UV에 의해 시험하였다.Two-step formulation method using lyophilized therapeutic agent: Approximately half of the required Capsicol® is weighed in a vial and dissolved in the appropriate amount of water. (Vortex, sonication, shaking) by adding PVP-10k (polyvinylpyrrolidone, MW = 10 kDa) and histidine HCl. The lyophilized wave pan nip 1 HCl (lyophilized from TFE or DMSO) was weighed and then added to the Capsytol® solution. If necessary, a small amount of hydrochloric acid (HCl) was added to adjust the pH of the solution to a pH of 2 or less. Additives such as triacetin or glycerol were added. The formulation was stirred and shaken at 37 [deg.] C or room temperature until the rippa nip was completely dissolved. Dissolution of pachopanip may take several hours. The pH of the Pazopanib-capricosol solution was then adjusted to pH 6-7 by addition of NaOH. The remaining Captisol® was then added and completely dissolved by shaking / vortexing the formulation at 37 ° C or room temperature. The pH was checked and adjusted as necessary prior to filtration of the formulation using a 0.2 [mu] m filter. The formulation was shaded at room temperature and stored. The content and purity of the preparation were tested by HPLC and UV.
실시예Example 5 5
치료제 방출 시험은 인큐베이터에서 37℃로 유지된, 유리체를 대표하는 유체로 PDS에 의해 방출된 치료제의 양을 측정함으로써 수행하였다. PDS는 포스페이트 완충 식염수를 함유하는 용기에 부유되어 있었다. 주기적으로, PDS를 새로운 용기로 옮기고 치료제의 농도를 이전 용기의 유체에서 측정하였다. 속도는 방출된 치료제의 양을 샘플 수집 기간으로 나누어 계산하였다. 누적 방출 %는 치료 장치 (PDS)에 초기에 충전된 치료제의 양으로 나눈 치료제의 누적 양으로부터 계산하였다. 반감기는 4주째의 누적 방출 %로부터 계산하였다.The therapeutic agent release test was performed by measuring the amount of therapeutic agent released by the PDS as a fluid representative of the vitreous, maintained at 37 [deg.] C in an incubator. The PDS was suspended in a container containing phosphate buffered saline. Periodically, the PDS was transferred to a new vessel and the concentration of the therapeutic agent was measured in the fluid of the previous vessel. The rate was calculated by dividing the amount of therapeutic released by the sample collection period. The cumulative release rate was calculated from the cumulative amount of therapeutic agent divided by the amount of therapeutic agent initially charged to the treatment device (PDS). The half-life was calculated from the cumulative% release at 4 weeks.
치료제 방출을 캅티솔®과 함께 제제화된 파조파닙 1HCl 또는 2HCl로 수행하였다. 제제를 23 μL의 저장소 부피를 갖는 치료 장치 (PDS)에 충전하였다. 파조파닙 샘플의 클로라이드 함량 비교가 표 2에 나타나 있다.XRD 결과는 표 3에 나타나 있다.Therapeutic release was performed with Papafanib® HCl or 2HCl formulated with Capsytol®. The formulation was filled into a Therapeutic Apparatus (PDS) with a storage volume of 23 μL. A comparison of the chloride content of the Pazopanib samples is shown in Table 2. The XRD results are shown in Table 3.
약물 방출 비교: 37℃에서 수용 유체 (PBS 완충액)로 PDS에 의해 방출된 치료제의 양을 측정함으로써 치료제 방출 속도를 시험하였다. 치료제 방출 시험은 인큐베이터에서 37℃로 유지된, 유리체를 대표하는 유체로 PDS에 의해 방출된 치료제의 양을 측정함으로써 수행하였다. PDS는 포스페이트 완충 식염수를 함유하는 용기에 부유되어 있었다. 주기적으로, PDS를 새로운 용기로 옮기고 치료제의 농도를 이전 용기의 유체에서 측정하였다. 속도는 방출된 치료제의 양을 샘플 수집 기간으로 나누어 계산하였다. 누적 방출 %는 치료 장치 (PDS)에 초기에 충전된 치료제의 양으로 나눈 치료제의 누적 양으로부터 계산하였다. 반감기는 4주째의 누적 방출 %로부터 계산하였다. 결과가 도 1에 도시되어 있으며 하기에 요약되었다.Drug release comparison: Therapeutic release rate was tested by measuring the amount of therapeutic released by PDS with aqueous fluid (PBS buffer) at 37 ° C. The therapeutic agent release test was performed by measuring the amount of therapeutic agent released by the PDS as a fluid representative of the vitreous, maintained at 37 [deg.] C in an incubator. The PDS was suspended in a container containing phosphate buffered saline. Periodically, the PDS was transferred to a new vessel and the concentration of the therapeutic agent was measured in the fluid of the previous vessel. The rate was calculated by dividing the amount of therapeutic released by the sample collection period. The cumulative release rate was calculated from the cumulative amount of therapeutic agent divided by the amount of therapeutic agent initially charged to the treatment device (PDS). The half-life was calculated from the cumulative% release at 4 weeks. The results are shown in FIG. 1 and are summarized below.
1. 캅티솔® 중의 파조파닙-2HCl (샘플-1) (PA-96):1. Pazopanib-2 HCl (Sample-1) (PA-96) in Captisol®:
*제제는 pH 6.5의 60.0 mg/mL의 파조파닙, 2.2:1의 캅티솔®, 1%의 PVP, 6 mg/ml의 히스티딘 HCl임*The formulation is 60.0 mg / mL of Pazopanib pH 6.5, 2.2: 1 of Capsol®, 1% of PVP, 6 mg / ml of histidine HCl
*반감기 = 53일;*Half-life = 53 days;
2. 캅티솔® 중의 파조파닙-1HCl (샘플-2) (PA-110):2. Pazopanib-1 HCl (Sample-2) (PA-110) in Captisol®:
*제제는 pH 6.5의 60.0 mg/mL의 파조파닙, 2.2:1의 캅티솔®, 1%의 PVP, 6 mg/ml의 히스티딘 HCl임*The formulation is 60.0 mg / mL of Pazopanib pH 6.5, 2.2: 1 of Capsol®, 1% of PVP, 6 mg / ml of histidine HCl
*반감기 = 99일; 방출 중에 가시적인 약물 침전*Half-life = 99 days; Visible drug deposition during release
3. 파조파닙-1HCl-TFE 동결건조 (PAL-18)3. Pazopanib-1 HCl-TFE lyophilization (PAL-18)
*제제는 pH 6.5의 36 mg/mL의 파조파닙, 4:1의 캅티솔®, 1%의 PVP, 25 mg/ml의 히스티딘 HCl임*The formulation is 36 mg / mL prazopanib at pH 6.5, 4: 1 capricol, 1% PVP, 25 mg / ml histidine HCl
*반감기 = 45일*Half-life = 45 days
4. 파조파닙-1HCl-DMSO 동결건조 (PAD-5)4. Pazopanib-1 HCl-DMSO lyophilization (PAD-5)
*제제는 pH 3.4의 50 mg/mL의 파조파닙, 3:1의 캅티솔®, 1%의 PVP, 6 mg/ml의 히스티딘 HCl임*The formulation is 50 mg / mL of Pazopapin at pH 3.4, 3: 1 of Capsysol®, 1% of PVP, 6 mg / ml of histidine HCl
*반감기 = 45일*Half-life = 45 days
실시예Example 6 6
침전 시험 - 비교용 결과: 본 시험은 약물 방출 시의, 즉 소량의 제제가 다량의 완충 용액으로 방출될 때의 조건을 모델링하기 위한 목적으로 수행되었다. 이 모델에서, 약물이 희석 (방출) 시에 침전된다면, 이는 장치의 폐색 및/또는 수용 유체 중의 고체 약물이 측정가능하지 않을 것이므로 (또한 아마도 생체내 조건 하에 방출될 때는 사용가능하지 않을 것임) 약물의 손실을 초래할 수 있다. 시험을 수행하기 위해, 제제를 포스페이트 완충 식염수 용액 (약 0.1% 아지드화나트륨)으로 330배 희석시키는데, 예를 들어 3 μL의 제제를 1 mL의 PBS 완충액에 첨가하였다. 용액을 37℃ 온도조절기에서 유지하고, 결정 성장/침전의 출현에 대하여 주기적으로 확인하였다.상이한 약물 공급원으로 제조된 제제는 표 5에 요약되어 있는 바와 같이, 희석 시에 침전에 대하여 상이한 안정성을 나타냈다.Precipitation Test - Comparative Results: This test was performed for the purpose of modeling the conditions at the time of drug release, that is, when a small amount of the preparation is released into a large amount of buffer solution. In this model, if the drug is precipitated at the time of dilution (release), it is possible that the obstruction of the device and / or the solid drug in the receiving fluid will not be measurable (and probably will not be available when released under in vivo conditions) Which may result in loss of < / RTI > To perform the test, the formulation is diluted 330 fold with phosphate buffered saline (about 0.1% sodium azide), for example 3 μL of the formulation is added to 1 mL of PBS buffer. The solution was maintained in a 37 ° C thermostat and periodically checked for the appearance of crystal growth / precipitation. Preparations prepared with different drug sources exhibited different stability against precipitation during dilution, as summarized in Table 5.
참고문헌 포함Include references
본원에서 언급된 특허 문헌 및 과학 논문 각각의 전체 개시내용은 모든 목적을 위해 참조로 포함된다. 본 개시내용에서 호스트 문헌은 충분한 특이성으로 확인되고 본 개시내용과 관련된 물질은 참고문헌의 문맥에 기반하여 해석된다. 공보 및 특허 문헌의 인용은 어떤 관련 선행기술을 인정하려는 것이 아니며, 그의 내용 또는 날짜에 대해서도 인정하는 것으로 간주되지 않는다. 본 발명이 여기서 명세서에 의해 기재되었으므로, 관련 기술분야의 통상의 기술자라면 본 발명이 다양한 실시양태로 실시될 수 있음을 알 것이고, 상기의 상세한 설명 및 실시예는 하기 청구범위를 제한하는 것이 아니라 예시 목적을 갖는다.The entire disclosure of each of the patent and scientific articles referred to herein is incorporated by reference for all purposes. In this disclosure, the host literature is identified with sufficient specificity and the materials associated with this disclosure are interpreted based on the context of the reference. The citation of publications and patent literature is not intended to acknowledge any relevant prior art and is not to be construed as acknowledging its content or date. It will be appreciated by those skilled in the relevant art that the present invention may be practiced in various embodiments and that the detailed description and examples are not intended to limit the scope of the following claims, Purpose.
다른 실시양태Other embodiments
본 발명이 그의 상세한 설명과 함께 기재되었지만, 상기의 상세한 설명은 첨부된 청구범위의 범주에 의해 한정되는, 본 발명의 범주를 제한하는 것이 아닌 예시하기 위한 것이다. 다른 측면, 장점 및 수정도 하기 청구범위의 범주 내에 포함된다.While the invention has been described in conjunction with the detailed description thereof, the foregoing description is intended to illustrate and not limit the scope of the invention, which is defined by the scope of the appended claims. Other aspects, advantages and modifications are also within the scope of the following claims.
Claims (38)
Translated fromKorean여기서 제약상 허용되는 염은 1가 또는 2가 염이고, 1종 이상의 제제화 작용제는 착물화제, 가용화제 및 완충제를 포함하고; 치료제의 염이 제제에 용해되어 있는 것인, 후안부의 안과 질환 또는 장애를 치료 및/또는 완화시키는 방법.Comprising delivering a pharmaceutically acceptable salt of a therapeutic agent having a low water solubility and a stable pharmaceutical formulation of one or more formulation agents from a delivery system comprising a storage chamber coupled to the porous structure, Controlled release from the reservoir through the porous structure of the formulation increases the half-life of the therapeutic agent in the vitreous;
Wherein the pharmaceutically acceptable salt is a monovalent or divalent salt, wherein the at least one formulation agent comprises a complexing agent, a solubilizing agent and a buffering agent; Wherein the salt of the therapeutic agent is dissolved in the formulation.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462035274P | 2014-08-08 | 2014-08-08 | |
| US62/035,274 | 2014-08-08 | ||
| PCT/US2015/043921WO2016022750A1 (en) | 2014-08-08 | 2015-08-06 | Stable and soluble formulations of receptor tyrosine kinase inhibitors, and methods of preparation thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20170040798Atrue KR20170040798A (en) | 2017-04-13 |
Family
ID=55264539
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020177006057ACeasedKR20170040798A (en) | 2014-08-08 | 2015-08-06 | Stable and soluble formulations of receptor tyrosine kinase inhibitors, and methods of preparation thereof |
Country Status (14)
| Country | Link |
|---|---|
| US (4) | US9474756B2 (en) |
| EP (1) | EP3177289A4 (en) |
| JP (2) | JP2017524034A (en) |
| KR (1) | KR20170040798A (en) |
| CN (1) | CN107106551A (en) |
| AU (1) | AU2015301054B2 (en) |
| BR (1) | BR112017002466A2 (en) |
| CA (1) | CA2957548A1 (en) |
| IL (1) | IL250447A0 (en) |
| MX (1) | MX2017001818A (en) |
| MY (1) | MY182793A (en) |
| RU (1) | RU2017105844A (en) |
| SG (1) | SG11201700943TA (en) |
| WO (1) | WO2016022750A1 (en) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104887389B (en) | 2009-01-29 | 2017-06-23 | 弗赛特影像4股份有限公司 | Posterior segment drug delivery |
| ES2972168T3 (en) | 2013-03-28 | 2024-06-11 | Forsight Vision4 Inc | Ophthalmic implant for administration of therapeutic substances |
| ES2803102T3 (en) | 2014-07-15 | 2021-01-22 | Forsight Vision4 Inc | Eye implant delivery device |
| WO2016022750A1 (en) | 2014-08-08 | 2016-02-11 | Forsight Vision4, Inc. | Stable and soluble formulations of receptor tyrosine kinase inhibitors, and methods of preparation thereof |
| WO2016077371A1 (en)* | 2014-11-10 | 2016-05-19 | Forsight Vision4, Inc. | Expandable drug delivery devices and method of use |
| US11432959B2 (en) | 2015-11-20 | 2022-09-06 | Forsight Vision4, Inc. | Porous structures for extended release drug delivery devices |
| MX2018012021A (en) | 2016-04-05 | 2019-01-24 | Forsight Vision4 Inc | Implantable ocular drug delivery devices. |
| TW201806928A (en)* | 2016-07-01 | 2018-03-01 | 第一三共股份有限公司 | Crystals of aminocarboxylic acid acid addition salts and the synthetic method thereof |
| US11083728B2 (en) | 2017-04-07 | 2021-08-10 | Syros Pharmaceuticals, Inc. | Compositions of cyclin dependent kinase 7 (CDK7) inhibitor |
| AU2018372806B2 (en) | 2017-11-21 | 2024-05-30 | Forsight Vision4, Inc. | Fluid exchange apparatus for expandable port delivery system and methods of use |
| ES2914305T3 (en)* | 2017-12-26 | 2022-06-09 | Ind Tech Res Inst | Composition to improve the solubility of poorly soluble substances, use thereof and complex formulation containing the same |
| CN117881411A (en) | 2021-06-01 | 2024-04-12 | 艾迪雅生物有限责任公司 | Extended release drug delivery systems for ocular drugs and methods of use |
| BR112023025185A2 (en)* | 2021-06-01 | 2024-02-27 | Eyedea Bio Llc | PROLONGED RELEASE DRUG DELIVERY SYSTEM FOR OCULAR DRUGS AND METHODS OF USE |
| USD1033637S1 (en) | 2022-01-24 | 2024-07-02 | Forsight Vision4, Inc. | Fluid exchange device |
| JP2025519987A (en)* | 2023-04-28 | 2025-07-01 | 維眸生物科技(浙江)有限公司 | Ophthalmic Compositions for Treating Non-Infectious Inflammatory Diseases - Patent application |
Family Cites Families (447)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2585815A (en) | 1947-01-16 | 1952-02-12 | Mclintock Duncan Menzies | Injection syringe |
| US2564977A (en) | 1949-01-19 | 1951-08-21 | Hu Quang Hsi | Medical injecting apparatus |
| US2886497A (en) | 1957-04-12 | 1959-05-12 | United States Steel Corp | Method for determining the permeability of steel to hydrogen |
| US3232117A (en) | 1962-09-14 | 1966-02-01 | Roger Gilmont Instr Inc | Micrometer buret |
| US3416530A (en) | 1966-03-02 | 1968-12-17 | Richard A. Ness | Eyeball medication dispensing tablet |
| US3618604A (en) | 1969-06-09 | 1971-11-09 | Alza Corp | Ocular insert |
| US3641237A (en) | 1970-09-30 | 1972-02-08 | Nat Patent Dev Corp | Zero order release constant elution rate drug dosage |
| US4034756A (en) | 1971-01-13 | 1977-07-12 | Alza Corporation | Osmotically driven fluid dispenser |
| US3831583A (en) | 1971-03-05 | 1974-08-27 | Univ California | Implantable bulb for inflation of surgical implements |
| US3995635A (en) | 1971-09-09 | 1976-12-07 | Alza Corporation | Ocular insert |
| US3828777A (en) | 1971-11-08 | 1974-08-13 | Alza Corp | Microporous ocular device |
| US3845201A (en) | 1972-04-24 | 1974-10-29 | S Loucas | Solid state ophthalmic medication delivery method |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US3914402A (en) | 1973-06-14 | 1975-10-21 | Alza Corp | Ophthalmic dosage form, for releasing medication over time |
| US4179497A (en) | 1973-12-17 | 1979-12-18 | Merck & Co., Inc. | Solid state ophthalmic medication |
| US3902495A (en) | 1974-01-28 | 1975-09-02 | Cavitron Corp | Flow control system |
| US3961628A (en) | 1974-04-10 | 1976-06-08 | Alza Corporation | Ocular drug dispensing system |
| US3949748A (en) | 1974-09-26 | 1976-04-13 | Oscar Malmin | Injection syringe having aspirating and metering capabilities |
| US3949750A (en) | 1974-10-07 | 1976-04-13 | Freeman Jerre M | Punctum plug and method for treating keratoconjunctivitis sicca (dry eye) and other ophthalmic aliments using same |
| US3926188A (en) | 1974-11-14 | 1975-12-16 | Alza Corp | Laminated drug dispenser |
| US4135514A (en) | 1974-12-23 | 1979-01-23 | Alza Corporation | Osmotic releasing system for administering ophthalmic drug to eye of animal |
| US4014335A (en) | 1975-04-21 | 1977-03-29 | Alza Corporation | Ocular drug delivery device |
| NL188266C (en) | 1975-07-29 | 1992-05-18 | Merck & Co Inc | PROCESS FOR THE PREPARATION OF AN ORGANIC IMPLANT. |
| US3977404A (en) | 1975-09-08 | 1976-08-31 | Alza Corporation | Osmotic device having microporous reservoir |
| US4034758A (en) | 1975-09-08 | 1977-07-12 | Alza Corporation | Osmotic therapeutic system for administering medicament |
| US4014333A (en) | 1975-09-22 | 1977-03-29 | Mcintyre David J | Instrument for aspirating and irrigating during ophthalmic surgery |
| US4077407A (en) | 1975-11-24 | 1978-03-07 | Alza Corporation | Osmotic devices having composite walls |
| US4014334A (en) | 1976-02-02 | 1977-03-29 | Alza Corporation | Laminated osmotic system for dispensing beneficial agent |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| US4111203A (en) | 1976-11-22 | 1978-09-05 | Alza Corporation | Osmotic system with means for improving delivery kinetics of system |
| US4111201A (en) | 1976-11-22 | 1978-09-05 | Alza Corporation | Osmotic system for delivering selected beneficial agents having varying degrees of solubility |
| US4160452A (en) | 1977-04-07 | 1979-07-10 | Alza Corporation | Osmotic system having laminated wall comprising semipermeable lamina and microporous lamina |
| US4256108A (en) | 1977-04-07 | 1981-03-17 | Alza Corporation | Microporous-semipermeable laminated osmotic system |
| US4164559A (en) | 1977-09-21 | 1979-08-14 | Cornell Research Foundation, Inc. | Collagen drug delivery device |
| US4186184A (en) | 1977-12-27 | 1980-01-29 | Alza Corporation | Selective administration of drug with ocular therapeutic system |
| US4220153A (en) | 1978-05-08 | 1980-09-02 | Pfizer Inc. | Controlled release delivery system |
| US4220152A (en) | 1978-05-08 | 1980-09-02 | Pfizer Inc. | Delivery system |
| US4200098A (en) | 1978-10-23 | 1980-04-29 | Alza Corporation | Osmotic system with distribution zone for dispensing beneficial agent |
| US4298000A (en) | 1978-11-08 | 1981-11-03 | Minnesota Mining And Manufacturing Company | Fluid dispensing device |
| US4300557A (en) | 1980-01-07 | 1981-11-17 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Method for treating intraocular malignancies |
| EP0033042B1 (en) | 1980-01-28 | 1984-08-22 | Merck & Co. Inc. | Ophthalmic inserts for lowering intraocular pressure comprising carbonic anhydrase inhibitors |
| US4309776A (en) | 1980-05-13 | 1982-01-12 | Ramon Berguer | Intravascular implantation device and method of using the same |
| US4326525A (en) | 1980-10-14 | 1982-04-27 | Alza Corporation | Osmotic device that improves delivery properties of agent in situ |
| US4327725A (en) | 1980-11-25 | 1982-05-04 | Alza Corporation | Osmotic device with hydrogel driving member |
| US4484922A (en) | 1981-06-25 | 1984-11-27 | Rosenwald Peter L | Occular device |
| US4439198A (en) | 1981-07-09 | 1984-03-27 | University Of Illinois Foundation | Biodegradable ocular insert for controlled delivery of ophthalmic medication |
| US4730013A (en) | 1981-10-08 | 1988-03-08 | Merck & Co., Inc. | Biosoluble ocular insert |
| US4475916A (en) | 1982-03-18 | 1984-10-09 | Merck & Co., Inc. | Osmotic drug delivery system |
| US4439196A (en) | 1982-03-18 | 1984-03-27 | Merck & Co., Inc. | Osmotic drug delivery system |
| US4519801A (en) | 1982-07-12 | 1985-05-28 | Alza Corporation | Osmotic device with wall comprising cellulose ether and permeability enhancer |
| US4673405A (en) | 1983-03-04 | 1987-06-16 | Alza Corporation | Osmotic system with instant drug availability |
| US4883459A (en) | 1983-07-29 | 1989-11-28 | Reynaldo Calderon | Retrograde perfusion |
| US4774091A (en) | 1983-10-14 | 1988-09-27 | Sumitomo Pharmaceuticals Company, Ltd. | Long-term sustained-release preparation |
| US4627850A (en) | 1983-11-02 | 1986-12-09 | Alza Corporation | Osmotic capsule |
| US4777049A (en) | 1983-12-01 | 1988-10-11 | Alza Corporation | Constant release system with pulsed release |
| US4634418A (en) | 1984-04-06 | 1987-01-06 | Binder Perry S | Hydrogel seton |
| US4851228A (en) | 1984-06-20 | 1989-07-25 | Merck & Co., Inc. | Multiparticulate controlled porosity osmotic |
| US4634427A (en) | 1984-09-04 | 1987-01-06 | American Hospital Supply Company | Implantable demand medication delivery assembly |
| US5049142A (en) | 1984-11-07 | 1991-09-17 | Herrick Robert S | Intracanalicular implant for horizontal canalicular blockade treatment of the eye |
| US5053030A (en) | 1984-11-07 | 1991-10-01 | Herrick Robert S | Intracanalicular implant for horizontal canalicular blockade treatment of the eye |
| US4712550A (en) | 1985-04-08 | 1987-12-15 | Sinnett Kevin B | Retinal tack |
| US4693886A (en) | 1985-04-22 | 1987-09-15 | Alza Corporation | Osmotic device with inert core |
| US4609374A (en) | 1985-04-22 | 1986-09-02 | Alza Corporation | Osmotic device comprising means for governing initial time of agent release therefrom |
| EP0201611A1 (en) | 1985-05-10 | 1986-11-20 | B. Braun-SSC AG | Two cannulae syringe |
| US4781675A (en) | 1985-11-27 | 1988-11-01 | White Thomas C | Infusion cannula |
| EP0228185B1 (en) | 1985-11-27 | 1990-07-25 | Thomas C. White | Tissue-implantable fluid-dissipating device |
| US4959217A (en) | 1986-05-22 | 1990-09-25 | Syntex (U.S.A.) Inc. | Delayed/sustained release of macromolecules |
| US5147647A (en) | 1986-10-02 | 1992-09-15 | Sohrab Darougar | Ocular insert for the fornix |
| US5322691A (en) | 1986-10-02 | 1994-06-21 | Sohrab Darougar | Ocular insert with anchoring protrusions |
| US4863457A (en) | 1986-11-24 | 1989-09-05 | Lee David A | Drug delivery device |
| EP0498471B1 (en) | 1986-12-23 | 1996-08-14 | The Liposome Company, Inc. | Liposomes comprising a guanidino aminoglycoside |
| US4853229A (en) | 1987-10-26 | 1989-08-01 | Alza Corporation | Method for adminstering tiny pills |
| JP2702953B2 (en) | 1988-01-30 | 1998-01-26 | オリンパス光学工業株式会社 | Chemical impregnated ceramics |
| US4865846A (en) | 1988-06-03 | 1989-09-12 | Kaufman Herbert E | Drug delivery system |
| US5174999A (en) | 1988-12-13 | 1992-12-29 | Alza Corporation | Delivery system comprising fluid ingress and drug egress |
| WO1990007575A1 (en) | 1988-12-30 | 1990-07-12 | Anderson David M | Stabilized microporous materials and hydrogel materials |
| US5141748A (en) | 1989-02-17 | 1992-08-25 | Hoffmann-La Roche, Inc. | Implant drug delivery device |
| US5098443A (en) | 1989-03-23 | 1992-03-24 | University Of Miami | Method of implanting intraocular and intraorbital implantable devices for the controlled release of pharmacological agents |
| US4979938A (en) | 1989-05-11 | 1990-12-25 | Iomed, Inc. | Method of iontophoretically treating acne, furuncles and like skin disorders |
| US5164188A (en) | 1989-11-22 | 1992-11-17 | Visionex, Inc. | Biodegradable ocular implants |
| US5171270A (en) | 1990-03-29 | 1992-12-15 | Herrick Robert S | Canalicular implant having a collapsible flared section and method |
| US5324280A (en) | 1990-04-02 | 1994-06-28 | Alza Corporation | Osmotic dosage system for delivering a formulation comprising liquid carrier and drug |
| US5128145A (en) | 1990-06-13 | 1992-07-07 | Alza Corporation | Dosage form for Parkinson's disease, spasticity and muscle spasms |
| US5238687A (en) | 1990-07-11 | 1993-08-24 | Alza Corporation | Delivery device with a protective sleeve |
| US5084021A (en) | 1990-11-02 | 1992-01-28 | Baldwin Brian E | Patient controlled infusion apparatus and method |
| US5378475A (en) | 1991-02-21 | 1995-01-03 | University Of Kentucky Research Foundation | Sustained release drug delivery devices |
| AU650113B2 (en) | 1991-04-05 | 1994-06-09 | Eli Lilly And Company | Sustained release capsule and formulations |
| US5334189A (en) | 1991-06-03 | 1994-08-02 | Wade Stephen E | Device for controlled diffusion of a chemical substance |
| US5282829A (en) | 1991-08-15 | 1994-02-01 | United States Surgical Corporation | Hollow body implants |
| FR2682090B1 (en) | 1991-10-03 | 1993-12-31 | Holzstoff Holding Sa | RESERVOIR SYSTEM FOR EXTENDED BROADCASTING OF AN ACTIVE INGREDIENT. |
| US5681572A (en) | 1991-10-18 | 1997-10-28 | Seare, Jr.; William J. | Porous material product and process |
| US5178635A (en) | 1992-05-04 | 1993-01-12 | Allergan, Inc. | Method for determining amount of medication in an implantable device |
| US6096756A (en) | 1992-09-21 | 2000-08-01 | Albert Einstein College Of Medicine Of Yeshiva University | Method of simultaneously enhancing analgesic potency and attenuating dependence liability caused by morphine and other bimodally-acting opioid agonists |
| US5336175A (en) | 1992-10-29 | 1994-08-09 | Mames Robert N | Method for the treatment of retinal detachments |
| US5519030A (en) | 1993-10-07 | 1996-05-21 | Senju Pharmaceutical Co., Ltd. | Method for prophylaxis and treatment of myopia |
| US5443505A (en) | 1993-11-15 | 1995-08-22 | Oculex Pharmaceuticals, Inc. | Biocompatible ocular implants |
| CA2140053C (en) | 1994-02-09 | 2000-04-04 | Joel S. Rosenblatt | Collagen-based injectable drug delivery system and its use |
| US5770076A (en) | 1994-03-07 | 1998-06-23 | The Regents Of The University Of California | Micromachined capsules having porous membranes and bulk supports |
| US5985328A (en) | 1994-03-07 | 1999-11-16 | Regents Of The University Of California | Micromachined porous membranes with bulk support |
| US5578042A (en) | 1994-03-14 | 1996-11-26 | Cumming; J. Stuart | Ophthalmic kit and method for lens insertion |
| US5466233A (en) | 1994-04-25 | 1995-11-14 | Escalon Ophthalmics, Inc. | Tack for intraocular drug delivery and method for inserting and removing same |
| AUPM897594A0 (en) | 1994-10-25 | 1994-11-17 | Daratech Pty Ltd | Controlled release container |
| ATE232089T1 (en) | 1994-11-10 | 2003-02-15 | Univ Kentucky Res Found | CONTROLLED RELEASE IMPLANTABLE REFILLABLE DEVICE FOR ADMINISTERING DRUGS IMMEDIATELY TO AN INTERNAL PART OF THE BODY |
| US5725493A (en) | 1994-12-12 | 1998-03-10 | Avery; Robert Logan | Intravitreal medicine delivery |
| US5554132A (en) | 1995-03-30 | 1996-09-10 | Abbott Laboratories | Hand grip for use with syringe |
| IL113723A (en) | 1995-05-14 | 2002-11-10 | Optonol Ltd | Intraocular implant |
| US6685940B2 (en) | 1995-07-27 | 2004-02-03 | Genentech, Inc. | Protein formulation |
| US5773019A (en) | 1995-09-27 | 1998-06-30 | The University Of Kentucky Research Foundation | Implantable controlled release device to deliver drugs directly to an internal portion of the body |
| US6641708B1 (en) | 1996-01-31 | 2003-11-04 | Board Of Regents, The University Of Texas System | Method and apparatus for fractionation using conventional dielectrophoresis and field flow fractionation |
| GB2310149A (en) | 1996-02-15 | 1997-08-20 | Nomix Chipman Ltd | Spray gun |
| US20090005864A1 (en) | 1996-03-18 | 2009-01-01 | Eggleston Harry C | Modular intraocular implant |
| US5951512A (en) | 1996-05-28 | 1999-09-14 | Horizon Medical Products, Inc. | Infusion port with modified drug reservoir |
| US5797898A (en) | 1996-07-02 | 1998-08-25 | Massachusetts Institute Of Technology | Microchip drug delivery devices |
| US5928662A (en) | 1996-07-31 | 1999-07-27 | Phillips; Andrew F. | Ocular drug delivery device |
| AU7533696A (en) | 1996-12-13 | 1998-06-18 | Ciba-Geigy Ag | New materials |
| EP0973499B1 (en) | 1997-03-31 | 2003-08-06 | Alza Corporation | Diffusional implantable delivery system |
| US7160687B1 (en) | 1997-05-29 | 2007-01-09 | Cellomics, Inc. | Miniaturized cell array methods and apparatus for cell-based screening |
| US5968008A (en) | 1997-08-04 | 1999-10-19 | Grams; Guenter A. | Cannula with parallel channels and sliding sheath |
| US5902598A (en) | 1997-08-28 | 1999-05-11 | Control Delivery Systems, Inc. | Sustained release drug delivery devices |
| US6331523B1 (en) | 1998-03-12 | 2001-12-18 | Genentech, Inc. | Method of enhancing the survival of retinal neurons and treating ocular diseases using FGF-5 |
| US6196993B1 (en) | 1998-04-20 | 2001-03-06 | Eyelab Group, Llc | Ophthalmic insert and method for sustained release of medication to the eye |
| US20020198174A1 (en) | 2001-05-07 | 2002-12-26 | Allergan Sales, Inc. | Disinfecting and solubilizing steroid compositions |
| FR2784296B1 (en) | 1998-09-18 | 2001-01-05 | Imedex Biomateriaux | DEVICE FOR FORMULATING AND DELIVERING A MIXTURE, PARTICULARLY FOR THE SURGICAL APPLICATION OF THIS MIXTURE |
| GB9822170D0 (en)* | 1998-10-13 | 1998-12-02 | Danbioyst Uk Ltd | Novel formulations of fexofenadine |
| US7973068B2 (en) | 1998-10-20 | 2011-07-05 | Omeros Corporation | Arthroscopic irrigation solution and method for peripheral vasoconstriction and inhibition of pain and inflammation |
| WO2000035531A1 (en) | 1998-12-14 | 2000-06-22 | Tre Esse Progettazione Biomedica S.R.L. | Catheter system for performing intramyocardiac therapeutic treatment |
| ME00275B (en) | 1999-01-13 | 2011-02-10 | Bayer Corp | ?-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| WO2000042012A1 (en) | 1999-01-13 | 2000-07-20 | Bayer Corporation | φ-CARBOXYARYL SUBSTITUTED DIPHENYL UREAS AS RAF KINASE INHIBITORS |
| IL144144A0 (en) | 1999-01-13 | 2002-05-23 | Bayer Ag | Omega-carboxy aryl substituted diphenyl ureas as p38 kinase inhibitors |
| DE19948783C2 (en) | 1999-02-18 | 2001-06-13 | Alcove Surfaces Gmbh | Implant |
| US7914442B1 (en) | 1999-03-01 | 2011-03-29 | Gazdzinski Robert F | Endoscopic smart probe and method |
| US6395300B1 (en) | 1999-05-27 | 2002-05-28 | Acusphere, Inc. | Porous drug matrices and methods of manufacture thereof |
| US6472162B1 (en) | 1999-06-04 | 2002-10-29 | Thermogenesis Corp. | Method for preparing thrombin for use in a biological glue |
| DK1187617T3 (en) | 1999-06-18 | 2004-04-13 | Alcon Mfg Ltd | Method of selecting the concentration of an amphipathic antihistamine drug by determining the drug's surface activity target |
| US7141581B2 (en) | 1999-07-02 | 2006-11-28 | Agouron Pharmaceuticals, Inc. | Indazole compounds and pharmaceutical compositions for inhibiting protein kinases, and methods for their use |
| TWI262914B (en) | 1999-07-02 | 2006-10-01 | Agouron Pharma | Compounds and pharmaceutical compositions for inhibiting protein kinases |
| US6551291B1 (en) | 1999-08-04 | 2003-04-22 | Johns Hopkins University | Non-traumatic infusion cannula and treatment methods using same |
| US6638263B1 (en) | 1999-10-12 | 2003-10-28 | Durect Corporation | Regulation of drug delivery through flow diversion |
| US6416777B1 (en) | 1999-10-21 | 2002-07-09 | Alcon Universal Ltd. | Ophthalmic drug delivery device |
| TR200201047T2 (en) | 1999-10-21 | 2002-07-22 | Alcon, Inc. | Medication delivery means. |
| US6331313B1 (en) | 1999-10-22 | 2001-12-18 | Oculex Pharmaceticals, Inc. | Controlled-release biocompatible ocular drug delivery implant devices and methods |
| US20020010203A1 (en) | 1999-12-22 | 2002-01-24 | Ken Lipson | Methods of modulating c-kit tyrosine protein kinase function with indolinone compounds |
| US20030212383A1 (en) | 2001-01-05 | 2003-11-13 | Dana Cote | System and methods for reducing intraocular pressure |
| US20050119737A1 (en) | 2000-01-12 | 2005-06-02 | Bene Eric A. | Ocular implant and methods for making and using same |
| MXPA02006889A (en) | 2000-01-12 | 2003-05-23 | Becton Dickinson Co | Systems and methods for reducing intraocular pressure. |
| ES2290117T3 (en) | 2000-02-15 | 2008-02-16 | Sugen, Inc. | PROTEIN QUINASE 2-INDOLIN INHIBITORS REPLACED WITH PIRROL. |
| DE10008826C2 (en) | 2000-02-25 | 2002-03-14 | Disetronic Licensing Ag | Micro perfusion device with collection container |
| US7077848B1 (en) | 2000-03-11 | 2006-07-18 | John Hopkins University | Sutureless occular surgical methods and instruments for use in such methods |
| US7169444B2 (en) | 2000-03-13 | 2007-01-30 | Seiko Epson Corporation | Method for treating surface of ink jet recording medium having recorded image |
| US7141152B2 (en) | 2000-03-16 | 2006-11-28 | Le Febre David A | Analyte species separation system |
| US20040208910A1 (en) | 2000-04-26 | 2004-10-21 | Control Delivery Systems, Inc. | Sustained release device and method for ocular delivery of adrenergic agents |
| US6375972B1 (en) | 2000-04-26 | 2002-04-23 | Control Delivery Systems, Inc. | Sustained release drug delivery devices, methods of use, and methods of manufacturing thereof |
| DE60135352D1 (en) | 2000-08-30 | 2008-09-25 | Univ Johns Hopkins | DEVICE FOR INTRA-OCCULAR ACTIVE AGGREGATION |
| ATE380042T1 (en) | 2000-09-06 | 2007-12-15 | Alcon Inc | COATING AGENT WITH ADJUSTABLE ADHESIVENESS FOR EYE IMPLANTS |
| US6303290B1 (en) | 2000-09-13 | 2001-10-16 | The Trustees Of The University Of Pennsylvania | Encapsulation of biomaterials in porous glass-like matrices prepared via an aqueous colloidal sol-gel process |
| CA2432000C (en) | 2000-12-21 | 2011-03-15 | Glaxo Group Limited | Pyrimidineamines as angiogenesis modulators |
| WO2002056863A2 (en) | 2000-12-29 | 2002-07-25 | Bausch & Lomb Incorporated | Sustained release drug delivery devices |
| CA2432203C (en) | 2001-01-03 | 2008-03-25 | Michael J. Brubaker | Sustained release drug delivery devices with multiple agents |
| EP1347742A1 (en) | 2001-01-03 | 2003-10-01 | Bausch & Lomb Incorporated | Sustained release drug delivery devices with prefabricated permeable plugs |
| WO2002053130A2 (en) | 2001-01-03 | 2002-07-11 | Bausch & Lomb Incorporated | Sustained release drug delivery devices with coated drug cores |
| EP1372602B1 (en) | 2001-01-09 | 2007-04-18 | Microchips, Inc. | Flexible microchip devices for ophthalmic and other applications |
| US7235576B1 (en) | 2001-01-12 | 2007-06-26 | Bayer Pharmaceuticals Corporation | Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US6995162B2 (en) | 2001-01-12 | 2006-02-07 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| US6878714B2 (en) | 2001-01-12 | 2005-04-12 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| US6872198B1 (en) | 2001-01-24 | 2005-03-29 | Arrow International, Inc. | Double-y-shaped multi-lumen catheter with selectively attachable hubs |
| JP2004520900A (en) | 2001-01-26 | 2004-07-15 | ボシュ・アンド・ロム・インコーポレイテッド | Improved manufacturing method of sustained release drug delivery device |
| US7181287B2 (en) | 2001-02-13 | 2007-02-20 | Second Sight Medical Products, Inc. | Implantable drug delivery device |
| FR2821672B1 (en) | 2001-03-02 | 2003-05-02 | Scp Dosagene R & D | MEANS FOR THE EXTRACTION OF PRODUCTS TO BE ANALYZED AND THEIR APPLICATIONS IN DIAGNOSIS AND ANALYSIS |
| US6713081B2 (en) | 2001-03-15 | 2004-03-30 | The United States Of America As Represented By The Department Of Health And Human Services | Ocular therapeutic agent delivery devices and methods for making and using such devices |
| EP2316394B1 (en) | 2001-06-12 | 2016-11-23 | The Johns Hopkins University | Reservoir device for intraocular drug delivery |
| MXPA04000209A (en) | 2001-07-10 | 2005-03-07 | Teva Pharma | Drug delivery system for zero order, zero order-biphasic, ascending or descending drug delivery. |
| IL144446A0 (en) | 2001-07-19 | 2002-05-23 | Prochon Biotech Ltd | Plasma protein matrices and methods for their preparation |
| PL204645B1 (en) | 2001-07-23 | 2010-01-29 | Alcon | Ophthalmic drug delivery device |
| WO2003009774A2 (en) | 2001-07-23 | 2003-02-06 | Alcon, Inc. | Ophthalmic drug delivery device |
| PT1420716E (en) | 2001-08-29 | 2012-11-21 | Ricardo A P De Carvalho | An implantable and sealable system for unidirectional delivery of therapeutic agents to targeted tissues |
| US7749528B2 (en) | 2001-08-29 | 2010-07-06 | Ricardo Azevedo Pontes De Carvalho | Implantable and sealable medical device for unidirectional delivery of therapeutic agents to tissues |
| WO2003028765A1 (en) | 2001-09-28 | 2003-04-10 | Santen Pharmaceutical Co., Ltd. | Injections for eye tissue containing drug bound to polyethylene glycol |
| AU2002341959A1 (en) | 2001-10-04 | 2003-04-14 | Case Western Reserve University | Drug delivery devices and methods |
| US8663687B2 (en) | 2001-10-12 | 2014-03-04 | Monosol Rx, Llc | Film compositions for delivery of actives |
| JP2005509883A (en) | 2001-11-15 | 2005-04-14 | アリックス,インコーポレーテッド | Sample tip |
| US20080108672A1 (en) | 2002-01-11 | 2008-05-08 | Bernd Riedl | Omega-Carboxyaryl Substituted Diphenyl Ureas As Raf Kinase Inhibitors |
| GB2399753B (en) | 2002-01-18 | 2006-04-19 | Michael E Snyder | Method of making a sustained release ophthalmological device |
| US8364229B2 (en) | 2003-07-25 | 2013-01-29 | Dexcom, Inc. | Analyte sensors having a signal-to-noise ratio substantially unaffected by non-constant noise |
| WO2003077972A2 (en) | 2002-03-11 | 2003-09-25 | Alcon, Inc. | Implantable drug delivery system |
| US7074426B2 (en) | 2002-03-27 | 2006-07-11 | Frank Kochinke | Methods and drug delivery systems for the treatment of orofacial diseases |
| US7968569B2 (en) | 2002-05-17 | 2011-06-28 | Celgene Corporation | Methods for treatment of multiple myeloma using 3-(4-amino-1-oxo-1,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione |
| US7939094B2 (en) | 2002-06-19 | 2011-05-10 | Boston Scientific Scimed, Inc. | Multiphase polymeric drug release region |
| PT1521572E (en) | 2002-07-15 | 2009-04-21 | Alcon Inc | Bioerodible film for ophthalmic drug delivery |
| US8425549B2 (en) | 2002-07-23 | 2013-04-23 | Reverse Medical Corporation | Systems and methods for removing obstructive matter from body lumens and treating vascular defects |
| US20040019325A1 (en) | 2002-07-29 | 2004-01-29 | Medrip Ltd. | Syringe Pump |
| US20070219632A1 (en) | 2002-08-02 | 2007-09-20 | David Castillejos | Method and intra-sclera implant for treatment of glaucoma and presbyopia |
| US7468065B2 (en) | 2002-09-18 | 2008-12-23 | Allergan, Inc. | Apparatus for delivery of ocular implants |
| PL223153B1 (en) | 2002-09-18 | 2016-10-31 | Allergan Inc | Methods and apparatus for delivery of ocular implants |
| CA2498489C (en) | 2002-09-29 | 2010-02-23 | Surmodics, Inc. | Method for subretinal administration of therapeutics including steroids;method for localizing pharmacodynamic action at the choroid and the retina; and related methods for treatment and/or prevention of retinal diseases |
| US7794437B2 (en) | 2003-01-24 | 2010-09-14 | Doheny Retina Institute | Reservoirs with subretinal cannula for subretinal drug delivery |
| WO2004073551A2 (en) | 2003-02-18 | 2004-09-02 | Massachusetts Eye And Ear Infirmary | Transscleral drug delivery device and related methods |
| US7452913B2 (en) | 2003-02-24 | 2008-11-18 | Pharmacia & Upjohn Company | Polymorphs of pyrrole substituted 2-indolinone protein kinase inhibitors |
| WO2004076647A2 (en) | 2003-02-26 | 2004-09-10 | The Regents Of The University Of California | Use of steady-state oxygen gradients to modulate animal cell functions |
| US20050074497A1 (en) | 2003-04-09 | 2005-04-07 | Schultz Clyde L. | Hydrogels used to deliver medicaments to the eye for the treatment of posterior segment diseases |
| US20050255144A1 (en) | 2003-04-09 | 2005-11-17 | Directcontact Llc | Methods and articles for the delivery of medicaments to the eye for the treatment of posterior segment diseases |
| US7094222B1 (en) | 2003-04-28 | 2006-08-22 | The United States Of America As Represented By The Secretary Of Navy | Syringe device for simultaneous infusion and withdrawal |
| AU2004237774B2 (en) | 2003-05-02 | 2009-09-10 | Surmodics, Inc. | Implantable controlled release bioactive agent delivery device |
| EP1671624B1 (en) | 2003-05-02 | 2010-08-11 | SurModics, Inc. | Delivery device for a controlled drug release of an active agent into the posterior section of the eye |
| US7297709B2 (en) | 2003-05-22 | 2007-11-20 | Abbott Laboratories | Indazole, benzisoxazole, and benzisothiazole kinase inhibitors |
| US20050163711A1 (en) | 2003-06-13 | 2005-07-28 | Becton, Dickinson And Company, Inc. | Intra-dermal delivery of biologically active agents |
| US20040260380A1 (en) | 2003-06-18 | 2004-12-23 | D-Crown Ltd | Devices for delivering multiple stenting structures in situ |
| WO2004112653A2 (en) | 2003-06-18 | 2004-12-29 | D-Crown Ltd. | Devices and methods for delivering and forming single and multiple stenting structures in situ |
| US20040260381A1 (en) | 2003-06-18 | 2004-12-23 | D-Crown Ltd | Devices and methods for forming stenting structures in situ |
| US8367410B2 (en) | 2003-06-20 | 2013-02-05 | Massachusetts Institute Of Technology | Application of electrical stimulation for functional tissue engineering in vitro and in vivo |
| AU2004258857A1 (en) | 2003-07-10 | 2005-02-03 | Alcon, Inc. | Ophthalmic drug delivery device |
| FI120333B (en) | 2003-08-20 | 2009-09-30 | Bioretec Oy | Porous medical agent and process for its manufacture |
| AU2004274026A1 (en) | 2003-09-18 | 2005-03-31 | Macusight, Inc. | Transscleral delivery |
| US20050181018A1 (en) | 2003-09-19 | 2005-08-18 | Peyman Gholam A. | Ocular drug delivery |
| AU2006225241B2 (en) | 2003-09-24 | 2008-07-17 | Tianda Pharmaceuticals (Australia) Pty Limited | Medication Holder |
| US7686016B2 (en) | 2003-09-24 | 2010-03-30 | Medi-Stream Pty Ltd Acn 111 815 715 | Medication holder |
| US7615141B2 (en) | 2003-10-03 | 2009-11-10 | University Of Washington | Electrochemical micromanufacturing system and method |
| US7211272B2 (en) | 2003-12-22 | 2007-05-01 | Bausch & Lomb Incorporated | Drug delivery device |
| EP1720608B1 (en) | 2004-02-12 | 2010-11-17 | NeoVista, Inc. | Apparatus for intraocular brachytherapy |
| US7276050B2 (en) | 2004-03-02 | 2007-10-02 | Alan Franklin | Trans-scleral drug delivery method and apparatus |
| US20050256499A1 (en) | 2004-03-03 | 2005-11-17 | Pettis Ronald J | Methods and devices for improving delivery of a substance to skin |
| WO2005110374A1 (en) | 2004-04-30 | 2005-11-24 | Allergan, Inc. | Intraocular drug delivery systems containing a therapeutic component, a cyclodextrin, and a polymeric component |
| US20060182783A1 (en) | 2004-04-30 | 2006-08-17 | Allergan, Inc. | Sustained release intraocular drug delivery systems |
| US20070059336A1 (en) | 2004-04-30 | 2007-03-15 | Allergan, Inc. | Anti-angiogenic sustained release intraocular implants and related methods |
| US8591885B2 (en) | 2004-04-30 | 2013-11-26 | Allergan, Inc. | Carbonic anhydrase inhibitor sustained release intraocular drug delivery systems |
| US20050244469A1 (en) | 2004-04-30 | 2005-11-03 | Allergan, Inc. | Extended therapeutic effect ocular implant treatments |
| US8685435B2 (en) | 2004-04-30 | 2014-04-01 | Allergan, Inc. | Extended release biodegradable ocular implants |
| EP1761283A2 (en) | 2004-06-07 | 2007-03-14 | California Institute Of Technology | Biodegradable drug-polymer delivery system |
| US20060110428A1 (en) | 2004-07-02 | 2006-05-25 | Eugene Dejuan | Methods and devices for the treatment of ocular conditions |
| US7117870B2 (en) | 2004-07-26 | 2006-10-10 | Clarity Corporation | Lacrimal insert having reservoir with controlled release of medication and method of manufacturing the same |
| EP1786451A4 (en) | 2004-08-06 | 2009-07-22 | Sopherion Therapeutics Inc | Anti-angiogenic peptides and methods of use thereof |
| WO2006031358A2 (en) | 2004-08-13 | 2006-03-23 | Hyperbranch Medical Technology, Inc. | Dendritic polymers, crosslinked gels, and their uses as ophthalmic sealants and lenses |
| US20060104969A1 (en) | 2004-08-16 | 2006-05-18 | Massachusetts Institute Of Technology | Compositions and methods for enhancing structural and functional nervous system reorganization and recovery |
| WO2006031388A2 (en) | 2004-08-20 | 2006-03-23 | Hyperbranch Medical Technology, Inc. | Dentritic polymers, crosslinked gels, and their uses in orthopedic applications |
| US20060052754A1 (en) | 2004-09-04 | 2006-03-09 | Fields Douglas W | Thumb trigger syringe pole |
| WO2006031658A2 (en) | 2004-09-10 | 2006-03-23 | Allergan, Inc. | Therapeutic lacrimal canalicular inserts and related methods |
| WO2006031532A2 (en) | 2004-09-10 | 2006-03-23 | Surmodics, Inc. | Methods, devices, and coatings for controlled active agent release |
| US20080038316A1 (en) | 2004-10-01 | 2008-02-14 | Wong Vernon G | Conveniently implantable sustained release drug compositions |
| EP2452670A1 (en) | 2004-10-01 | 2012-05-16 | Ramscor, Inc. | Conveniently implantable sustained release drug compositions |
| WO2006043965A1 (en) | 2004-10-14 | 2006-04-27 | Allergan, Inc. | Therapeutic ophthalmic compositions containing retinal friendly excipients and related methods |
| US20080207502A1 (en) | 2004-10-14 | 2008-08-28 | Sopherion Therapeutics, Inc. | Anti-Angiogenic Peptides and Methods of Use Thereof |
| JO3000B1 (en) | 2004-10-20 | 2016-09-05 | Genentech Inc | Antibody Formulations. |
| WO2006050221A2 (en) | 2004-10-29 | 2006-05-11 | The Regents Of The University Of California | Porous photonic crystals for drug delivery to the eye |
| US20060129215A1 (en) | 2004-12-09 | 2006-06-15 | Helmus Michael N | Medical devices having nanostructured regions for controlled tissue biocompatibility and drug delivery |
| US20060128653A1 (en)* | 2004-12-10 | 2006-06-15 | Chunlin Tang | Pharmaceutical formulation of decitabine |
| US20060154981A1 (en) | 2005-01-12 | 2006-07-13 | Alcon, Inc. | Method of reducing intraocular pressure and treating glaucoma |
| WO2006082588A2 (en) | 2005-02-07 | 2006-08-10 | Pharmalight Inc. | Method and device for ophthalmic administration of active pharmaceutical ingredients |
| US8663639B2 (en) | 2005-02-09 | 2014-03-04 | Santen Pharmaceutical Co., Ltd. | Formulations for treating ocular diseases and conditions |
| TW200640443A (en) | 2005-02-23 | 2006-12-01 | Alcon Inc | Methods for treating ocular angiogenesis, retinal edema, retinal ischemia, and diabetic retinopathy using selective RTK inhibitors |
| US20060233858A1 (en) | 2005-03-08 | 2006-10-19 | Allergan, Inc. | Systems and methods providing targeted intraocular drug delivery |
| KR20070121754A (en) | 2005-03-21 | 2007-12-27 | 마커사이트, 인코포레이티드 | Drug delivery system for the treatment of a disease or condition |
| US20070077270A1 (en) | 2005-03-28 | 2007-04-05 | Clemson University | Delivery devices and methods for long-term, targeted delivery of therapeutic agents to the eye and ear |
| EP1875560A4 (en) | 2005-04-05 | 2011-02-09 | Univ Ohio State | DIFFUSION DELIVERY SYSTEM AND METHODS OF MAKING |
| US20100168535A1 (en) | 2006-04-12 | 2010-07-01 | Mark Ries Robinson | Methods and apparatuses related to blood analyte measurement system |
| US20060258994A1 (en) | 2005-05-12 | 2006-11-16 | Avery Robert L | Implantable delivery device for administering pharmacological agents to an internal portion of a body |
| WO2006127962A2 (en) | 2005-05-25 | 2006-11-30 | Becton, Dickinson And Comapny | Particulate formulations for intradermal delivery of biologically active agents |
| AU2006259262A1 (en) | 2005-06-17 | 2006-12-28 | Dynamis Therapeutics, Inc. | Treatment of inflammatory conditions |
| US7893040B2 (en) | 2005-07-22 | 2011-02-22 | Oculis Ehf | Cyclodextrin nanotechnology for ophthalmic drug delivery |
| US8663673B2 (en) | 2005-07-29 | 2014-03-04 | Surmodics, Inc. | Devices, articles, coatings, and methods for controlled active agent release or hemocompatibility |
| EP1912574A4 (en) | 2005-08-10 | 2010-08-04 | Insight Instr Inc | Tool for extracting vitreous samples from an eye |
| US20070212397A1 (en) | 2005-09-15 | 2007-09-13 | Roth Daniel B | Pharmaceutical delivery device and method for providing ocular treatment |
| EP1924309A1 (en) | 2005-09-16 | 2008-05-28 | (OSI) Eyetech Inc. | Ophthalmic syringe |
| US20070071756A1 (en) | 2005-09-26 | 2007-03-29 | Peyman Gholam A | Delivery of an agent to ameliorate inflammation |
| US20080167600A1 (en) | 2005-09-26 | 2008-07-10 | Peyman Gholam A | Device for delivery of an agent to the eye and other sites |
| US20070072933A1 (en) | 2005-09-26 | 2007-03-29 | Peyman Gholam A | Delivery of an ocular agent |
| US20080003219A1 (en) | 2005-09-26 | 2008-01-03 | Minu, L.L.C. | Delivery of an ocular agent |
| WO2007038453A2 (en) | 2005-09-26 | 2007-04-05 | Advanced Ocular Systems Limited | Use of an anti-vascular endothelial growth factor (vegf) agent to ameliorate inflammation |
| TW200732347A (en) | 2005-10-06 | 2007-09-01 | Trophogen Inc | VEGF analogs and methods of use |
| US8168584B2 (en) | 2005-10-08 | 2012-05-01 | Potentia Pharmaceuticals, Inc. | Methods of treating age-related macular degeneration by compstatin and analogs thereof |
| CA2625359A1 (en) | 2005-10-11 | 2007-04-19 | Blake Podaima | Smart medical compliance method and system |
| US20080125406A1 (en) | 2005-10-14 | 2008-05-29 | Robin Alan L | Method for Treating Primary and Secondary Forms of Glaucoma |
| KR20080059280A (en) | 2005-10-14 | 2008-06-26 | 알콘, 인코퍼레이티드 | Methods of treating primary and secondary forms of glaucoma |
| CN107929731A (en) | 2005-11-04 | 2018-04-20 | 健泰科生物技术公司 | Utilize complement pathway inhibitors to treat ocular diseases |
| ATE482708T1 (en) | 2005-11-29 | 2010-10-15 | Glaxosmithkline Llc | TREATMENT OF NEOVASCULAR EYE DISEASES, SUCH AS MACULAR DEGENERATION, ANGIOID STRIPES, UVEITIS AND MACULA EDEMA |
| JP4721425B2 (en) | 2005-12-01 | 2011-07-13 | キヤノン株式会社 | Fluid moving method and fluid moving device |
| CN101336315B (en) | 2005-12-07 | 2012-12-19 | 特拉维夫大学拉莫特有限公司 | Drug-delivering composite structures |
| US20070131611A1 (en) | 2005-12-13 | 2007-06-14 | General Electric Company | Membrane-based article and associated method |
| US20070131610A1 (en) | 2005-12-13 | 2007-06-14 | General Electric Company | Membrane-based apparatus and associated method |
| US20080108664A1 (en) | 2005-12-23 | 2008-05-08 | Liu Belle B | Solid-state form of AMG 706 and pharmaceutical compositions thereof |
| WO2007076448A2 (en)* | 2005-12-23 | 2007-07-05 | Alcon, Inc. | Pharmaceutical composition for delivery of receptor tyrosine kinase inhibiting (rtki) compounds to the eye |
| WO2007087061A2 (en) | 2006-01-17 | 2007-08-02 | Transcend Medical, Inc. | Glaucoma treatment device |
| WO2007084582A2 (en) | 2006-01-17 | 2007-07-26 | Forsight Labs, Llc | Drug delivery treatment device |
| US20080216736A1 (en) | 2006-01-19 | 2008-09-11 | Takeda San Diego, Inc. | Microfluidic device with diffusion between adjacent lumens |
| US7989631B2 (en) | 2006-02-10 | 2011-08-02 | Amgen Inc. | Hydrate forms of AMG706 |
| WO2007100986A2 (en) | 2006-02-24 | 2007-09-07 | Rosetta Inpharmatics Llc | Extraction and diagnostic fluid devices, systems and methods of use |
| WO2007101204A1 (en) | 2006-02-27 | 2007-09-07 | Alcon Research, Ltd. | Method of treating glaucoma |
| EP1832301A3 (en) | 2006-03-08 | 2007-12-05 | Sahajanand Medical Technologies PVT. ltd | Coatings for implantable medical devices |
| EP2316505B1 (en) | 2006-03-14 | 2017-01-18 | University Of Southern California | Mems device for delivery of therapeutic agents |
| WO2007117394A2 (en) | 2006-03-31 | 2007-10-18 | Dynamis Therapeutics, Inc. | Compositions and methods related to fructosamine-3-kinase inhibitors |
| NZ572193A (en) | 2006-03-31 | 2011-10-28 | Quadra Logic Tech Inc | Nasolacrimal drainage system implants for drug therapy with non-fluid swellable retention structure around drug core |
| US7547300B2 (en) | 2006-04-12 | 2009-06-16 | Icu Medical, Inc. | Vial adaptor for regulating pressure |
| EP2014322A4 (en) | 2006-04-18 | 2012-09-26 | Zhongshan Botai Pharmaceutic Instr Co Ltd | A sterile drug-mixing syringe |
| US8197435B2 (en) | 2006-05-02 | 2012-06-12 | Emory University | Methods and devices for drug delivery to ocular tissue using microneedle |
| US7918814B2 (en) | 2006-05-02 | 2011-04-05 | Georgia Tech Research Corporation | Method for drug delivery to ocular tissue using microneedle |
| US7981096B2 (en) | 2006-05-12 | 2011-07-19 | David Castillejos | Optic nerve head implant and medication delivery system |
| US20070270750A1 (en) | 2006-05-17 | 2007-11-22 | Alcon, Inc. | Drug delivery device |
| US20080124372A1 (en) | 2006-06-06 | 2008-05-29 | Hossainy Syed F A | Morphology profiles for control of agent release rates from polymer matrices |
| EP2037882B1 (en) | 2006-06-28 | 2014-12-10 | SurModics, Inc. | Combination degradable and non-degradable matrices for active agent delivery |
| TW200815045A (en) | 2006-06-29 | 2008-04-01 | Jazz Pharmaceuticals Inc | Pharmaceutical compositions of ropinirole and methods of use thereof |
| US8104331B2 (en) | 2006-07-19 | 2012-01-31 | Noab Biodiscoveries Inc. | Method and device to extract components contained in a fluid |
| WO2008011125A2 (en) | 2006-07-20 | 2008-01-24 | Neurosystec Corporation | Devices, systems and methods for ophthalmic drug delivery |
| US20080069854A1 (en) | 2006-08-02 | 2008-03-20 | Inframat Corporation | Medical devices and methods of making and using |
| US20080097335A1 (en) | 2006-08-04 | 2008-04-24 | Allergan, Inc. | Ocular implant delivery assemblies |
| JP2008054521A (en) | 2006-08-29 | 2008-03-13 | Canon Inc | Cell culture treatment apparatus and cell culture treatment method |
| WO2008033924A2 (en) | 2006-09-12 | 2008-03-20 | Board Of Regents Of The University Of Nebraska | Methods and compositions for targeted delivery of therapeutic agents |
| US20080066739A1 (en) | 2006-09-20 | 2008-03-20 | Lemahieu Edward | Methods and systems of delivering medication via inhalation |
| JP5694664B2 (en) | 2006-09-29 | 2015-04-01 | サーモディクス,インコーポレイティド | Biodegradable ocular implant and method for treating ocular diseases |
| WO2008045272A2 (en) | 2006-10-06 | 2008-04-17 | Dynamis Therapeutics, Inc. | Compositions and methods for skin lightening |
| US9022970B2 (en) | 2006-10-16 | 2015-05-05 | Alcon Research, Ltd. | Ophthalmic injection device including dosage control device |
| US8591957B2 (en) | 2006-10-25 | 2013-11-26 | Revalesio Corporation | Methods of therapeutic treatment of eyes and other human tissues using an oxygen-enriched solution |
| US8609148B2 (en) | 2006-10-25 | 2013-12-17 | Revalesio Corporation | Methods of therapeutic treatment of eyes |
| US8784898B2 (en) | 2006-10-25 | 2014-07-22 | Revalesio Corporation | Methods of wound care and treatment |
| US8784897B2 (en) | 2006-10-25 | 2014-07-22 | Revalesio Corporation | Methods of therapeutic treatment of eyes |
| EP2083876A4 (en) | 2006-10-25 | 2012-09-19 | Revalesio Corp | Methods of wound care and treatment |
| EP2088976B1 (en) | 2006-11-10 | 2019-07-03 | Glaukos Corporation | Uveoscleral shunt |
| US20080111282A1 (en) | 2006-11-10 | 2008-05-15 | Baojun Xie | Process for Making Three Dimensional Objects From Dispersions of Polymer Colloidal Particles |
| US20100286791A1 (en) | 2006-11-21 | 2010-11-11 | Goldsmith David S | Integrated system for the ballistic and nonballistic infixion and retrieval of implants |
| US20090263346A1 (en) | 2006-12-05 | 2009-10-22 | David Taft | Systems and methods for delivery of drugs |
| JP2010511713A (en) | 2006-12-05 | 2010-04-15 | ランデック コーポレイション | Drug delivery |
| WO2008073295A2 (en) | 2006-12-07 | 2008-06-19 | Surmodics, Inc. | Latent stabilization of bioactive agents releasable from implantable medical articles |
| US20080147021A1 (en) | 2006-12-15 | 2008-06-19 | Jani Dharmendra M | Drug delivery devices |
| AR063619A1 (en) | 2006-12-18 | 2009-02-04 | Alcon Res Ltd | DEVICES AND METHODS FOR THE ADMINISTRATION OF AN OPHTHALMIC DRUG |
| EP2097044A4 (en) | 2006-12-26 | 2012-10-10 | Quadra Logic Tech Inc | DRUG RELIEF IMPLANTS FOR INHIBITING OPTICAL DEFECTS |
| CA2674891C (en) | 2007-01-09 | 2016-03-15 | Fovea Pharmaceuticals | Apparatus for intra-ocular injection |
| AU2008206953A1 (en) | 2007-01-19 | 2008-07-24 | Cinvention Ag | Porous, non-degradable implant made by powder molding |
| UY30883A1 (en) | 2007-01-31 | 2008-05-31 | Alcon Res | PUNCTURAL PLUGS AND METHODS OF RELEASE OF THERAPEUTIC AGENTS |
| GB0702864D0 (en) | 2007-02-14 | 2007-03-28 | Benoist Girard Sas | Prosthetic implant for use without bone cement |
| EP2236518B1 (en) | 2007-03-14 | 2014-08-06 | Alexion Cambridge Corporation | Humaneered anti-factor B antibody |
| WO2008116165A2 (en) | 2007-03-21 | 2008-09-25 | Next Safety, Inc. | Methods and systems of delivering medication via inhalation |
| US20080249501A1 (en) | 2007-04-09 | 2008-10-09 | Medtronic Vascular, Inc. | Methods for Simultaneous Injection and Aspiration of Fluids During a Medical Procedure |
| MX2009009738A (en) | 2007-04-30 | 2009-09-24 | Alcon Res Ltd | Treatment of age-related macular degeneration using inhibitors of complement factor d. |
| US20080286338A1 (en) | 2007-05-15 | 2008-11-20 | Boston Foundation For Sight | Drug delivery system with scleral lens |
| US8231892B2 (en) | 2007-05-24 | 2012-07-31 | Allergan, Inc. | Biodegradable drug delivery system |
| CN101815724B (en) | 2007-05-29 | 2015-02-25 | 安吉奥开米公司 | Aprotinin-like polypeptides for delivery of agents conjugated thereto to tissues |
| US9365634B2 (en) | 2007-05-29 | 2016-06-14 | Angiochem Inc. | Aprotinin-like polypeptides for delivering agents conjugated thereto to tissues |
| CN101327356A (en) | 2007-06-21 | 2008-12-24 | 庄臣及庄臣视力保护公司 | Dacryon plug for conveying promoting agent |
| ES2493641T3 (en) | 2007-06-28 | 2014-09-12 | Cydex Pharmaceuticals, Inc. | Nasal administration of aqueous corticosteroid solutions |
| US20090036827A1 (en) | 2007-07-31 | 2009-02-05 | Karl Cazzini | Juxtascleral Drug Delivery and Ocular Implant System |
| US9808557B2 (en) | 2007-08-10 | 2017-11-07 | Trustees Of Tufts College | Tubular silk compositions and methods of use thereof |
| CN101984745B (en) | 2007-09-07 | 2013-08-14 | Qlt股份有限公司 | Drug Cores for Sustained Release of Therapeutics |
| CN101861135A (en) | 2007-09-07 | 2010-10-13 | Qlt栓塞输送公司 | Lacrimal implant testing |
| WO2009035567A2 (en) | 2007-09-07 | 2009-03-19 | Qlt Plug Delivery, Inc | Insertion and extraction tools for lacrimal implants |
| EP2190287B1 (en)* | 2007-09-10 | 2014-10-29 | Curis, Inc. | Tartrate salts or complexes of quinazoline based egfr inhibitors containing a zinc binding moiety |
| US8974809B2 (en) | 2007-09-24 | 2015-03-10 | Boston Scientific Scimed, Inc. | Medical devices having a filter insert for controlled diffusion |
| US8308755B2 (en) | 2007-09-24 | 2012-11-13 | Ethicon Endo-Surgery, Inc. | Elliptical retractor |
| US20090081272A1 (en) | 2007-09-24 | 2009-03-26 | John Clarke | Medical devices having a metal particulate composition for controlled diffusion |
| US20090214601A1 (en) | 2007-09-28 | 2009-08-27 | Chappa Ralph A | Porous Drug Delivery Devices and Related Methods |
| US20090093752A1 (en) | 2007-10-05 | 2009-04-09 | Tyco Healthcare Group Lp | Seal anchor for use in surgical procedures |
| US7960564B2 (en) | 2007-10-19 | 2011-06-14 | Abbott Laboratories | Crystalline chemotherapeutic |
| US7943782B2 (en) | 2007-10-19 | 2011-05-17 | Abbott Laboratories | Crystalline chemotherapeutic |
| US7772404B2 (en) | 2007-10-19 | 2010-08-10 | Abbott Laboratories | Crystalline form 2 of the chemotherapeutic N-[4-(3-amino-1H-indazol-4-yl)phenyl]-N′-(2-fluoro-5-methylphenyl)urea |
| US20100009008A1 (en) | 2007-10-25 | 2010-01-14 | Revalesio Corporation | Bacteriostatic or bacteriocidal compositions and methods |
| US20100303918A1 (en) | 2007-10-25 | 2010-12-02 | Revalesio Corporation | Compositions and methods for treating asthma and other lung disorders |
| US10125359B2 (en) | 2007-10-25 | 2018-11-13 | Revalesio Corporation | Compositions and methods for treating inflammation |
| WO2010062628A1 (en) | 2008-10-27 | 2010-06-03 | Revalesio Corporation | Compositions and methods for treating asthma and other lung disorders |
| US20100004189A1 (en) | 2007-10-25 | 2010-01-07 | Revalesio Corporation | Compositions and methods for treating cystic fibrosis |
| US20100008997A1 (en) | 2007-10-25 | 2010-01-14 | Revalesio Corporation | Compositions and methods for treating asthma and other lung disorders |
| US20090227018A1 (en) | 2007-10-25 | 2009-09-10 | Revalesio Corporation | Compositions and methods for modulating cellular membrane-mediated intracellular signal transduction |
| US20090263495A1 (en) | 2007-10-25 | 2009-10-22 | Revalesio Corporation | Bacteriostatic or bacteriocidal compositions and methods |
| US9523090B2 (en) | 2007-10-25 | 2016-12-20 | Revalesio Corporation | Compositions and methods for treating inflammation |
| CN101909869B (en) | 2007-10-25 | 2014-12-17 | 利发利希奥公司 | Bacteriostatic or bactericidal compositions and methods |
| US20100303917A1 (en) | 2007-10-25 | 2010-12-02 | Revalesio Corporation | Compositions and methods for treating cystic fibrosis |
| US20100310665A1 (en) | 2007-10-25 | 2010-12-09 | Revalesio Corporation | Bacteriostatic or bacteriocidal compositions and methods |
| MX2010004366A (en) | 2007-11-05 | 2010-05-05 | Bausch & Lomb | Water-immiscible materials as vehicles for drug delivery. |
| US8114883B2 (en) | 2007-12-04 | 2012-02-14 | Landec Corporation | Polymer formulations for delivery of bioactive materials |
| WO2009086112A2 (en) | 2007-12-20 | 2009-07-09 | University Of Southern California | Apparatus and methods for delivering therapeutic agents |
| US8414646B2 (en) | 2007-12-27 | 2013-04-09 | Forsight Labs, Llc | Intraocular, accommodating lens and methods of use |
| WO2009089094A2 (en) | 2008-01-03 | 2009-07-16 | University Of Southern California | Implantable drug-delivery devices, and apparatus and methods for refilling the devices |
| WO2009089409A2 (en) | 2008-01-10 | 2009-07-16 | Bausch & Lomb Incorporated | Intravitreal injection system having coaxial cannulae and use thereof |
| WO2009094466A2 (en) | 2008-01-22 | 2009-07-30 | University Of Florida Research Foundation, Inc. | Contact lenses for extended release of bioactive agents containing diffusion attenuators |
| WO2009097468A2 (en) | 2008-01-29 | 2009-08-06 | Kliman Gilbert H | Drug delivery devices, kits and methods therefor |
| CA2716720A1 (en)* | 2008-02-28 | 2009-09-03 | Takeda Pharmaceutical Company Limited | Pharmaceutical composition |
| US8201752B2 (en) | 2008-03-10 | 2012-06-19 | Vapore, Inc. | Low energy vaporization of liquids: apparatus and methods |
| TNSN08110A1 (en) | 2008-03-11 | 2009-07-14 | Rekik Raouf Dr | Drug delivery to the anterior and posterior segment of the eye from drops |
| US20090240208A1 (en) | 2008-03-19 | 2009-09-24 | Warsaw Orthopedic, Inc. | Microparticle delivery syringe and needle for placing particle suspensions and removing vehicle fluid |
| EP2257326A2 (en) | 2008-03-21 | 2010-12-08 | Novartis AG | Powder dispersion apparatus |
| CA3138128C (en) | 2008-03-31 | 2024-02-13 | Passport Technologies, Inc. | Permeant delivery system and methods for use thereof |
| PL2285439T3 (en) | 2008-04-04 | 2014-05-30 | Nektar Therapeutics | Aerosolization device |
| US8801665B2 (en) | 2008-04-10 | 2014-08-12 | Henry Ford Health System | Apparatus and method for controlled depth of injection into myocardial tissue |
| US20090258069A1 (en) | 2008-04-15 | 2009-10-15 | John Burnier | Delivery of LFA-1 antagonists to the gastrointestinal system |
| CN104623741A (en) | 2008-04-30 | 2015-05-20 | 马缇医疗股份有限公司 | Composite lacrimal insert and related methods |
| EP2285347A4 (en) | 2008-05-01 | 2011-09-21 | Revalesio Corp | Compositions and methods for treating digestive disorders |
| MX2010012212A (en) | 2008-05-08 | 2011-09-27 | Minipumps Llc | Implantable drug-delivery devices, and apparatus and methods for filling the devices. |
| EP2296621A1 (en) | 2008-05-20 | 2011-03-23 | Yale University | Biodegradable sustained-release polymeric microparticulates comprising a hydrophobic drug and determined for ophthalmic use |
| CA2724607A1 (en) | 2008-05-30 | 2009-12-31 | Fairfield Clinical Trials, Llc | Method and composition for skin inflammation and discoloration |
| US10517839B2 (en) | 2008-06-09 | 2019-12-31 | Cornell University | Mast cell inhibition in diseases of the retina and vitreous |
| CA2769307C (en) | 2008-07-11 | 2017-12-12 | Intellicyt Corporation | Multi-sample particle analyzer system and method for high throughput screening |
| US8821870B2 (en) | 2008-07-18 | 2014-09-02 | Allergan, Inc. | Method for treating atrophic age related macular degeneration |
| JP2011528605A (en) | 2008-07-21 | 2011-11-24 | アルスタシス,インコーポレイテッド | Device, method, and kit for forming a tube in tissue |
| US20100022943A1 (en) | 2008-07-25 | 2010-01-28 | Medtronic Vascular, Inc. | Hydrodynamic Thrombectomy Catheter |
| US20100023033A1 (en) | 2008-07-25 | 2010-01-28 | Medtronic Vescular, Inc. | Hydrodynamic Thrombectomy Catheter |
| US8242106B2 (en)* | 2008-08-01 | 2012-08-14 | Ventirx Pharmaceuticals, Inc. | Toll-like receptor agonist formulations and their use |
| WO2010021993A1 (en) | 2008-08-19 | 2010-02-25 | Cytori Therapeutics, Inc. | Methods of using adipose tissue-derived cells in the treatment of the lymphatic system and malignant disease |
| US7678078B1 (en) | 2008-10-21 | 2010-03-16 | KMG Pharma LLC | Intravitreal injection device, system and method |
| US8221353B2 (en) | 2008-10-21 | 2012-07-17 | KMG Pharma, Inc | Intravitreal injection device and system |
| WO2010062394A2 (en) | 2008-11-26 | 2010-06-03 | Surmodics, Inc. | Implantable ocular drug delivery device and methods |
| DE102008054431B3 (en) | 2008-12-09 | 2010-06-17 | Pari Pharma Gmbh | Aerosol therapy device |
| US20100158980A1 (en) | 2008-12-18 | 2010-06-24 | Casey Kopczynski | Drug delivery devices for delivery of therapeutic agents |
| BRPI0923810A2 (en) | 2009-01-02 | 2015-07-14 | Alcon Res Ltd | In-Place Replenished Ophthalmic Implant |
| US8545554B2 (en) | 2009-01-16 | 2013-10-01 | Allergan, Inc. | Intraocular injector |
| CN104887389B (en) | 2009-01-29 | 2017-06-23 | 弗赛特影像4股份有限公司 | Posterior segment drug delivery |
| US8623395B2 (en) | 2010-01-29 | 2014-01-07 | Forsight Vision4, Inc. | Implantable therapeutic device |
| WO2010093945A2 (en) | 2009-02-13 | 2010-08-19 | Glaukos Corporation | Uveoscleral drug delivery implant and methods for implanting the same |
| PT3395372T (en) | 2009-02-20 | 2022-05-05 | Enhanx Biopharm Inc | Glutathione-based drug delivery system |
| WO2010101989A1 (en) | 2009-03-03 | 2010-09-10 | Alcon Research, Ltd. | PHARMACEUTICAL COMPOSITION FOR DELIVERY OF RECEPTOR TYROSINE KINASE INHIBITING (RTKi) COMPOUNDS TO THE EYE |
| US8424367B2 (en) | 2009-03-04 | 2013-04-23 | University Of South Carolina | Systems and methods for measurement of gas permeation through polymer films |
| US8815292B2 (en) | 2009-04-27 | 2014-08-26 | Revalesio Corporation | Compositions and methods for treating insulin resistance and diabetes mellitus |
| WO2010125416A1 (en) | 2009-04-27 | 2010-11-04 | Raouf Rekik | Drug delivery to the anterior and posterior segments of the eye |
| MX2011011333A (en) | 2009-04-27 | 2011-11-18 | Revalesio Corp | Compositions and methods for treating insulin resistance and diabetes mellitus. |
| EP2432476A4 (en)* | 2009-05-01 | 2013-03-20 | Ophthotech Corp | METHODS OF TREATING OR PREVENTING OPHTHALMOLOGICAL DISEASES |
| US20110125178A1 (en) | 2009-05-15 | 2011-05-26 | Michael Drews | Devices, methods and kits for forming tracts in tissue |
| WO2010135369A1 (en) | 2009-05-18 | 2010-11-25 | Dose Medical Corporation | Drug eluting ocular implant |
| CN102596097B (en) | 2009-06-03 | 2015-05-20 | 弗赛特实验室有限责任公司 | an eye insert |
| WO2011008896A2 (en) | 2009-07-14 | 2011-01-20 | Board Of Regents, The University Of Texas System | Therapeutic methods using controlled delivery devices having zero order kinetics |
| US20110033933A1 (en) | 2009-07-15 | 2011-02-10 | Morteza Gharib | Method applying hemodynamic forcing and klf2 to initiate the growth and development of cardiac valves |
| WO2011020049A1 (en) | 2009-08-14 | 2011-02-17 | Genentech, Inc. | Biological markers for monitoring patient response to vegf antagonists |
| JP2013502440A (en) | 2009-08-27 | 2013-01-24 | バイオノミックス リミテッド | Treatment of macular degeneration |
| WO2011028850A1 (en) | 2009-09-01 | 2011-03-10 | Northwestern University | Delivery of therapeutic agents using oligonucleotide-modified nanoparticles as carriers |
| CA2811959A1 (en) | 2009-09-21 | 2011-03-24 | Harvard Bioscience, Inc. | Methods and apparatus for introducing cells at a tissue site |
| WO2011069053A1 (en) | 2009-12-04 | 2011-06-09 | Teva Pharmaceutical Industries Ltd. | Process for the preparation of pazopanip hcl and crystalline forms of pazopanib hcl |
| EP2512389B1 (en) | 2009-12-16 | 2015-09-02 | Allergan, Inc. | Intracameral devices for sustained delivery |
| CA2785468A1 (en) | 2009-12-23 | 2011-06-30 | Psivida Us, Inc. | Sustained release delivery devices |
| UY33164A (en) | 2010-01-06 | 2011-08-31 | Glaxo Wellcome Mfg Pte Ltd | TREATMENT METHOD |
| US10166142B2 (en) | 2010-01-29 | 2019-01-01 | Forsight Vision4, Inc. | Small molecule delivery with implantable therapeutic device |
| UY33367A (en)* | 2010-05-05 | 2011-10-31 | Glaxo Wellcome Mfg Pte Ltd | PHARMACEUTICAL COMPOSITIONS AND METHODS FOR THEIR DEVELOPMENT |
| PT2600812T (en)* | 2010-08-05 | 2021-11-09 | Forsight Vision4 Inc | Implantable therapeutic device |
| LT2600930T (en) | 2010-08-05 | 2021-04-12 | Forsight Vision4, Inc. | Injector apparatus for drug delivery |
| WO2012019047A2 (en) | 2010-08-05 | 2012-02-09 | Forsight Vision4, Inc. | Subconjunctival implant for posterior segment drug delivery |
| US10617557B2 (en) | 2010-08-05 | 2020-04-14 | Forsight Vision4, Inc. | Combined drug delivery methods and apparatus |
| WO2012042421A1 (en) | 2010-09-29 | 2012-04-05 | Pfizer Inc. | Method of treating abnormal cell growth |
| AU2011308504B2 (en)* | 2010-10-01 | 2016-06-02 | The Trustees Of The University Of Pennsylvania | Therapeutic use of a TLR agonist and combination therapy |
| US20140033800A1 (en) | 2010-11-11 | 2014-02-06 | Forsight Vision4, Inc. | Methods and apparatus to determine diffusion properties of porous structures for drug delivery |
| WO2012068549A2 (en) | 2010-11-19 | 2012-05-24 | Forsight Vision4, Inc. | Therapeutic agent formulations for implanted devices |
| WO2012103060A1 (en) | 2011-01-27 | 2012-08-02 | Glaxo Wellcome Manufacturing Pte Ltd | Method of administration and treatment |
| TWI544922B (en)* | 2011-05-19 | 2016-08-11 | 愛爾康研究有限公司 | High concentration europart ingot ophthalmic composition |
| US10398592B2 (en) | 2011-06-28 | 2019-09-03 | Forsight Vision4, Inc. | Diagnostic methods and apparatus |
| AU2012302051B2 (en)* | 2011-08-30 | 2017-04-27 | Astex Pharmaceuticals, Inc. | Decitabine derivative formulations |
| HRP20210676T1 (en) | 2011-09-16 | 2021-05-28 | Forsight Vision4, Inc. | Fluid exchange apparatus |
| WO2013116061A1 (en) | 2012-02-03 | 2013-08-08 | Forsight Vision4, Inc. | Insertion and removal methods and apparatus for therapeutic devices |
| JP2014521602A (en)* | 2012-04-04 | 2014-08-28 | ポラリス グループ | Treatment method using arginine deiminase |
| US10251778B2 (en) | 2012-08-06 | 2019-04-09 | Baylor College Of Medicine | Therapeutics dispensing device and methods of making same |
| MX2015005230A (en)* | 2012-10-26 | 2015-08-14 | Lupin Ltd | Stable pharmaceutical composition of peginterferon alpha-2b. |
| WO2014152959A1 (en)* | 2013-03-14 | 2014-09-25 | Forsight Vision4, Inc. | Systems for sustained intraocular delivery of low solubility compounds from a port delivery system implant |
| ES2972168T3 (en) | 2013-03-28 | 2024-06-11 | Forsight Vision4 Inc | Ophthalmic implant for administration of therapeutic substances |
| WO2015085234A1 (en) | 2013-12-06 | 2015-06-11 | Forsight Vision4, Inc. | Implantable therapeutic devices |
| ES2803102T3 (en) | 2014-07-15 | 2021-01-22 | Forsight Vision4 Inc | Eye implant delivery device |
| WO2016022750A1 (en) | 2014-08-08 | 2016-02-11 | Forsight Vision4, Inc. | Stable and soluble formulations of receptor tyrosine kinase inhibitors, and methods of preparation thereof |
| WO2016077371A1 (en) | 2014-11-10 | 2016-05-19 | Forsight Vision4, Inc. | Expandable drug delivery devices and method of use |
- 2015
- 2015-08-06WOPCT/US2015/043921patent/WO2016022750A1/enactiveApplication Filing
- 2015-08-06KRKR1020177006057Apatent/KR20170040798A/ennot_activeCeased
- 2015-08-06JPJP2017527197Apatent/JP2017524034A/enactivePending
- 2015-08-06RURU2017105844Apatent/RU2017105844A/ennot_activeApplication Discontinuation
- 2015-08-06EPEP15830652.2Apatent/EP3177289A4/ennot_activeWithdrawn
- 2015-08-06AUAU2015301054Apatent/AU2015301054B2/ennot_activeCeased
- 2015-08-06MYMYPI2017700417Apatent/MY182793A/enunknown
- 2015-08-06USUS14/819,682patent/US9474756B2/ennot_activeExpired - Fee Related
- 2015-08-06BRBR112017002466Apatent/BR112017002466A2/ennot_activeApplication Discontinuation
- 2015-08-06CNCN201580054818.4Apatent/CN107106551A/enactivePending
- 2015-08-06SGSG11201700943TApatent/SG11201700943TA/enunknown
- 2015-08-06MXMX2017001818Apatent/MX2017001818A/enunknown
- 2015-08-06CACA2957548Apatent/CA2957548A1/ennot_activeAbandoned
- 2016
- 2016-09-20USUS15/270,493patent/US9895369B2/enactiveActive
- 2017
- 2017-02-05ILIL250447Apatent/IL250447A0/enunknown
- 2018
- 2018-01-25USUS15/880,180patent/US10363255B2/enactiveActive
- 2019
- 2019-06-07USUS16/434,966patent/US10765677B2/ennot_activeExpired - Fee Related
- 2020
- 2020-02-04JPJP2020016975Apatent/JP2020079275A/enactivePending
Also Published As
| Publication number | Publication date |
|---|---|
| JP2020079275A (en) | 2020-05-28 |
| IL250447A0 (en) | 2017-03-30 |
| RU2017105844A3 (en) | 2019-03-13 |
| CN107106551A (en) | 2017-08-29 |
| US9895369B2 (en) | 2018-02-20 |
| US20190365757A1 (en) | 2019-12-05 |
| MX2017001818A (en) | 2017-05-30 |
| EP3177289A4 (en) | 2018-03-21 |
| WO2016022750A1 (en) | 2016-02-11 |
| US9474756B2 (en) | 2016-10-25 |
| MY182793A (en) | 2021-02-05 |
| SG11201700943TA (en) | 2017-03-30 |
| US20170071938A1 (en) | 2017-03-16 |
| US20180147204A1 (en) | 2018-05-31 |
| RU2017105844A (en) | 2018-09-11 |
| CA2957548A1 (en) | 2016-02-11 |
| JP2017524034A (en) | 2017-08-24 |
| US10765677B2 (en) | 2020-09-08 |
| AU2015301054B2 (en) | 2020-05-14 |
| AU2015301054A1 (en) | 2017-03-30 |
| US20160038488A1 (en) | 2016-02-11 |
| US10363255B2 (en) | 2019-07-30 |
| BR112017002466A2 (en) | 2017-12-05 |
| EP3177289A1 (en) | 2017-06-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10765677B2 (en) | Stable and soluble formulations of receptor tyrosine kinase inhibitors, and methods of preparation thereof | |
| EP2968113B1 (en) | Systems for sustained intraocular delivery of low solubility compounds from a port delivery system implant | |
| US20210077481A1 (en) | Pharmaceutical composition | |
| WO2021195163A1 (en) | Ocular implant containing a tyrosine kinase inhibitor | |
| US20110178145A1 (en) | Alpha-2 adrenergic agonist having long duration ofintraocular pressure-lowering effect | |
| JP6931257B2 (en) | A novel eye drop composition for treating dry eye containing rebamipide and a method for solubilizing and stabilizing it. | |
| JP6522592B2 (en) | Topical aqueous ophthalmic composition containing 1H-indole-1-carboxamide derivatives and its use for the treatment of ocular diseases | |
| WO2007013590A1 (en) | Non-invasive drug delivery system targeting posterior eye tissue using solid composition | |
| EP4099979A1 (en) | Mucoadhesive solid or semisolid ocular delivery systems based on preactivated thiomers | |
| WO2022065404A1 (en) | Pharmaceutical composition for otic administration | |
| HK1242223A1 (en) | Stable and soluble formulations of receptor tyrosine kinase inhibitors, and methods of preparation thereof | |
| BR122024022585A2 (en) | OCULAR IMPLANT CONTAINING A TYROSINE KINASE INHIBITOR AND ITS USE | |
| CN114949229A (en) | Ophthalmic medicinal composition containing non-steroidal anti-inflammatory drug and preparation method and application thereof | |
| CN115501175A (en) | A biodegradable hydrogel composition for multiple drug delivery and its preparation method and application | |
| HK40007038A (en) | Medicinal composition comprising tivozanib |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application | Patent event date:20170303 Patent event code:PA01051R01D Comment text:International Patent Application | |
| PG1501 | Laying open of application | ||
| A201 | Request for examination | ||
| PA0201 | Request for examination | Patent event code:PA02012R01D Patent event date:20200805 Comment text:Request for Examination of Application | |
| E902 | Notification of reason for refusal | ||
| PE0902 | Notice of grounds for rejection | Comment text:Notification of reason for refusal Patent event date:20220125 Patent event code:PE09021S01D | |
| E601 | Decision to refuse application | ||
| PE0601 | Decision on rejection of patent | Patent event date:20220406 Comment text:Decision to Refuse Application Patent event code:PE06012S01D Patent event date:20220125 Comment text:Notification of reason for refusal Patent event code:PE06011S01I |