JP4072597B2 - Sustained formulation - Google Patents
Sustained formulationDownload PDFInfo
- Publication number
- JP4072597B2 JP4072597B2JP35197295AJP35197295AJP4072597B2JP 4072597 B2JP4072597 B2JP 4072597B2JP 35197295 AJP35197295 AJP 35197295AJP 35197295 AJP35197295 AJP 35197295AJP 4072597 B2JP4072597 B2JP 4072597B2
- Authority
- JP
- Japan
- Prior art keywords
- water
- drug
- tablet
- polyvinyl alcohol
- sustained
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000000203mixtureSubstances0.000titleclaimsdescription90
- 238000009472formulationMethods0.000titleclaimsdescription21
- 230000002459sustained effectEffects0.000titleclaimsdescription9
- 239000003814drugSubstances0.000claimsdescription111
- 229940079593drugDrugs0.000claimsdescription110
- 229920002451polyvinyl alcoholPolymers0.000claimsdescription68
- 229920003169water-soluble polymerPolymers0.000claimsdescription55
- 238000002360preparation methodMethods0.000claimsdescription53
- HYIMSNHJOBLJNT-UHFFFAOYSA-NnifedipineChemical compoundCOC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=OHYIMSNHJOBLJNT-UHFFFAOYSA-N0.000claimsdescription51
- FWLKKPKZQYVAFR-LVEZLNDCSA-N(e)-but-2-enedioic acid;1-(2-ethoxyethyl)-2-(4-methyl-1,4-diazepan-1-yl)benzimidazoleChemical compoundOC(=O)\C=C\C(O)=O.OC(=O)\C=C\C(O)=O.N=1C2=CC=CC=C2N(CCOCC)C=1N1CCCN(C)CC1FWLKKPKZQYVAFR-LVEZLNDCSA-N0.000claimsdescription49
- 229960000325emedastineDrugs0.000claimsdescription49
- 239000004372Polyvinyl alcoholSubstances0.000claimsdescription48
- 229960001597nifedipineDrugs0.000claimsdescription48
- 239000007962solid dispersionSubstances0.000claimsdescription48
- 239000003405delayed action preparationSubstances0.000claimsdescription43
- 229920003088hydroxypropyl methyl cellulosePolymers0.000claimsdescription39
- 235000010979hydroxypropyl methyl celluloseNutrition0.000claimsdescription39
- 239000001866hydroxypropyl methyl celluloseSubstances0.000claimsdescription38
- UFVKGYZPFZQRLF-UHFFFAOYSA-Nhydroxypropyl methyl celluloseChemical compoundOC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1UFVKGYZPFZQRLF-UHFFFAOYSA-N0.000claimsdescription38
- FAPWRFPIFSIZLT-UHFFFAOYSA-MSodium chlorideChemical compound[Na+].[Cl-]FAPWRFPIFSIZLT-UHFFFAOYSA-M0.000claimsdescription34
- 229920003132hydroxypropyl methylcellulose phthalatePolymers0.000claimsdescription31
- 229920000642polymerPolymers0.000claimsdescription31
- 239000002775capsuleSubstances0.000claimsdescription30
- 239000011248coating agentSubstances0.000claimsdescription27
- 238000000576coating methodMethods0.000claimsdescription27
- 239000001856Ethyl celluloseSubstances0.000claimsdescription26
- 229920001249ethyl cellulosePolymers0.000claimsdescription26
- 235000019325ethyl celluloseNutrition0.000claimsdescription26
- ZZSNKZQZMQGXPY-UHFFFAOYSA-NEthyl celluloseChemical compoundCCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1ZZSNKZQZMQGXPY-UHFFFAOYSA-N0.000claimsdescription24
- 150000003839saltsChemical class0.000claimsdescription22
- 229920003176water-insoluble polymerPolymers0.000claimsdescription21
- 239000001509sodium citrateSubstances0.000claimsdescription20
- HRXKRNGNAMMEHJ-UHFFFAOYSA-Ktrisodium citrateChemical compound[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=OHRXKRNGNAMMEHJ-UHFFFAOYSA-K0.000claimsdescription20
- 229940038773trisodium citrateDrugs0.000claimsdescription20
- PMZURENOXWZQFD-UHFFFAOYSA-LSodium SulfateChemical compound[Na+].[Na+].[O-]S([O-])(=O)=OPMZURENOXWZQFD-UHFFFAOYSA-L0.000claimsdescription19
- 229910052938sodium sulfateInorganic materials0.000claimsdescription19
- 235000011152sodium sulphateNutrition0.000claimsdescription19
- 239000011780sodium chlorideSubstances0.000claimsdescription17
- 229940031704hydroxypropyl methylcellulose phthalateDrugs0.000claimsdescription14
- JQSAYKKFZOSZGJ-UHFFFAOYSA-N1-[bis(4-fluorophenyl)methyl]-4-[(2,3,4-trimethoxyphenyl)methyl]piperazineChemical compoundCOC1=C(OC)C(OC)=CC=C1CN1CCN(C(C=2C=CC(F)=CC=2)C=2C=CC(F)=CC=2)CC1JQSAYKKFZOSZGJ-UHFFFAOYSA-N0.000claimsdescription4
- 229950007692lomerizineDrugs0.000claimsdescription4
- 238000011049fillingMethods0.000claimsdescription2
- 230000002688persistenceEffects0.000claimsdescription2
- 238000007127saponification reactionMethods0.000claimsdescription2
- 230000005923long-lasting effectEffects0.000claims4
- 239000006185dispersionSubstances0.000claims1
- 239000006104solid solutionSubstances0.000claims1
- 239000003826tabletSubstances0.000description271
- 239000000243solutionSubstances0.000description93
- LFQSCWFLJHTTHZ-UHFFFAOYSA-NEthanolChemical compoundCCOLFQSCWFLJHTTHZ-UHFFFAOYSA-N0.000description78
- 239000000047productSubstances0.000description53
- YMWUJEATGCHHMB-UHFFFAOYSA-NDichloromethaneChemical compoundClCClYMWUJEATGCHHMB-UHFFFAOYSA-N0.000description39
- 229920002153Hydroxypropyl cellulosePolymers0.000description38
- 235000010977hydroxypropyl celluloseNutrition0.000description38
- HQKMJHAJHXVSDF-UHFFFAOYSA-Lmagnesium stearateChemical compound[Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=OHQKMJHAJHXVSDF-UHFFFAOYSA-L0.000description38
- XLYOFNOQVPJJNP-UHFFFAOYSA-NwaterSubstancesOXLYOFNOQVPJJNP-UHFFFAOYSA-N0.000description30
- 239000007941film coated tabletSubstances0.000description24
- -1methoxyl groupChemical group0.000description23
- 239000001863hydroxypropyl celluloseSubstances0.000description21
- IMROMDMJAWUWLK-UHFFFAOYSA-NEthenolChemical compoundOC=CIMROMDMJAWUWLK-UHFFFAOYSA-N0.000description20
- 235000019359magnesium stearateNutrition0.000description19
- 239000007788liquidSubstances0.000description18
- 230000036470plasma concentrationEffects0.000description18
- 235000012239silicon dioxideNutrition0.000description18
- 239000012085test solutionSubstances0.000description18
- 239000012046mixed solventSubstances0.000description17
- RMAQACBXLXPBSY-UHFFFAOYSA-Nsilicic acidChemical compoundO[Si](O)(O)ORMAQACBXLXPBSY-UHFFFAOYSA-N0.000description17
- 238000010998test methodMethods0.000description17
- 239000011230binding agentSubstances0.000description16
- 238000009505enteric coatingMethods0.000description15
- 239000002702enteric coatingSubstances0.000description15
- 238000007922dissolution testMethods0.000description14
- GUBGYTABKSRVRQ-QKKXKWKRSA-NLactoseNatural productsOC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1OGUBGYTABKSRVRQ-QKKXKWKRSA-N0.000description13
- 239000008101lactoseSubstances0.000description13
- 239000003960organic solventSubstances0.000description13
- 230000036325urinary excretionEffects0.000description13
- QTBSBXVTEAMEQO-UHFFFAOYSA-NAcetic acidChemical compoundCC(O)=OQTBSBXVTEAMEQO-UHFFFAOYSA-N0.000description12
- 238000004090dissolutionMethods0.000description12
- 238000012360testing methodMethods0.000description12
- VEXZGXHMUGYJMC-UHFFFAOYSA-NHydrochloric acidChemical compoundClVEXZGXHMUGYJMC-UHFFFAOYSA-N0.000description11
- 239000012530fluidSubstances0.000description11
- DOOTYTYQINUNNV-UHFFFAOYSA-NTriethyl citrateChemical compoundCCOC(=O)CC(O)(C(=O)OCC)CC(=O)OCCDOOTYTYQINUNNV-UHFFFAOYSA-N0.000description10
- 239000011148porous materialSubstances0.000description10
- NLJMYIDDQXHKNR-UHFFFAOYSA-Ksodium citrateChemical compoundO.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=ONLJMYIDDQXHKNR-UHFFFAOYSA-K0.000description10
- 239000000126substanceSubstances0.000description10
- OKKJLVBELUTLKV-UHFFFAOYSA-NMethanolChemical compoundOCOKKJLVBELUTLKV-UHFFFAOYSA-N0.000description9
- 238000010828elutionMethods0.000description9
- 238000000034methodMethods0.000description9
- OHHDIOKRWWOXMT-UHFFFAOYSA-Ntrazodone hydrochlorideChemical compound[H+].[Cl-].ClC1=CC=CC(N2CCN(CCCN3C(N4C=CC=CC4=N3)=O)CC2)=C1OHHDIOKRWWOXMT-UHFFFAOYSA-N0.000description9
- 229960002301trazodone hydrochlorideDrugs0.000description9
- 230000000694effectsEffects0.000description8
- 238000005507sprayingMethods0.000description8
- 230000000052comparative effectEffects0.000description7
- 238000010586diagramMethods0.000description7
- 210000001035gastrointestinal tractAnatomy0.000description7
- 238000002156mixingMethods0.000description7
- 239000002904solventSubstances0.000description7
- CSCPPACGZOOCGX-UHFFFAOYSA-NAcetoneChemical compoundCC(C)=OCSCPPACGZOOCGX-UHFFFAOYSA-N0.000description6
- 150000008064anhydridesChemical class0.000description6
- 229920003086cellulose etherPolymers0.000description6
- KRKNYBCHXYNGOX-UHFFFAOYSA-Ncitric acidChemical compoundOC(=O)CC(O)(C(O)=O)CC(O)=OKRKNYBCHXYNGOX-UHFFFAOYSA-N0.000description6
- 239000000546pharmaceutical excipientSubstances0.000description6
- 230000008961swellingEffects0.000description6
- 238000009492tablet coatingMethods0.000description6
- 239000002700tablet coatingSubstances0.000description6
- 210000002700urineAnatomy0.000description6
- 239000002253acidSubstances0.000description5
- 230000015572biosynthetic processEffects0.000description5
- 238000001035dryingMethods0.000description5
- 238000004128high performance liquid chromatographyMethods0.000description5
- 239000011159matrix materialSubstances0.000description5
- HEDRZPFGACZZDS-UHFFFAOYSA-NChloroformChemical compoundClC(Cl)ClHEDRZPFGACZZDS-UHFFFAOYSA-N0.000description4
- KFZMGEQAYNKOFK-UHFFFAOYSA-NIsopropanolChemical compoundCC(C)OKFZMGEQAYNKOFK-UHFFFAOYSA-N0.000description4
- 238000010521absorption reactionMethods0.000description4
- 229920002678cellulosePolymers0.000description4
- 239000001913celluloseSubstances0.000description4
- 235000010980celluloseNutrition0.000description4
- 238000004817gas chromatographyMethods0.000description4
- 239000008187granular materialSubstances0.000description4
- 239000007902hard capsuleSubstances0.000description4
- 239000012528membraneSubstances0.000description4
- WEVYAHXRMPXWCK-UHFFFAOYSA-NAcetonitrileChemical compoundCC#NWEVYAHXRMPXWCK-UHFFFAOYSA-N0.000description3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-NSilicium dioxideChemical compoundO=[Si]=OVYPSYNLAJGMNEJ-UHFFFAOYSA-N0.000description3
- 230000027288circadian rhythmEffects0.000description3
- 238000010276constructionMethods0.000description3
- 230000003247decreasing effectEffects0.000description3
- 230000003111delayed effectEffects0.000description3
- 201000010099diseaseDiseases0.000description3
- 208000037265diseases, disorders, signs and symptomsDiseases0.000description3
- 238000005469granulationMethods0.000description3
- 230000003179granulationEffects0.000description3
- 229920000639hydroxypropylmethylcellulose acetate succinatePolymers0.000description3
- 238000004519manufacturing processMethods0.000description3
- 239000002207metaboliteSubstances0.000description3
- 229920000609methyl cellulosePolymers0.000description3
- 239000001923methylcelluloseSubstances0.000description3
- 235000010981methylcelluloseNutrition0.000description3
- 239000001267polyvinylpyrrolidoneSubstances0.000description3
- 229920000036polyvinylpyrrolidonePolymers0.000description3
- 235000013855polyvinylpyrrolidoneNutrition0.000description3
- 239000001069triethyl citrateSubstances0.000description3
- VMYFZRTXGLUXMZ-UHFFFAOYSA-Ntriethyl citrateNatural productsCCOC(=O)C(O)(C(=O)OCC)C(=O)OCCVMYFZRTXGLUXMZ-UHFFFAOYSA-N0.000description3
- 235000013769triethyl citrateNutrition0.000description3
- 229920002261Corn starchPolymers0.000description2
- RTZKZFJDLAIYFH-UHFFFAOYSA-NDiethyl etherChemical compoundCCOCCRTZKZFJDLAIYFH-UHFFFAOYSA-N0.000description2
- NBIIXXVUZAFLBC-UHFFFAOYSA-NPhosphoric acidChemical compoundOP(O)(O)=ONBIIXXVUZAFLBC-UHFFFAOYSA-N0.000description2
- 238000011481absorbance measurementMethods0.000description2
- 239000007864aqueous solutionSubstances0.000description2
- 239000008280bloodSubstances0.000description2
- 210000004369bloodAnatomy0.000description2
- 235000021152breakfastNutrition0.000description2
- 125000002057carboxymethyl groupChemical group[H]OC(=O)C([H])([H])[*]0.000description2
- 238000007906compressionMethods0.000description2
- 230000006835compressionEffects0.000description2
- 229920001577copolymerPolymers0.000description2
- 239000008120corn starchSubstances0.000description2
- 230000007423decreaseEffects0.000description2
- 238000001514detection methodMethods0.000description2
- 239000003480eluentSubstances0.000description2
- 125000001495ethyl groupChemical group[H]C([H])([H])C([H])([H])*0.000description2
- 230000002496gastric effectEffects0.000description2
- 239000000499gelSubstances0.000description2
- 229940031705hydroxypropyl methylcellulose 2910Drugs0.000description2
- CGIGDMFJXJATDK-UHFFFAOYSA-NindomethacinChemical compoundCC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1CGIGDMFJXJATDK-UHFFFAOYSA-N0.000description2
- JEIPFZHSYJVQDO-UHFFFAOYSA-Niron(III) oxideInorganic materialsO=[Fe]O[Fe]=OJEIPFZHSYJVQDO-UHFFFAOYSA-N0.000description2
- YOBAEOGBNPPUQV-UHFFFAOYSA-Niron;trihydrateChemical compoundO.O.O.[Fe].[Fe]YOBAEOGBNPPUQV-UHFFFAOYSA-N0.000description2
- 238000005259measurementMethods0.000description2
- 239000011259mixed solutionSubstances0.000description2
- AJDUTMFFZHIJEM-UHFFFAOYSA-Nn-(9,10-dioxoanthracen-1-yl)-4-[4-[[4-[4-[(9,10-dioxoanthracen-1-yl)carbamoyl]phenyl]phenyl]diazenyl]phenyl]benzamideChemical compoundO=C1C2=CC=CC=C2C(=O)C2=C1C=CC=C2NC(=O)C(C=C1)=CC=C1C(C=C1)=CC=C1N=NC(C=C1)=CC=C1C(C=C1)=CC=C1C(=O)NC1=CC=CC2=C1C(=O)C1=CC=CC=C1C2=OAJDUTMFFZHIJEM-UHFFFAOYSA-N0.000description2
- XNGIFLGASWRNHJ-UHFFFAOYSA-Lphthalate(2-)Chemical compound[O-]C(=O)C1=CC=CC=C1C([O-])=OXNGIFLGASWRNHJ-UHFFFAOYSA-L0.000description2
- 239000007974sodium acetate bufferSubstances0.000description2
- 238000013268sustained releaseMethods0.000description2
- 239000012730sustained-release formSubstances0.000description2
- 230000001225therapeutic effectEffects0.000description2
- URAYPUMNDPQOKB-UHFFFAOYSA-NtriacetinChemical compoundCC(=O)OCC(OC(C)=O)COC(C)=OURAYPUMNDPQOKB-UHFFFAOYSA-N0.000description2
- 239000001043yellow dyeSubstances0.000description2
- TURGQPDWYFJEDY-UHFFFAOYSA-N1-hydroperoxypropaneChemical compoundCCCOOTURGQPDWYFJEDY-UHFFFAOYSA-N0.000description1
- LCCVJQVUSQPANR-UHFFFAOYSA-N5-methoxycarbonyl-2,6-dimethyl-4-(2-nitrophenyl)pyridine-3-carboxylic acidChemical compoundCOC(=O)C1=C(C)N=C(C)C(C(O)=O)=C1C1=CC=CC=C1[N+]([O-])=OLCCVJQVUSQPANR-UHFFFAOYSA-N0.000description1
- GUBGYTABKSRVRQ-XLOQQCSPSA-NAlpha-LactoseChemical compoundO[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1OGUBGYTABKSRVRQ-XLOQQCSPSA-N0.000description1
- RYGMFSIKBFXOCR-UHFFFAOYSA-NCopperChemical compound[Cu]RYGMFSIKBFXOCR-UHFFFAOYSA-N0.000description1
- FBPFZTCFMRRESA-KVTDHHQDSA-ND-MannitolChemical compoundOC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)COFBPFZTCFMRRESA-KVTDHHQDSA-N0.000description1
- FDJCVHVKXFIEPJ-JCNFZFLDSA-NDelapril hydrochlorideChemical compoundCl.C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1FDJCVHVKXFIEPJ-JCNFZFLDSA-N0.000description1
- LCGLNKUTAGEVQW-UHFFFAOYSA-NDimethyl etherChemical compoundCOCLCGLNKUTAGEVQW-UHFFFAOYSA-N0.000description1
- LYCAIKOWRPUZTN-UHFFFAOYSA-NEthylene glycolChemical compoundOCCOLYCAIKOWRPUZTN-UHFFFAOYSA-N0.000description1
- 229920003152Eudragit® RS polymerPolymers0.000description1
- GHASVSINZRGABV-UHFFFAOYSA-NFluorouracilChemical compoundFC1=CNC(=O)NC1=OGHASVSINZRGABV-UHFFFAOYSA-N0.000description1
- HEFNNWSXXWATRW-UHFFFAOYSA-NIbuprofenChemical compoundCC(C)CC1=CC=C(C(C)C(O)=O)C=C1HEFNNWSXXWATRW-UHFFFAOYSA-N0.000description1
- DMPRDSPPYMZQBT-CEAXSRTFSA-NIfenprodil tartrateChemical compoundOC(=O)[C@H](O)[C@@H](O)C(O)=O.C1CC(CC=2C=CC=CC=2)CCN1C(C)C(O)C1=CC=C(O)C=C1.C1CC(CC=2C=CC=CC=2)CCN1C(C)C(O)C1=CC=C(O)C=C1DMPRDSPPYMZQBT-CEAXSRTFSA-N0.000description1
- 229930195725MannitolNatural products0.000description1
- 229920000168Microcrystalline cellulosePolymers0.000description1
- SNIOPGDIGTZGOP-UHFFFAOYSA-NNitroglycerinChemical compound[O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=OSNIOPGDIGTZGOP-UHFFFAOYSA-N0.000description1
- 239000000006NitroglycerinSubstances0.000description1
- DBMJMQXJHONAFJ-UHFFFAOYSA-MSodium laurylsulphateChemical compound[Na+].CCCCCCCCCCCCOS([O-])(=O)=ODBMJMQXJHONAFJ-UHFFFAOYSA-M0.000description1
- 229920002472StarchPolymers0.000description1
- 229930006000SucroseNatural products0.000description1
- CZMRCDWAGMRECN-UGDNZRGBSA-NSucroseChemical compoundO[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1CZMRCDWAGMRECN-UGDNZRGBSA-N0.000description1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-NTitan oxideChemical compoundO=[Ti]=OGWEVSGVZZGPLCZ-UHFFFAOYSA-N0.000description1
- YWXYYJSYQOXTPL-JGWLITMVSA-N[(3r,3ar,6s,6as)-3-hydroxy-2,3,3a,5,6,6a-hexahydrofuro[3,2-b]furan-6-yl] nitrateChemical compound[O-][N+](=O)O[C@H]1CO[C@@H]2[C@H](O)CO[C@@H]21YWXYYJSYQOXTPL-JGWLITMVSA-N0.000description1
- YKTSYUJCYHOUJP-UHFFFAOYSA-N[O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-]Chemical compound[O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-]YKTSYUJCYHOUJP-UHFFFAOYSA-N0.000description1
- 238000002835absorbanceMethods0.000description1
- 125000002777acetyl groupChemical group[H]C([H])([H])C(*)=O0.000description1
- 239000000654additiveSubstances0.000description1
- 125000003158alcohol groupChemical group0.000description1
- 229960000473altretamineDrugs0.000description1
- 229910000147aluminium phosphateInorganic materials0.000description1
- 229920003144amino alkyl methacrylate copolymerPolymers0.000description1
- 229940035676analgesicsDrugs0.000description1
- 239000000730antalgic agentSubstances0.000description1
- 239000000043antiallergic agentSubstances0.000description1
- 239000000935antidepressant agentSubstances0.000description1
- 229940005513antidepressantsDrugs0.000description1
- 239000002246antineoplastic agentSubstances0.000description1
- 230000002051biphasic effectEffects0.000description1
- FAKRSMQSSFJEIM-RQJHMYQMSA-NcaptoprilChemical compoundSC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=OFAKRSMQSSFJEIM-RQJHMYQMSA-N0.000description1
- 229960000830captoprilDrugs0.000description1
- 239000003795chemical substances by applicationSubstances0.000description1
- 239000007931coated granuleSubstances0.000description1
- 239000008119colloidal silicaSubstances0.000description1
- 238000013329compoundingMethods0.000description1
- 238000000748compression mouldingMethods0.000description1
- 238000013267controlled drug releaseMethods0.000description1
- 238000007796conventional methodMethods0.000description1
- MTHSVFCYNBDYFN-UHFFFAOYSA-Ndiethylene glycolChemical compoundOCCOCCOMTHSVFCYNBDYFN-UHFFFAOYSA-N0.000description1
- 230000001079digestive effectEffects0.000description1
- 238000011978dissolution methodMethods0.000description1
- 229960001253domperidoneDrugs0.000description1
- FGXWKSZFVQUSTL-UHFFFAOYSA-NdomperidoneChemical compoundC12=CC=CC=C2NC(=O)N1CCCN(CC1)CCC1N1C2=CC=C(Cl)C=C2NC1=OFGXWKSZFVQUSTL-UHFFFAOYSA-N0.000description1
- 239000002552dosage formSubstances0.000description1
- 238000001647drug administrationMethods0.000description1
- 230000005264electron captureEffects0.000description1
- 150000002148estersChemical class0.000description1
- FSXVSUSRJXIJHB-UHFFFAOYSA-Methyl prop-2-enoate;methyl 2-methylprop-2-enoate;trimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azanium;chlorideChemical compound[Cl-].CCOC(=O)C=C.COC(=O)C(C)=C.CC(=C)C(=O)OCC[N+](C)(C)CFSXVSUSRJXIJHB-UHFFFAOYSA-M0.000description1
- VJJPUSNTGOMMGY-MRVIYFEKSA-NetoposideChemical compoundCOC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1VJJPUSNTGOMMGY-MRVIYFEKSA-N0.000description1
- 229960005420etoposideDrugs0.000description1
- 229960002949fluorouracilDrugs0.000description1
- 210000004051gastric juiceAnatomy0.000description1
- 125000002791glucosyl groupChemical groupC1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)*0.000description1
- 239000001087glyceryl triacetateSubstances0.000description1
- 235000013773glyceryl triacetateNutrition0.000description1
- 229960003711glyceryl trinitrateDrugs0.000description1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-NhexamethylmelamineChemical compoundCN(C)C1=NC(N(C)C)=NC(N(C)C)=N1UUVWYPNAQBNQJQ-UHFFFAOYSA-N0.000description1
- 150000004677hydratesChemical class0.000description1
- 229940031702hydroxypropyl methylcellulose 2208Drugs0.000description1
- 229960001680ibuprofenDrugs0.000description1
- 229960000204ifenprodil tartrateDrugs0.000description1
- 239000012729immediate-release (IR) formulationSubstances0.000description1
- 238000001727in vivoMethods0.000description1
- 229960000905indomethacinDrugs0.000description1
- 239000004615ingredientSubstances0.000description1
- 238000007689inspectionMethods0.000description1
- 238000011835investigationMethods0.000description1
- 238000004811liquid chromatographyMethods0.000description1
- 239000000314lubricantSubstances0.000description1
- 229960003511macrogolDrugs0.000description1
- 239000000594mannitolSubstances0.000description1
- 235000010355mannitolNutrition0.000description1
- 230000007721medicinal effectEffects0.000description1
- 229960004503metoclopramideDrugs0.000description1
- TTWJBBZEZQICBI-UHFFFAOYSA-NmetoclopramideChemical compoundCCN(CC)CCNC(=O)C1=CC(Cl)=C(N)C=C1OCTTWJBBZEZQICBI-UHFFFAOYSA-N0.000description1
- 235000019813microcrystalline celluloseNutrition0.000description1
- 239000008108microcrystalline celluloseSubstances0.000description1
- 229940016286microcrystalline celluloseDrugs0.000description1
- 239000007932molded tabletSubstances0.000description1
- AIKVCUNQWYTVTO-UHFFFAOYSA-Nnicardipine hydrochlorideChemical compoundCl.COC(=O)C1=C(C)NC(C)=C(C(=O)OCCN(C)CC=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1AIKVCUNQWYTVTO-UHFFFAOYSA-N0.000description1
- 229960002289nicardipine hydrochlorideDrugs0.000description1
- 229940079468nifedipine 20 mgDrugs0.000description1
- 239000006186oral dosage formSubstances0.000description1
- 239000002245particleSubstances0.000description1
- 230000000737periodic effectEffects0.000description1
- 239000012466permeateSubstances0.000description1
- 239000004014plasticizerSubstances0.000description1
- 229920006316polyvinylpyrrolidinePolymers0.000description1
- 230000002265preventionEffects0.000description1
- 230000003449preventive effectEffects0.000description1
- 230000033764rhythmic processEffects0.000description1
- 239000000377silicon dioxideSubstances0.000description1
- 238000004513sizingMethods0.000description1
- 235000019333sodium laurylsulphateNutrition0.000description1
- 239000008107starchSubstances0.000description1
- 235000019698starchNutrition0.000description1
- 239000013589supplementSubstances0.000description1
- 230000001629suppressionEffects0.000description1
- 208000024891symptomDiseases0.000description1
- 239000000454talcSubstances0.000description1
- 229910052623talcInorganic materials0.000description1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-NtegafurChemical compoundO=C1NC(=O)C(F)=CN1[C@@H]1OCCC1WFWLQNSHRPWKFK-ZCFIWIBFSA-N0.000description1
- 229960001674tegafurDrugs0.000description1
- OGIDPMRJRNCKJF-UHFFFAOYSA-Ntitanium oxideInorganic materials[Ti]=OOGIDPMRJRNCKJF-UHFFFAOYSA-N0.000description1
- 229960002622triacetinDrugs0.000description1
- 239000003643water by typeSubstances0.000description1
Images
















Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Description
Translated fromJapanese【0001】
【発明の属する技術分野】
本発明は、新規な持続性製剤に関する。さらに詳しくは、(イ)薬物と水溶性高分子との混合物、またはそれらからなる固体分散体、(ロ)ポリビニルアルコール、および(ハ)クエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムからなる群より選択される1種または2種以上の塩とを含有する素錠が、(ニ)水不溶性高分子と、水溶性高分子および/または腸溶性高分子とからなる皮膜で被覆された持続性製剤に関する。
【0002】
【従来の技術】
薬物の血漿中濃度を長時間一定に保つことは、疾患の予防および治療効果を高めるとともに、薬剤の投与回数を軽減し、患者のコンプライアンスを向上させる点から多くの利点を有している。このために、製剤からの薬物放出速度をコントロールした種々の持続性製剤が開発され、臨床上広く用いられている。
【0003】
例えば、特公平6−11699号には、速放性の形態で薬物(ニフェジピン)を含有する芯を、親水性ゲル形成重合体および薬物からなる皮膜で圧縮コーティングした錠剤が開示されている。しかしながら、この錠剤は1日1回投与型製剤として適度な持続性が得られてはいるが、必ずしも十分に満足しうるものではない上、製剤を調製するために二段階の圧縮が必要である等製造工程が煩雑であるという問題点を有している。
【0004】
また、上記の持続性製剤以外にも、薬物を含有する芯物質あるいは素錠に水不溶性高分子と水溶性高分子の皮膜を被覆して製造するメンブランコーティング顆粒やメンブランコーティング錠、あるいは薬物を水不溶性高分子またはワックス等とともに打錠して製造するマトリックス錠等が種々検討されている。しかし、これらの製剤では、時間の経過とともに薬物の放出速度が低下するために、生体内に投与された際に、経時的に薬物の血漿中濃度が低下しやすくなり、長時間に渡って薬物の血漿中濃度を持続化させることが困難であるという問題点を有している。
【0005】
また、近年、生体の周期的リズム(サーカディアンリズム)と疾患との関連が注目され、薬物治療においても、サーカディアンリズムを考慮する必要性が重要視されるようになってきている。
【0006】
以上のことから、さらに種々の工夫が為された持続性製剤の検討が行われている。
【0007】
【発明が解決しようとする課題】
本発明の目的は、長時間に渡って薬物が持続的に放出され、1日1〜2回投与により十分な予防および治療効果が期待できる持続性製剤を提供することにある。
【0008】
【課題を解決するための手段】
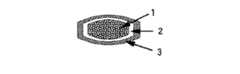
本発明者等は、上記目的を達成するために鋭意研究を行った結果、(イ)薬物と水溶性高分子との混合物、またはそれらからなる固体分散体、(ロ)ポリビニルアルコール、および(ハ)クエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムからなる群より選択される1種または2種以上の塩とを含有する素錠を、(ニ)水不溶性高分子と、水溶性高分子および/または腸溶性高分子とからなる皮膜で被覆することにより製造される持続性製剤(図1および図5)が、本発明の目的に適うことを見い出し、本発明を完成した。
【0009】
本発明によれば、本発明の持続性製剤を生体に投与した際に、水不溶性高分子と水溶性高分子からなる皮膜(以下、このような皮膜を水溶性ポア形成皮膜という)、または水不溶性高分子と腸溶性高分子からなる皮膜(以下、このような皮膜を腸溶性ポア形成皮膜という)、あるいは水不溶性高分子、水溶性高分子および腸溶性高分子からなる皮膜(以下、このような皮膜を水溶性腸溶性ポア形成皮膜という)中の水溶性高分子および/または腸溶性高分子が消化管液に溶解して、皮膜中に細孔(ポア)が形成され、その細孔を通して素錠表面およびその近傍の薬物が徐々に放出される。一方、それに伴い、素錠中のポリビニルアルコールが徐々に膨潤して膨潤力が生じ、その膨潤力により皮膜が破裂し、素錠中の薬物がフリーに放出される。ただし、素錠のマトリックスはポリビニルアルコールの膨潤に伴いゲルマトリックス状となってそれに含まれる薬物は徐々に放出される。
【0010】
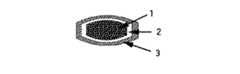
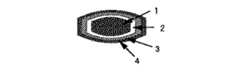
本発明の持続性製剤においては、要すれば、薬物血漿中濃度をさらに持続化させるために、上記持続性製剤の表面に腸溶性高分子からなる皮膜(以下、このような皮膜を腸溶性皮膜という)を被覆することができる(図2および図6参照)。また、投与初期の薬物の血漿中濃度を確保するために、要すれば、前記持続性製剤あるいは上記の腸溶性皮膜を被覆した持続性製剤の表面に、薬物と水溶性高分子との混合物、またはそれらからなる固体分散体を被覆することができる(図3、図4、図7および図8参照)。
【0011】
【発明の実施の形態】
本発明に用いる水溶性高分子としては、水溶性セルロースエーテルおよびポリビニルピロリドン等が挙げられる。
【0012】
上記水溶性セルロースエーテルとは、セルロース中のブドウ糖残基であるアルコール基の一部をメチルエーテル、ヒドロキシプロピルエーテルおよび/またはヒドロキシエチルエーテル等の形に置換したもので、かつ水に可溶性のものである。
【0013】
上記水溶性セルロースエーテルとしては、メチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース等が挙げられる。
【0014】
上記のメチルセルロースは、メトキシル基の含量が26.0〜33.0%のものが好ましく、第十二改正日本薬局方(第一法規出版社発行、1991年、以下局方と略記する)の1070頁に記載のメチルセルロースが好適に使用される。これは例えばメトローズSMなる商品名(信越化学工業社製)のものが入手できる。ヒドロキシプロピルセルロース(以下、HPCともいう)は、ヒドロキシプロポキシル基の含量が53.4〜77.5%のものが好ましく、局方の1023頁に記載のヒドロキシプロピルセルロースが好適に使用される。これは例えば日曹HPCなる商品名(日本曹達社製)のものが入手できる。ヒドロキシプロピルメチルセルロース(以下、HPMCともいう)としては、メトキシル基およびヒドロキシプロポキシル基の含量がそれぞれ19.0〜30.0%、4.0〜12.0%のものが好ましく、局方の1025頁および1027頁に記載のヒドロキシプロピルメチルセルロース2208、ヒドロキシプロピルメチルセルロース2906およびヒドロキシプロピルメチルセルロース2910が好適に使用される。これらは例えばそれぞれメトローズ90SH、メトローズ65SHおよびTC−5なる商品名(いずれも信越化学工業社製)のものが入手できる。
【0015】
上記のポリビニルピロリドンは、分子量約25000〜約1200000のものが使用し得るが、分子量約40000のものが好ましく、第十二改正日本薬局方第二追補(第一法規出版社発行、1995年)の75頁に記載のポリビニルピロリドンK30が好適に使用される。
【0016】
上記水溶性高分子は、素錠中および皮膜中のいずれにも用いることができ、それら水溶性高分子を適宜混合して使用することもできる。例えば水溶性セルロースエーテルの1種を単独で、あるいは2種以上の混合物として、もしくは水溶性セルロースエーテルの1種または2種以上とポリビニルピロリドンとの混合物として用いることができるが、皮膜中には水溶性セルロースエーテルの1種または2種以上のみで用いることが好ましい。また、皮膜中には、これら水溶性高分子と後述する腸溶性高分子を適宜混合して使用することもできる。
【0017】
上記水溶性高分子の中でも、メトキシル基およびヒドロキシプロポキシル基の含量がそれぞれ28.0〜30.0%、7.0〜12.0%のヒドロキシプロピルメチルセルロースが好ましく、局方の1027頁に記載のヒドロキシプロピルメチルセルロース2910が特に好適に使用される。
【0018】
素錠中に用いる水溶性高分子は、薬物の吸収をコントロールするために配合されるものであって、薬物の吸収性の程度に応じて配合形態も変えてもよい。例えば、消化管からの吸収が良好な薬物の場合には、薬物と水溶性高分子とを単に混合して混合物として用いるか、もしくは薬物と他の(ロ)成分および(ハ)成分との混合物を造粒する際に水溶性高分子の水溶液を結合剤として用いてもよい。さらに、上記造粒に際して薬物単独の代わりに薬物と水溶性高分子との混合物を用いてもよい。一方、薬物が水難溶性薬物等の消化管からの吸収が悪い場合には、薬物と水溶性高分子の有機溶媒溶液から溶媒を除去して得られる固体分散体として用いるか、もしくはその一部を上記と同様にして水溶性高分子との混合物またはそれを結合剤として造粒物として用いることができる。水難溶性薬物を固体分散体とすることにより、消化管からの薬物吸収性が改善しうる。
【0019】
本発明に用いるポリビニルアルコールは、例えば、医薬品添加物規格(薬事日報社発行、1993年)の304頁および307頁にそれぞれ記載の完全けん化物あるいは部分けん化物のいずれも使用できるが、これらの中でも水と接触したときに膨潤力の大きなものが好適に使用される。これは例えばポリビニルアルコ−ルのけん化度が93.5〜97.5%のものを挙げることができる。これらは例えばクラレポバールPVA−613、クラレポバールPVA−CST、クラレポバールPVA−CSTS、クラレポバールPVA−CSなる商品名(いずれも株式会社クラレ製)のものが入手できる。
【0020】
本発明に用いるクエン酸三ナトリウム、硫酸ナトリウムまたは塩化ナトリウムは、それぞれそれらの無水物およびその水和物を使用することができる。また、これらを適宜混合して使用することもできる。
【0021】
本発明に用いる水不溶性高分子としては、エチルセルロース、アクリル酸エチル−メタアクリル酸メチル−メタアクリル酸塩化トリメチルアンモニウムエチル共重合体等が挙げられる。
【0022】
上記エチルセルロースは、エトキシル基の含量が46.5〜51.0%のものが好ましく、医薬品添加物規格の74頁に記載のエチルセルロースが好適に使用される。これは例えばエトセルなる商品名(ダウ・ケミカル社製)のものが入手できる。アクリル酸エチル−メタアクリル酸メチル−メタアクリル酸塩化トリメチルアンモニウムエチル共重合体は、医薬品添加物規格の45頁に記載のアミノアルキルメタアクリレート コポリマーRSが好適に使用される。これは例えばオイドラギットRSなる商品名(レーム・ファーマ社製)のものが入手できる。
【0023】
上記の水不溶性高分子の中でも、エトキシル基の含量が46.5〜51.0%のエチルセルロースが特に好ましい。
【0024】
本発明に用いる腸溶性高分子としては、ヒドロキシプロピルメチルセルロースフタレ−ト(以下、HPMCPともいう)、ヒドロキシプロピルメチルセルロースアセテートサクシネート、カルボキシメチルエチルセルロース等が挙げられる。
【0025】
ヒドロキシプロピルメチルセルロースフタレ−トとしては、メトキシル基、ヒドロキシプロポキシル基およびカルボキシベンゾイル基の含量がそれぞれ18.0〜24.0%、5.0〜10.0%、21.0〜35.0%のものが好ましく、局方の1028頁および1029頁に記載のヒドロキシプロピルメチルセルロースフタレ−ト200731、ヒドロキシプロピルメチルセルロースフタレ−ト220824が好適に使用される。これらは例えばそれぞれHPMCP HP−55、HPMCP HP−50なる商品名(いずれも信越化学工業社製)のものが入手できる。ヒドロキシプロピルメチルセルロースアセテートサクシネートは、ヒドロキシプロピルメチルセルロースの酢酸およびモノコハク酸の混合エステルで、メトキシル基、ヒドロキシプロポキシル基、アセチル基およびサクシノイル基がそれぞれ12.0〜28.0%、4.0〜23.0%、2.0〜16.0%、4.0〜28.0%のものが好ましく、医薬品添加物規格の257頁に記載のヒドロキシプロピルメチルセルロースアセテートサクシネートが好適に使用される。これらは例えば信越エーコート(AQOAT)なる商品名(信越化学工業社製)のものが入手できる。カルボキシメチルエチルセルロースとしては、セルロースのカルボキシメチルおよびエチルの混合エーテルで、カルボキシメチル基、エトキシル基がそれぞれ8.9〜14.9%、32.5〜43.0%のものが好ましく、医薬品添加物規格の105頁に記載のカルボキシメチルエチルセルロースが好適に使用される。これらは例えばCMECなる商品名(フロイント産業社製)のものが入手できる。また、これらの腸溶性高分子を適宜混合して使用することもできる。
【0026】
上記の腸溶性高分子の中でも、メトキシル基、ヒドロキシプロポキシル基およびカルボキシベンゾイル基の含量がそれぞれ18.0〜22.0%、5.0〜9.0%、27.0〜35.0%のヒドロキシプロピルメチルセルロースフタレ−トが好ましく、局方の1028頁に記載のヒドロキシプロピルメチルセルロースフタレ−ト200731が特に好適に使用される。
【0027】
本発明の持続性製剤は、以下の製造方法により製造することができる。まず、(イ)成分中の薬物または薬物と水溶性高分子と、(ロ)成分のポリビニルアルコール、および(ハ)成分のクエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムからなる群より選択される1種または2種以上の塩とを混合した後、これら成分(イ)、(ロ)および(ハ)の混合物を水溶性高分子の水溶液を結合剤として用いて造粒し、薬物含有の造粒物を製造するか、成分(ロ)および(ハ)の混合物に、薬物と水溶性高分子とを有機溶媒に溶解した溶液(この溶液を固体分散体溶液という)を噴霧、乾燥するか、もしくは固体分散体溶液を成分(ロ)および(ハ)の混合物に加えて練合した後に溶媒を留去し、粉砕して薬物含有の造粒物を製造する。
【0028】
次に、上記の薬物含有の造粒物を打錠して素錠を調製する。
【0029】
さらに、上記素錠表面に、水不溶性高分子と水溶性高分子とを、水および/または有機溶媒に分散または溶解させた液(以下、この液を水溶性ポア形成皮膜液という)を噴霧、乾燥して水溶性ポア形成皮膜を被覆させた錠剤(以下、これを水溶性ポア形成皮膜被覆錠という)を調製するか、水不溶性高分子と腸溶性高分子とを、水および/または有機溶媒に分散または溶解させた液(以下、この液を腸溶性ポア形成皮膜液という)を噴霧、乾燥して腸溶性ポア形成皮膜を被覆させた錠剤(以下、これを腸溶性ポア形成皮膜被覆錠という)を調製するか、あるいは水不溶性高分子、水溶性高分子および腸溶性高分子とを、水および/または有機溶媒に分散または溶解させた液(以下、この液を水溶性腸溶性ポア形成皮膜液という)を噴霧、乾燥して水溶性腸溶性ポア形成皮膜を被覆させた錠剤(以下、これを水溶性腸溶性ポア形成皮膜被覆錠という)を調製することにより、本発明の持続性製剤を製造することができる。
【0030】
本発明の持続性製剤においては、要すれば、前記持続性製剤の表面に、腸溶性皮膜を上記と同様の方法で、腸溶性高分子を、水および/または有機溶媒に分散または溶解させた液(以下、この液を腸溶性皮膜液という)を噴霧、乾燥して被覆して錠剤(以下、このような錠剤を腸溶性皮膜被覆錠という)を調製することにより、薬物の血漿中濃度をさらに持続化させることが可能な製剤とすることもできる。また、要すれば、前記水溶性ポア形成皮膜被覆錠、腸溶性ポア形成皮膜被覆錠、水溶性腸溶性ポア形成皮膜被覆錠あるいは腸溶性皮膜被覆錠の表面に、薬物と水溶性高分子との混合物、またはそれらからなる固体分散体を上記と同様の方法で被覆することにより、投与初期の薬物の血漿中濃度を確保することもできる。
【0031】
ポリビニルアルコール、およびクエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムからなる群より選択される1種または2種以上の塩からなる前記混合物には、医薬品に使用される通常の賦形剤(例えば、でんぷん、白糖、乳糖、マンニトール、結晶セルロース等)の1種または2種以上を適宜混合することができる。また、前記造粒物に、滑沢剤(例えば、ステアリン酸マグネシウム、タルク等)および流動化剤(例えば、軽質無水ケイ酸、含水二酸化ケイ素、合成ケイ酸アルミニウム等)の1種または2種以上を適宜混合して、素錠を調製することができる。
【0032】
素錠の直径は通常2〜10mmであり、好ましくは2〜6mm、より好ましくは5mmである。
【0033】
素錠に配合する場合の水溶性高分子の配合量は、水溶性高分子を結合剤として用いる場合には、薬物1重量部に対して、通常0.01〜20重量部、好ましくは0.05〜5重量部である。また、前記のポリビニルアルコール、およびクエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムからなる群より選択される1種または2種以上の塩からなる混合物に、水溶性高分子を混合して用いる場合は、薬物1重量部に対して、通常0.01〜150重量部、好ましくは0.1〜30重量部である。
【0034】
一方、水溶性高分子を固体分散体の調製に用いる場合には、水溶性高分子の配合量は、薬物1重量部に対して、通常1〜7重量部、好ましくは1.5〜3重量部である。
【0035】
固体分散体溶液に用いる有機溶媒としては、薬物と水溶性高分子のいずれも溶解させることのできる溶媒であれば特に限定されず、例えばメタノール、エタノール、イソプロパノール、アセトン、クロロホルム、ジクロルメタンの1種または2種以上を混合して用いることができる。有機溶媒の使用量は、薬物と水溶性高分子とを室温で溶解するのに要する量の1.2〜3倍量が適当である。
【0036】
素錠中のポリビニルアルコ−ルの配合量は、薬物1重量部に対して、通常0.1〜400重量部、好ましくは1〜50重量部である。クエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムからなる群より選択される1種または2種以上の塩の配合量は、薬物1重量部に対して、通常0.01〜300重量部、好ましくは0.1〜50重量部である。
【0037】
皮膜形成用の水不溶性高分子と、水溶性高分子および/または腸溶性高分子の使用量は、素錠重量に対して、通常5〜20重量%が適当である。水不溶性高分子と水溶性高分子および/または腸溶性高分子の配合比率は、水不溶性高分子1重量部に対して、水溶性高分子および腸溶性高分子を単独で用いる場合も併用する場合もいずれもその総量として通常0.4〜0.8重量部が適当である。
【0038】
水溶性ポア形成皮膜液、腸溶性ポア形成皮膜液、水溶性腸溶性ポア形成皮膜液および腸溶性皮膜液には、水不溶性高分子と、水溶性高分子および/または腸溶性高分子の他に、可塑剤(例えば、トリアセチン、クエン酸トリエチル、マクロゴ−ル6000等)等の添加剤の1種または2種以上を適宜混合することができる。その配合量は、水不溶性高分子と、水溶性高分子および/または腸溶性高分子の総量に対して、5〜40重量%である。
【0039】
水溶性ポア形成皮膜液、腸溶性ポア形成皮膜液または水溶性腸溶性ポア形成皮膜液に用いる有機溶媒としては、特に限定はなく、例えば、メタノール、エタノール、イソプロパノール、アセトン、クロロホルム、ジクロルメタンの1種または2種以上を混合して用いることができるが、水と有機溶媒との混合溶媒を用いる場合には両溶媒が混じり合う有機溶媒を選択する必要がある。
【0040】
腸溶性皮膜被覆錠とする場合に、腸溶性高分子の使用量は、水溶性ポア形成皮膜被覆錠重量、腸溶性ポア形成皮膜被覆錠重量または水溶性腸溶性ポア形成皮膜被覆錠重量に対して、4〜12重量%が適当である。
【0041】
投与初期の薬物の血漿中濃度を確保するために、薬物を、結合剤または固体分散体溶液を使用して錠剤表面に被覆する場合、薬物を含有する皮膜あるいは固体分散体皮膜には、薬物量として本発明の持続性製剤に使用する薬物全量の1〜30重量%、好ましくは5〜25重量%を配合する。
【0042】
なお、薬物が光に対して不安定な場合、薬物の安定性を保持するために、例えばヒドロキシプロピルメチルセルロースと酸化チタン、三二酸化鉄、黄色三二酸化鉄、食用黄色色素4号または食用黄色色素5号等を水および/または有機溶媒に溶解または分散した液を、本発明の持続性製剤に噴霧、乾燥して遮光皮膜を被覆することもできる。
【0043】
上述の方法は錠剤の製造法について記載したが、本発明の持続性製剤は錠剤に限られず、他の剤形とすることもでき、例えば、直径2〜6mmの素錠を使用して製造されたものは、通常の方法によりカプセルに充填し、カプセル剤とすることもできる。
【0044】
本発明の持続性製剤に含有させる薬物としては、血漿中濃度を長時間に渡って持続化させることにより治療効果を上げることができる薬物であれば、特に限定されるものではなく、それら薬物としては、循環器用薬(例えば、ニフェジピン、塩酸ロメリジン、塩酸ニカルジピン、ニトログリセリン、硝酸イソソルビド、カプトプリル、塩酸デラプリル、酒石酸イフェンプロジル)、消化器官用薬(例えば、ドンペリドン、メトクロプラミド)、抗アレルギー剤(例えば、フマル酸エメダスチン)、抗うつ剤(例えば、塩酸トラゾドン)、鎮痛剤(例えば、インドメタシン、イブプロフェン)、制ガン剤(例えば、アルトレタミン、フトラフール、フルオロウラシル、エトポシド)等を挙げることができる。これら薬物の配合量は、それら各薬物について知られている薬効を奏する単位用量であればよく、例えば、1日1回投与用には1日用量を配合する。
【0045】
上記薬物の中でも、特に水難溶性薬物のニフェジピンを含有させた本発明の持続性製剤は、経口吸収性および血漿中濃度の持続性に優れ、薬物の1日1回投与用製剤として有用である。
【0046】
本発明の持続性製剤は、各種疾患の予防および治療を目的として、通常成人1日当たり薬物投与量を1日1〜2回経口投与する。
【0047】
【発明の効果】
本発明の持続性製剤からの薬物の放出は、少なくとも以下に示した二段階で起こる。
【0048】
まず、第一段階として、水不溶性高分子と、水溶性高分子および/または腸溶性高分子とからなる水溶性ポア形成皮膜、腸溶性ポア形成皮膜あるいは水溶性腸溶性ポア形成皮膜中の、水溶性高分子および/または腸溶性高分子が消化管液に溶解することにより、皮膜中に細孔が形成される。このようにして生成した細孔より、消化管液が素錠中に浸透し、素錠中の薬物が溶解して徐々に放出される。この際、素錠中に水溶性高分子を添加することにより、薬物の放出を抑制することも可能である。また、薬物が徐々に放出されると同時に、素錠中に浸透した消化管液によって(ロ)成分のポリビニルアルコ−ルが膨潤し、薬物放出が助長されるが、この場合、素錠中に配合されている(ハ)成分たるクエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムがポリビニルアルコ−ルの膨潤抑制作用を示し、それらの塩が消化管液に溶解し、その溶液が皮膜の外に徐々に拡散することにより、素錠中の塩の濃度が低下し、しかる後にポリビニルアルコールが消化管液によって膨潤し始める。
【0049】
第二段階として、ポリビニルアルコールの膨潤力が大きくなると皮膜が破裂し、素錠中の薬物がフリーに放出される。この場合、素錠中のポリビニルアルコ−ルがマトリックスを形成して、その中に薬物が包含されているため、そのマトリックスがゲル化し、表面から徐々に崩壊するのに伴い、薬物が徐々に放出される。
【0050】
以上のことから、製剤全体としての薬物放出パターンは、放出初期の速度は遅く後期は速い二相性のパターンや、放出初期と後期の放出速度が遅く中期は速いシグモイド型のパターン、あるいは薬物の放出速度が一定である0次型のパターンを示す。
【0051】
本発明の持続性製剤においては、素錠中へのポリビニルアルコ−ルの配合量、水溶性高分子の配合量や、ポリビニルアルコ−ルの膨潤抑制作用を有する、クエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムからなる群より選択される1種または2種以上の塩の配合量あるいは水不溶性高分子と、水溶性高分子および/または腸溶性高分子の配合比率や皮膜の被覆量等を調節することにより、皮膜の細孔を通して薬物が放出される第一段階の放出速度と、皮膜の破裂時間および皮膜が破裂した後に薬物が放出される第二段階の放出速度を任意に調節することができ、それによって上記のような種々の放出パターンを有する製剤が調製し得る。従って、本発明の持続性製剤においては、薬物に応じて、それに適する放出パターンを適宜選択することができるため、本発明の持続性製剤は1日1〜2回の経口投与型製剤に適した持続性製剤となり得る。
【0052】
上記持続性製剤の表面に、腸溶性皮膜を被覆すること、あるいは上記持続性製剤または腸溶性皮膜被覆錠に、薬物と水溶性高分子との混合物、またはそれらからなる固体分散体を被覆することによって、薬物の血漿中濃度をさらに持続化させることや投与初期の薬物の血漿中濃度を確保できる。
【0053】
これらのことから、本発明の持続性製剤を、生体のサーカディアンリズムを考慮した製剤とすることも可能である。
【0054】
さらに、本発明製剤の一実施態様は小型の錠剤形態であるので、症状や年齢等に合わせた服用量の調整が容易であるばかりか、それらをカプセル中に充填することで、カプセル剤として一度に服用することもできる(後記試験例1〜3参照)。
【0055】
以下に試験例を挙げて、本発明の効果を詳細に説明する。なお、薬物としてはフマル酸エメダスチン、塩酸ロメリジン、塩酸トラゾドンおよびニフェジピンを用いて評価した。
【0056】
〔試験例1〕(溶出試験)
(1)試料
実施例1の錠剤〜実施例18の錠剤、実施例20の錠剤、実施例22の錠剤および実施例24の錠剤(表1〜表4参照。単位はmg)
【0057】
【表1】
【表2】
【表3】
【表4】
局方の溶出試験法第2法(パドル法)に従って試験を行った。試験液は局方の崩壊試験法・第1液、第2液または酢酸・酢酸ナトリウム緩衝液(pH4.0)のいずれかを用いた。試験液の温度は37℃とし、パドルの回転数は50rpmとした。フマル酸エメダスチン、塩酸トラゾドンあるいはニフェジピンの溶出率は、それぞれ280nm、312nm、325nmにおける吸光度を測定することにより求めた。また、塩酸ロメリジンの溶出率は下記条件の液体クロマトグラフ法(以下HPLC法という)により求めた。
HPLC法の条件:
・カラム:L−columnTMODS[150mm×4.6mm、化学品検査協会製]
・溶離液:メタノール3容と、リン酸でpH2.5に調整した0.5W/V%ラウリル硫酸ナトリウム水溶液1容の混合溶液
・カラム温度:50℃
・流速:1.4ml/分
・検出方法:UV225nmにおける吸光度測定
(3)試験結果
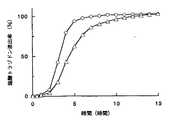
結果(3〜6例の平均値)を図9〜19に示す。
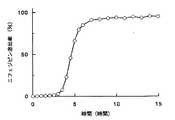
図9は、薬物としてフマル酸エメダスチンを用いて、試験液(pH)の影響を検討した図である。図9から明らかなように、いずれの試験液中においても、フマル酸エメダスチンの溶出は1時間目から始まり、約5時間後に皮膜が破裂した後は溶出速度が速くなり、溶出率は、試験液の影響をほとんど受けなかった。
【0061】
図10は、薬物としてフマル酸エメダスチンを用いて、素錠中のポリビニルアルコールの量を変化させた時の溶出速度に及ぼす影響を検討した図である。図10から明らかなように、ポリビニルアルコールの量を増加させるに伴い、皮膜の破裂時間が速くなり、ひいては溶出速度が速くなった。
【0062】
図11は、薬物としてフマル酸エメダスチンを用いて、素錠中の塩、すなわちクエン酸三ナトリウムの量を変化させた時の溶出速度に及ぼす影響を検討した図である。図11から明らかなように、クエン酸三ナトリウムの量を減少させるに伴い、皮膜の破裂時間が速くなり、ひいては溶出速度が速くなった。
【0063】
図12および図13は、薬物としてフマル酸エメダスチンを用いて、水溶性ポア形成皮膜の被覆量を変化させた時の溶出速度に及ぼす影響を検討した図である。図12および図13から明らかなように、水溶性ポア形成皮膜の被覆量を増加させるに伴い、皮膜の破裂時間が遅くなり、ひいては溶出速度が遅くなった。
【0064】
図14は、薬物としてフマル酸エメダスチンを用いて、素錠中への水溶性高分子の添加の有無の溶出速度に及ぼす影響を検討した図である。図14から明らかなように、水溶性高分子を添加することにより、溶出速度が遅くなった。
【0065】
図15および図16は、薬物として塩酸ロメリジンを用いて、水溶性ポア形成皮膜の被覆量を変化させた時の溶出速度に及ぼす影響を検討した図である。図15および図16から明らかなように、水溶性ポア形成皮膜の被覆量を増加させるに伴い、皮膜の破裂時間が遅くなり、ひいては溶出速度が遅くなった。
【0066】
図17は、薬物として塩酸トラゾドンを用いて、素錠の直径を8mmとして水溶性ポア形成皮膜の被覆量を変化させた時の溶出速度に及ぼす影響を検討した図である。図17から明らかなように、素錠の直径を大きくした場合も水溶性ポア形成皮膜の被覆量を増加させるに伴い、皮膜の破裂時間が遅くなり、ひいては溶出速度が遅くなった。
【0067】
図18は、薬物としてニフェジピンを用いた場合の水溶性ポア形成皮膜被覆錠(実施例18)の溶出試験結果である。図18から明らかなように、実施例18の持続性製剤からのニフェジピンの溶出は、3時間目から始まり、3.5時間で皮膜が破裂した後はニフェジピンが急激に溶出された。
【0068】
図19は、薬物としてニフェジピンを用いた場合の水溶性ポア形成皮膜被覆錠の表面に腸溶性皮膜を施した錠剤(実施例20)、水溶性ポア形成皮膜被覆錠の表面に腸溶性皮膜を施し、さらにその表面に固体分散体皮膜を被覆した錠剤(実施例22)および腸溶性ポア形成皮膜被覆錠の表面に固体分散体皮膜を被覆した錠剤(実施例24)の溶出試験結果〔試験液:局方に記載の崩壊試験法・第2液(pH6.8)〕である。実施例22の持続性製剤からの第2液(pH6.8)中でのニフェジピンの溶出は1時間目から始まり、2時間後に皮膜が破裂した後はニフェジピンが急激に溶出された。なお、第1液(pH1.2)中では、10時間目までニフェジピンは全く溶出されなかった。実施例22の持続性製剤からのニフェジピンの溶出は、試験開始後直ちに始まり、2時間後に皮膜が破裂した後はニフェジピンが急激に溶出された。実施例24の持続性製剤からのニフェジピンの溶出は試験開始後直ちに始まり、3時間後に皮膜が破裂した後は薬物が急激に溶出された。なお、実施例22および実施例24の持続性製剤のいずれの場合も、第1液(pH1.2)中では、最外層に被覆した固体分散体皮膜中のニフェジピンが徐々に溶出されるのみで、10時間目まで皮膜は破裂しなかった。
【0069】
〔試験例2〕(尿中排泄速度の測定)
本発明製剤の吸収性試験は、薬物としてニフェジピンを用い、ニフェジピンの主代謝物である2,6−ジメチル−4−(2−ニトロフェニル)−3,5−ピリジンジカルボン酸モノメチルエステル(以下、主代謝物という)の尿中排泄速度推移を指標にして検討した。なお、ニフェジピンの主代謝物の尿中濃度は下記条件のHPLC法により定量した。
HPLC法の条件:
・カラム:Nova−PakTMC18[150mm×3.9mm、Waters社製]
・溶離液:0.05M酢酸5容とアセトニトリル1容の混合溶液
・カラム温度:25℃
・流速:1.0ml/分
・検出方法:UV290nmに於ける吸光度測定
(1)試料
実施例19のカプセル剤、実施例21のカプセル剤、実施例23のカプセル剤、実施例25のカプセル剤および比較例1の錠剤
(2)試験方法
3〜4人の健常成人ボランティアに朝食摂取30分後に各試料(ニフェジピン20mg相当量)を経口投与し、投与後0〜1、1〜2、2〜4、4〜6、6〜8、8〜10、10〜12、12〜15、15〜22および22〜24時間の尿を採取し、さらに実施例21、23および25のカプセル剤については投与後24〜27、27〜30および30〜33時間の尿を採取して、ニフェジピンの主代謝物の尿中濃度をHPLC法により測定し、各時間毎の尿中排泄速度を求めた。
(3)試験結果
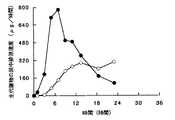
薬物としてニフェジピンを用いて、実施例18の水溶性ポア形成皮膜被覆錠4個をカプセルに充填した実施例19のカプセル剤と比較例1の錠剤をヒトに投与した際の、尿中から排泄される主代謝物の尿中排泄速度を比較した結果を図20に示す。図20から明らかなように、本発明の持続性製剤(カプセル剤)を投与した際の尿中排泄速度は、比較例1の錠剤(持続性製剤)を投与した場合の尿中排泄速度に比べて長時間持続化された。
【0070】
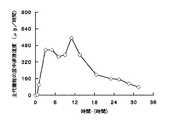
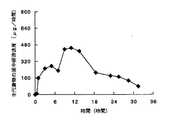
さらに、水溶性ポア形成皮膜被覆錠の表面に腸溶性皮膜を被覆した実施例20の錠剤4個をカプセルに充填した実施例21のカプセル剤、水溶性ポア形成皮膜被覆錠の表面に腸溶性皮膜を被覆し、さらにその表面に固体分散体皮膜を被覆した実施例22の錠剤4個をカプセルに充填した実施例23のカプセル剤、および腸溶性ポア形成皮膜被覆錠の表面に固体分散体皮膜を被覆した実施例24の錠剤4個をカプセルに充填した実施例25のカプセル剤をヒトに投与した際の、尿中から排泄される主代謝物の尿中排泄速度を測定した結果を図21〜図23に示す。図21〜図23から明らかなように、水溶性ポア形成皮膜被覆錠の表面に腸溶性皮膜を被覆した本発明の持続性製剤をカプセルに充填して投与した際の尿中排泄速度が長時間持続化された。また、水溶性ポア形成皮膜被覆錠、腸溶性皮膜被覆錠の表面に固体分散体皮膜を被覆した本発明の持続性製剤をカプセルに充填して投与した場合、初期の尿中排泄速度が確保され、しかも長時間持続化された。
【0071】
〔試験例3〕(血漿中濃度の測定)
ニフェジピンの血漿中濃度は、下記条件のガスクロマトグラフ法(以下、GC法という)により定量した。
GC法の条件:
・検出器:電子捕獲型検出器
・カラム:3%OV−17(3mm×3.1m、ジーエルサイエンス社製)
・カラム温度:270℃
(1)試料
実施例19のカプセル剤
(2)試験方法
6人の健常成人ボランティアに朝食摂取30分後に試料(ニフェジピン20mg相当量)を経口投与し、投与後一定時間(2、4、6、8、10、12、15、24、28、32、36、48時間)経過毎に採血し、得られた血液を遠心分離し血漿を得た。各時間毎に得られた血漿中のニフェジピンの濃度をGC法により定量した。
(3)試験結果
本発明の水溶性ポア形成皮膜被覆錠をヒトに投与した際の、ニフェジピンの血漿中濃度(平均値)の経時的な推移を図24に示す。
図24から明らかなように、本発明の持続性製剤をヒトに経口投与した時、ニフェジピンの血漿中濃度は、長時間持続化された。
従って、本発明製剤は水難溶性の薬物の経口吸収性および血漿中濃度の持続性に優れ、薬物の1日1〜2回投与用製剤として有用である。
【0072】
【実施例】
以下に実施例および比較例を挙げて本発明を説明する。
実施例1
フマル酸エメダスチン(特公平2−24821号参照)98g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)734.7g、クエン酸三ナトリウム・2水和物(100メッシュ篩通過品)244.9gおよび乳糖98gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)24.4gを水463gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し、薬物含有の造粒物を得た。この造粒物1114gにステアリン酸マグネシウム11.3gおよび軽質無水ケイ酸11.3gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠753g(約15100個)を得た。
次に、エチルセルロース(商品名:エトセル standard−10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)0.43重量部、クエン酸トリエチル(商品名:シトロフレックス2、ファイザー社製)0.29重量部を、エチルセルロースとヒドロキシプロピルメチルセルロースの濃度が5重量%となるように水とエタノール〔1:4(重量比)〕の混合溶媒に溶解し水溶性ポア形成皮膜液とした。上記の素錠350g(約7000個)を錠剤コーティング装置(商品名:ハイコーター・HCT−MINI、フロイント産業社製)に入れ、この水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、水溶性ポア形成皮膜を被覆して1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0073】
実施例2
フマル酸エメダスチン72g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)495g、クエン酸三ナトリウム・2水和物(100メッシュ篩通過品)180gおよび乳糖117gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gを水342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し薬物含有の造粒物を得た。この造粒物855gにステアリン酸マグネシウム10.5gおよび軽質無水ケイ酸7gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠737g(約14700個)を得た。
次に、この素錠350g(約7000個)に、実施例1と同様に水溶性ポア形成皮膜液を噴霧、乾燥し、1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0074】
実施例3
フマル酸エメダスチン152g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)950g、クエン酸三ナトリウム・2水和物(100メッシュ篩通過品)380gおよび乳糖342gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)38gを水722gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し、薬物含有の造粒物を得た。この造粒物1800gにステアリン酸マグネシウム18.4gおよび軽質無水ケイ酸18.4gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠1680g(約33600個)を得た。
次に、この素錠350g(約7000個)に、実施例1と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0075】
実施例4
フマル酸エメダスチン72g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)450g、クエン酸三ナトリウム・2水和物(100メッシュ篩通過品)36gおよび乳糖306gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gを水342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物855gにステアリン酸マグネシウム10.5gおよび軽質無水ケイ酸7gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠720g(約14400個)を得た。
次に、この素錠350g(約7000個)に、実施例1と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0076】
実施例5
フマル酸エメダスチン72g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)450g、クエン酸三ナトリウム・2水和物(100メッシュ篩通過品)54gおよび乳糖288gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gを水342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物855gにステアリン酸マグネシウム10.5gおよび軽質無水ケイ酸7gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠740g(約14800個)を得た。
次に、この素錠350g(約7000個)に、実施例1と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0077】
実施例6
フマル酸エメダスチン72g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)540g、クエン酸三ナトリウム・2水和物(100メッシュ篩通過品)180gおよび乳糖72gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gを水342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物860gにステアリン酸マグネシウム10.5gおよび軽質無水ケイ酸7gを混合し、混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠708g(約14200個)を得た。
次に、エチルセルロース(商品名:エトセル standard−10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)0.43重量部、クエン酸トリエチル(商品名:シトロフレックス2、ファイザー社製)0.29重量部を、エチルセルロースとヒドロキシプロピルセルロースの濃度が5重量%となるようにエタノールに溶解し水溶性ポア形成皮膜液とした。上記の素錠350g(約7000個)を錠剤コーティング装置(商品名:ハイコーター・HCT−MINI、フロイント産業社製)に入れ、この水溶性ポア形成皮膜液を素錠に噴霧、乾燥し水溶性ポア形成皮膜を被覆して1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0078】
実施例7
実施例6と同様に、フマル酸エメダスチンを含有する混合物を直径5mmの杵で1錠当たり50mgに打錠し、素錠708g(約14200個)を得た。
次に、この素錠350g(約7000個)に、1錠当たりの被覆量を1mg増量する以外は実施例6と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が57mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0079】
実施例8
フマル酸エメダスチン72g、ポリビニルアルコール(商品名:クラレポバールPVA−CS、株式会社クラレ製、100メッシュ篩通過品)540g、塩化ナトリウム(100メッシュ篩通過品)180gおよび乳糖72gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gを水342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物850gにステアリン酸マグネシウム10.4gおよび軽質無水ケイ酸7gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠755g(約15100個)を得た。
次に、この素錠350g(約7000個)に、実施例1と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0080】
実施例9
実施例8と同様に、フマル酸エメダスチンを含有する混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠755g(約15100個)を得た。
次に、この素錠350g(約7000個)に対し、1錠当たりの被覆量を1mg増量する以外は、実施例1と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が57mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0081】
実施例10
フマル酸エメダスチン72g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)540g、硫酸ナトリウム・無水物(100メッシュ篩通過品)180gおよび乳糖72gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gをエタノール342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物850gにステアリン酸マグネシウム10.4gおよび軽質無水ケイ酸7gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠770g(約15400個)を得た。
次に、この素錠350g(約7000個)に、実施例1と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0082】
実施例11
フマル酸エメダスチン72g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)540g、硫酸ナトリウム・無水物(100メッシュ篩通過品)180gおよびヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)72gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース18gをエタノール342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物850gにステアリン酸マグネシウム10.4gおよび軽質無水ケイ酸7gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠760g(約15200個)を得た。
次に、この素錠350g(約7000個)に、実施例1と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0083】
実施例12
塩酸ロメリジン(特公平3−13232号参照)90g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)360g、硫酸ナトリウム・無水物(100メッシュ篩通過品)360gおよび乳糖54gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gをエタノール342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物850gにステアリン酸マグネシウム12.1gおよび軽質無水ケイ酸5.2gを混合し、混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠750g(約15000個)を得た。
次に、エチルセルロース(商品名:エトセル standard−10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)0.49重量部、クエン酸トリエチル(商品名:シトロフレックス2、ファイザー社製)0.30重量部を、エチルセルロースとヒドロキシプロピルメチルセルロースの濃度が5重量%となるように水とエタノール〔1:4(重量比)〕の混合溶媒に溶解し水溶性ポア形成皮膜液とした。上記の素錠350g(約7000個)を錠剤コーティング装置(商品名:ハイコーター・HCT−MINI、フロイント産業社製)に入れ、この水溶性ポア形成皮膜液を素錠に噴霧、乾燥し水溶性ポア形成皮膜を被覆して1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりに塩酸ロメリジン5mgを含有する。
【0084】
実施例13
実施例12と同様に、塩酸ロメリジンを含有する混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠750g(約15000個)を得た。
次に、この素錠350g(約7000個)に、1錠当たりの被覆量を1mg増量する以外は、実施例12と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が57mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりに塩酸ロメリジン5mgを含有する。
【0085】
実施例14
塩酸ロメリジン90g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)450g、クエン酸三ナトリウム・2水和物(100メッシュ篩通過品)270gおよび乳糖57.6gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gを水342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物855gにステアリン酸マグネシウム7gおよび軽質無水ケイ酸7gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠735g(約14700個)を得た。
次に、エチルセルロース(商品名:エトセル standard−10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)0.47重量部、クエン酸トリエチル(商品名:シトロフレックス2、ファイザー社製)0.29重量部を、エチルセルロースとヒドロキシプロピルメチルセルロースの濃度が5重量%となるように水とエタノール〔1:4(重量比)〕の混合溶媒に溶解し水溶性ポア形成皮膜液とした。上記の素錠350g(約7000個)を錠剤コーティング装置(商品名:ハイコーター・HCT−MINI、フロイント産業社製)に入れ、この水溶性ポア形成皮膜液を素錠に噴霧、乾燥し水溶性ポア形成皮膜を被覆して1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりに塩酸ロメリジン5mgを含有する。
【0086】
実施例15
実施例14と同様に、塩酸ロメリジンを含有する混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠750g(約15000個)を得た。
次に、この素錠350g(約7000個)に、1錠当たりの被覆量を1mg増量する以外は、実施例16と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が57mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりに塩酸ロメリジン5mgを含有する。
【0087】
実施例16
塩酸トラゾドン(特公昭44−7341号参照)337.5g、ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)360gおよびクエン酸三ナトリウム・2水和物(100メッシュ篩通過品)180gの混合物を流動層造粒機中で流動させ、ヒドロキシプロピルセルロース(商品名:日曹HPC、日本曹達社製)18gを水342gに溶解した溶液を結合剤として噴霧した後、乾燥し、18メッシュの篩で整粒し造粒物を得た。この造粒物866gにステアリン酸マグネシウム8.7gおよび軽質無水ケイ酸4.4gを混合し、この混合物を直径8mmの杵を用いてロータリー式打錠機で1錠当たり202mgに打錠し、素錠750g(約3700個)を得た。
次に、エチルセルロース(商品名:エトセル standard−10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)0.47重量部、クエン酸トリエチル(商品名:シトロフレックス2、ファイザー社製)0.29重量部を、エチルセルロースとヒドロキシプロピルメチルセルロースの濃度が5重量%となるように水とエタノール〔1:4(重量比)〕の混合溶媒に溶解し水溶性ポア形成皮膜液とした。上記の素錠350g(約1700個)を錠剤コーティング装置(商品名:ハイコーター・HCT−MINI、フロイント産業社製)に入れ、この水溶性ポア形成皮膜液を素錠に噴霧、乾燥し水溶性ポア形成皮膜を被覆して1錠当たりの重量が213mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりに塩酸トラゾドン75mgを含有する。
【0088】
実施例17
実施例16と同様に、塩酸トラゾドンを含有する混合物を直径8mmの杵を用いてロータリー式打錠機で1錠当たり202mgに打錠し、素錠750g(約3700個)を得た。
次に、この素錠350g(約1700個)に、1錠当たりの被覆量を7mg増量する以外は、実施例16と同様に水溶性ポア形成皮膜液を素錠に噴霧、乾燥し、1錠当たりの重量が220mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりに塩酸トラゾドン75mgを含有する。
【0089】
実施例18
ニフェジピン101.4gおよびヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)152gをジクロルメタン821gとエタノール547gの混合溶媒に溶解し固体分散体溶液とする。ポリビニルアルコール(商品名:クラレポバールPVA−CSTS、株式会社クラレ製)810.8gおよびクエン酸三ナトリウム・2水和物(100メッシュ篩通過品)135.8gの混合物を流動層造粒機中で流動させ、固体分散体溶液を噴霧した後、乾燥し、30メッシュの篩で整粒し造粒物を得た。この造粒物592gにステアリン酸マグネシウム3gおよび軽質無水ケイ酸5gを混合し、混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり60mgに打錠し、素錠460g(約7600個)を得た。
次に、エチルセルロース(商品名:エトセル standard−10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)0.6重量部を、これら両者の総和の濃度が5重量%となるようにジクロルメタンとエタノール〔1:1(重量比)〕の混合溶媒に溶解し水溶性ポア形成皮膜液とした。上記の素錠350g(約5800個)にこの水溶性ポア形成皮膜液を噴霧、乾燥し、水溶性ポア形成皮膜を被覆して1錠当たりの重量が65mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにニフェジピン5mgを含有する。
【0090】
実施例19
実施例18の持続性製剤4個を2号硬カプセルに充填し、カプセル剤とした。
【0091】
実施例20
ニフェジピン338gおよびヒドロキシプロピルメチルセルロース(商品名:TC−5E:信越化学工業社製)507gをジクロルメタン2740gとエタノール1824gの混合溶媒に溶解し固体分散体溶液とする。ポリビニルアルコール(クラレポバールPVA−CSTS、株式会社クラレ製)2703gおよびクエン酸三ナトリウム・2水和物(100メッシュ通過品)453gの混合物を流動層造粒機中で流動させ、固体分散体溶液を噴霧した後、乾燥し、30メッシュの篩で整粒し造粒物を得た。この造粒物3552gにステアリン酸マグネシウム18gおよび軽質無水ケイ酸30gを混合し、混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり60mgに打錠し、素錠3420g(約57000個)を得た。
次に、エチルセルロース(商品名:エトセル standard-10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5E:信越化学工業社製)0.7重量部を、これら両者の総和の濃度が5重量%となるようにジクロルメタンとエタノール〔1:1(重量比)〕の混合溶媒に溶解し水溶性ポア形成皮膜液とした。上記の素錠350g(約5800個)にこの水溶性ポア形成皮膜液を噴霧、乾燥し、水溶性ポア形成皮膜を被覆し1錠当たりの重量が65mgの錠剤を得た後、さらにヒドロキシプロピルメチルセルロースフタレート〔商品名:HPMCP(HP−50)、信越化学工業社製〕の濃度が5重量%となるようにジクロルメタンとエタノール〔1:1(重量比)〕の混合溶媒に溶解した腸溶性皮膜液を噴霧、乾燥し、腸溶性皮膜を被覆して1錠当たりの重量が68mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにニフェジピン5mgを含有する。
【0092】
実施例21
実施例20の持続性製剤4個を2号硬カプセルに充填し、カプセル剤とした。
【0093】
実施例22
ニフェジピン300gおよびヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)450gをジクロルメタン2432gとエタノール1624gの混合溶媒に溶解し固体分散体溶液とする。ポリビニルアルコール(クラレポバールPVA−CSTS、株式会社クラレ製)3200gおよびクエン酸三ナトリウム・無水物(100メッシュ通過品)536gの混合物を流動層造粒機中で流動させ、固体分散体溶液を噴霧、乾燥した後、30メッシュの篩で整粒し造粒物を得た。この造粒物4190gにステアリン酸マグネシウム22.4gおよび軽質無水ケイ酸37.4gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で打錠し、1錠当たりの重量が56.88mgの素錠(1錠当たりにニフェジピン3.75mgを含む)4000g(約70000個)を得た。
次に、エチルセルロース(商品名:エトセル standard-10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)0.7重量部を、これら両者の総和の濃度が5重量%となるようにジクロルメタンとエタノール〔1:1(重量比)〕の混合溶媒に溶解し水溶性ポア形成皮膜液とした。上記の素錠350g(約6200個)をコーティングパンに入れ、この水溶性ポア形成皮膜液を素錠に噴霧、乾燥し皮膜を被覆して1錠当たりの重量が61.38mgの水溶性ポア形成皮膜被覆錠を得た後、さらにヒドロキシプロピルメチルセルロースフタレート〔商品名:HPMCP(HP−50)、信越化学工業社製〕の濃度が5重量%となるようにジクロルメタンとエタノール〔1:1(重量比)〕の混合溶媒に溶解した腸溶性皮膜液を、上記の錠剤に噴霧、乾燥し、腸溶性皮膜を被覆して1錠当たりの重量が64.38mgの腸溶性皮膜被覆錠を得た。
次いで、ニフェジピン1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5RW、信越化学工業社製)1.5重量部を、ヒドロキシプロピルメチルセルロースの濃度が5重量%となるようにジクロルメタンとエタノール〔6:4(重量比)〕の混合溶媒に溶解し固体分散体溶液とした。上記腸溶性皮膜被覆錠に、この固体分散体溶液を噴霧、乾燥し固体分散体皮膜を被覆(1錠当たりニフェジピン1.25mg対応量)して1錠当たりの重量が67.5mgの本発明の持続性製剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにニフェジピン5mgを含有する。
【0094】
実施例23
実施例22の持続性製剤4個を2号硬カプセルに充填し、カプセル剤とした。
【0095】
実施例24
ニフェジピン90gおよびヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)135.6gをジクロルメタン732gとエタノール488gの混合溶媒に溶解し固体分散体溶液とする。ポリビニルアルコール(クラレポバールPVA−CSTS、株式会社クラレ製)600g、クエン酸三ナトリウム・無水物(100メッシュ通過品)160.8gおよび結晶セルロース(商品名:アビセルPH301、旭化成工業社製)の混合物を流動層造粒機中で流動させ、固体分散体溶液を噴霧、乾燥した後、30メッシュの篩で整粒し造粒物を得た。この造粒物1088gにステアリン酸マグネシウム6.4gおよび軽質無水ケイ酸10.7gを混合し、この混合物を直径5mmの杵を用いてロータリー式打錠機で打錠し、1錠当たりの重量が51.9mgの素錠(1錠当たりにニフェジピン3.75mgを含有する)800g(約15400個)を得た。次に、エチルセルロース(商品名:エトセル standard−10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロースフタレート〔商品名:HPMCP(HP−55)、信越化学工業社製〕0.67重量部、クエン酸トリエチル(商品名:シトロフレックス2、ファイザー社製)0.33重量部を、エチルセルロースとヒドロキシプロピルメチルセルロースフタレートの総和の濃度が5重量%となるように水とエタノール〔1:4(重量比)〕の混合溶媒に溶解し腸溶性ポア形成皮膜液とした。上記の素錠350g(約6700個)をコーティングパンに入れ、この腸溶性ポア形成皮膜液を素錠に噴霧、乾燥し、腸溶性ポア形成皮膜を被覆して1錠当たりの重量が56.7mgの腸溶性ポア形成皮膜被覆錠を得た。
次いで、ニフェジピン1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5RW、信越化学工業社製)1.5重量部を、ヒドロキシプロピルメチルセルロースの濃度が5重量%となるようにジクロルメタンとエタノール〔6:4(重量比)〕の混合溶媒に溶解し固体分散体溶液とした。上記腸溶性ポア形成皮膜被覆錠に、この固体分散体溶液を噴霧、乾燥し固体分散体皮膜を被覆(1錠当たりニフェジピン1.25mg対応量)して1錠当たりの重量が59.85mgの本発明の持続性製剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにニフェジピン5mgを含有する。
【0096】
実施例25
実施例24の持続性製剤4個を2号硬カプセルに充填し、カプセル剤とした。
【0097】
実施例26
実施例8と同様に、フマル酸エメダスチンを含有する混合物を直径5mmの杵を用いてロータリー式打錠機で1錠当たり50mgに打錠し、素錠755g(約15100個)を得た。
次に、エチルセルロース(商品名:エトセル standard−10、ダウ・ケミカル社製)1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5E、信越化学工業社製)0.21重量部、ヒドロキシプロピルメチルセルロースフタレート〔商品名:HPMCP(HP−55)、信越化学工業社製〕0.21重量部、クエン酸トリエチル(商品名:シトロフレックス2、ファイザー社製)0.29重量部を、エチルセルロース、ヒドロキシプロピルメチルセルロースおよびヒドロキシプロピルメチルセルロースフタレートの濃度が5重量%となるように水とエタノール〔1:4(重量比)〕の混合溶媒に溶解し水溶性腸溶性ポア形成皮膜液とした。上記の素錠350g(約7000個)を錠剤コーティング装置(商品名:ハイコーター・HCT−MINI、フロイント産業社製)に入れ、この水溶性腸溶性ポア形成皮膜液を素錠に噴霧、乾燥し、水溶性腸溶性ポア形成皮膜を被覆して1錠当たりの重量が56mgの錠剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0098】
実施例27
実施例1と同様に、フマル酸エメダスチンを含有する混合物を打錠して素錠を調製し、その素錠表面に水溶性ポア形成皮膜液を噴霧、乾燥して得た、1錠当たり56mgの水溶性ポア形成皮膜被覆錠200g(約3560個)の表面に、さらにヒドロキシプロピルメチルセルロースフタレート〔商品名:HPMCP(HP−50)、信越化学工業社製〕の濃度が5重量%となるようにエタノールと水〔4:1(重量比)〕の混合溶媒に溶解した腸溶性皮膜液を噴霧、乾燥し、腸溶性皮膜を被覆して1錠当たりの重量が59mgの腸溶性皮膜被覆錠を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン4mgを含有する。
【0099】
実施例28
実施例1と同様に、フマル酸エメダスチンを含有する混合物を打錠して素錠を調製し、その素錠表面に水溶性ポア形成皮膜液を噴霧、乾燥して得た、1錠当たり56mgの水溶性ポア形成皮膜被覆錠100g(約1780個)の表面に、さらにフマル酸エメダスチン1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5MW、信越化学工業社製)2重量部を、ヒドロキシプロピルメチルセルロースの濃度が5重量%となるようにエタノールと水〔1:1(重量比)〕の混合溶媒に溶解した固体分散体溶液を噴霧、乾燥し固体分散体皮膜を被覆(1錠当たりフマル酸エメダスチン1mg対応量)して1錠当たりの重量が59mgの本発明の持続性製剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン5mgを含有する。
【0100】
実施例29
実施例27と同様に、フマル酸エメダスチンを含有する混合物を打錠して素錠を調製し、その素錠表面に水溶性ポア形成皮膜液および腸溶性皮膜液を順次噴霧、乾燥して得た、1錠当たり59mgの腸溶性皮膜被覆錠100g(約1690個)の表面に、さらにフマル酸エメダスチン1重量部に対して、ヒドロキシプロピルメチルセルロース(商品名:TC−5MW、信越化学工業社製)2重量部を、ヒドロキシプロピルメチルセルロースの濃度が5重量%となるようにエタノールと水〔1:1(重量比)〕の混合溶媒に溶解した固体分散体溶液を噴霧、乾燥し固体分散体皮膜を被覆(1錠当たりフマル酸エメダスチン1mg対応量)して1錠当たりの重量が62mgの本発明の持続性製剤を得た。このようにして製造した本発明の持続性製剤は1錠当たりにフマル酸エメダスチン5mgを含有する。
【0101】
比較例1
特公平6−11699号に記載の実施例1に準じて調製した。
A)芯部
結晶ニフェジピン〔平均粒子寸法3.3μm(空気透過法により測定)〕16.5gをラクトース194.0gおよびトウモロコシ澱粉75.0gと混合し、この混合物をトウモロコシ澱粉5.0gおよび熱水70.0gのペースト中で粒状化し、次いで乾燥した。この粒状物を篩にかけ、微結晶セルロース25.0gおよびステアリン酸マグネシウム1.0gと混合した。この混合物を直径6mmを有する重さ63.3mgの素錠に圧縮した。この芯部にヒドロキシプロピルメチルセルロースフタレート〔商品名:HPMCP(HP−55)、信越化学工業社製〕の有機溶媒溶液を用いることにより耐胃液性のコーティングを施した。この被覆された錠剤は重さが70.3mgであった。
B)コーティングに対する粒状物
ニフェジピン8.4gをラクトース20.0g、コロイド状シリカ0.8g、M型ヒドロキシプロピルセルロース〔商品名:日曹HPC(HPC−M)、日本曹達社製〕35.0g、L型ヒドロキシプロピルセルロース〔商品名:日曹HPC(HPC−L)、日本曹達社製〕87.4gおよびクエン酸16.0gと混合し、この混合物をL型ヒドロキシプロピルセルロース1.0gの溶液と共に流動層造粒機で粒状化した。次いで乾燥し且つ篩にかけた粒状物をステアリン酸マグネシウム1.4gと混合した。
【0102】
これらの粒状物およびA)に記述した芯部を、圧縮成形機により、直径10mmを有する重さ410mgの圧縮成形錠剤に圧縮した。
【図面の簡単な説明】
【図1】 本発明の持続性製剤(実施例1〜17および実施例26)の断面図を示す。
【図2】 本発明の持続性製剤(実施例27)の断面図を示す。
【図3】 本発明の持続性製剤(実施例28)の断面図を示す。
【図4】 本発明の持続性製剤(実施例29)の断面図を示す。
【図5】 本発明の持続性製剤(実施例18)の断面図を示す。
【図6】 本発明の持続性製剤(実施例20)の断面図を示す。
【図7】 本発明の持続性製剤(実施例24)の断面図を示す。
【図8】 本発明の持続性製剤(実施例22)の断面図を示す。
【図9】 実施例1の錠剤からのフマル酸エメダスチンの溶出試験結果を示すグラフであり、◇印は試験液として局方に記載の崩壊試験法・第1液(pH1.2)を使用した場合、◆印は試験液として局方に記載の酢酸・酢酸ナトリウム緩衝液(pH4.0)を使用した場合、△印は試験液として局方に記載の崩壊試験法・第2液(pH6.8)を使用した場合を示す。
【図10】 実施例1〜3の錠剤からのフマル酸エメダスチンの溶出試験〔試験液:局方に記載の崩壊試験法・第1液(pH1.2)〕結果を示すグラフであり、◇印は実施例1の錠剤、○印は実施例2の錠剤、△印は実施例3の錠剤を示す。
【図11】 実施例3〜5の錠剤からのフマル酸エメダスチンの溶出試験〔試験液:局方に記載の崩壊試験法・第1液(pH1.2)〕結果を示すグラフであり、△印は実施例3の錠剤、○印は実施例4の錠剤、◇印は実施例5の錠剤を示す。
【図12】 実施例6〜7の錠剤からのフマル酸エメダスチンの溶出試験〔試験液:局方に記載の崩壊試験法・第1液(pH1.2)〕結果を示すグラフであり、○印は実施例6の錠剤、△印は実施例7の錠剤を示す。
【図13】 実施例8〜9の錠剤からのフマル酸エメダスチンの溶出試験〔試験液:局方に記載の崩壊試験法・第1液(pH1.2)〕結果を示すグラフであり、○印は実施例8の錠剤、△印は実施例9の錠剤を示す。
【図14】 実施例10〜11の錠剤からのフマル酸エメダスチンの溶出試験〔試験液:局方に記載の崩壊試験法・第1液(pH1.2)〕結果を示すグラフであり、△印は実施例10の錠剤、●印は実施例11の錠剤を示す。
【図15】 実施例12〜13の錠剤からの塩酸ロメリジンの溶出試験〔試験液:局方に記載の崩壊試験法・第1液(pH1.2)〕結果を示すグラフであり、○印は実施例12の錠剤、△印は実施例13の錠剤を示す。
【図16】 実施例14〜15の錠剤からの塩酸ロメリジンの溶出試験〔試験液:局方に記載の崩壊試験法・第1液(pH1.2)〕結果を示すグラフであり、●印は実施例14の錠剤、△印は実施例15の錠剤を示す。
【図17】 実施例16〜17の錠剤からの塩酸トラゾドンの溶出試験〔試験液:局方に記載の崩壊試験法・第1液(pH1.2)〕結果を示すグラフであり、○印は実施例16の錠剤、△印は実施例17の錠剤を示す。
【図18】 実施例18の錠剤からのニフェジピンの溶出試験〔試験液:局方に記載の崩壊試験法・第2液(pH6.8)〕結果を示すグラフである。
【図19】 実施例20の錠剤、実施例22の錠剤および実施例24の錠剤からのニフェジピンの溶出試験〔試験液:局方に記載の崩壊試験法・第2液(pH6.8)〕結果を示すグラフであり、△印は実施例20の錠剤、◇印は実施例22の錠剤、◆印は実施例24の錠剤を示す。
【図20】 実施例19のカプセル剤および比較例1の錠剤をヒトに経口投与後のニフェジピン尿中排泄速度の経時的な推移を示すグラフであり、○印は実施例19のカプセル剤、●印は比較例1の錠剤を示す。
【図21】 実施例21のカプセル剤をヒトに経口投与後のニフェジピン尿中排泄速度の経時的な推移を示すグラフである。
【図22】 実施例23のカプセル剤をヒトに経口投与後のニフェジピン尿中排泄速度の経時的な推移を示すグラフである。
【図23】 実施例25のカプセル剤をヒトに経口投与後のニフェジピン尿中排泄速度の経時的な推移を示すグラフである。
【図24】 実施例19のカプセル剤をヒトに経口投与後のニフェジピン血漿中濃度の経時的な推移を示すグラフである。
【符号の説明】
1:(イ)薬物と水溶性高分子との混合物、またはそれらからなる固体分散体、
(ロ)ポリビニルアルコール、および(ハ)クエン酸三ナトリウム、硫酸ナトリウムおよび塩化ナトリウムからなる群より選択される1種または2種以上の塩とを含有する素錠
図1〜図4の素錠においては、薬物は黒色の不連続相で示され、薬物以外の成分は白色の連続相で示される。また、図5〜図8の素錠においては、薬物と水溶性高分子からなる固体分散体は黒色の連続相で示され、固体分散体以外の成分は白色の不連続相で示される。
2: 水不溶性高分子と、水溶性高分子および/または腸溶性高分子からなる皮膜
3:腸溶性高分子からなる皮膜
4:薬物と水溶性高分子との混合物、またはそれらからなる固体分散体皮膜[0001]
BACKGROUND OF THE INVENTION
The present invention relates to a novel sustained-release preparation. More specifically, it is selected from the group consisting of (a) a mixture of a drug and a water-soluble polymer, or a solid dispersion comprising them, (b) polyvinyl alcohol, and (c) trisodium citrate, sodium sulfate and sodium chloride. And (d) a sustained-release preparation coated with a film comprising a water-insoluble polymer, a water-soluble polymer and / or an enteric polymer. .
[0002]
[Prior art]
Keeping the plasma concentration of a drug constant for a long time has many advantages from the viewpoint of improving the prevention and treatment effect of the disease, reducing the number of administration of the drug, and improving patient compliance. For this reason, various sustained-release preparations with controlled drug release rates from the preparation have been developed and widely used clinically.
[0003]
For example, Japanese Patent Publication No. 6-11699 discloses a tablet in which a core containing a drug (nifedipine) in an immediate release form is compression-coated with a film made of a hydrophilic gel-forming polymer and a drug. However, although this tablet has a moderate persistence as a once-daily formulation, it is not always satisfactory and requires two-stage compression to prepare the formulation. There is a problem that the manufacturing process is complicated.
[0004]
In addition to the above-mentioned sustained-release preparations, membrane-coated granules or membrane-coated tablets produced by coating a core substance or uncoated tablet containing a drug with a film of a water-insoluble polymer and a water-soluble polymer, or a drug containing water. Various studies have been made on matrix tablets produced by tableting with an insoluble polymer or wax. However, in these preparations, the release rate of the drug decreases with time, so when administered in vivo, the plasma concentration of the drug tends to decrease over time, and the drug over a long period of time. It has a problem that it is difficult to maintain the plasma concentration of.
[0005]
In recent years, attention has been paid to the relationship between a periodic rhythm (circadian rhythm) of a living body and a disease, and the necessity of considering circadian rhythm in drug treatment has become important.
[0006]
In view of the above, investigations into sustained-release preparations that have been further devised have been conducted.
[0007]
[Problems to be solved by the invention]
An object of the present invention is to provide a sustained-release preparation in which a drug is continuously released over a long period of time and a sufficient preventive and therapeutic effect can be expected by administration once or twice a day.
[0008]
[Means for Solving the Problems]
As a result of intensive studies to achieve the above object, the present inventors have found that (a) a mixture of a drug and a water-soluble polymer, or a solid dispersion comprising them, (b) polyvinyl alcohol, and (c) ) Uncoated tablets containing one or more salts selected from the group consisting of trisodium citrate, sodium sulfate and sodium chloride, (d) a water-insoluble polymer, a water-soluble polymer and / or The inventors have found that a sustained-release preparation (FIGS. 1 and 5) produced by coating with a film composed of an enteric polymer meets the purpose of the present invention, thereby completing the present invention.
[0009]
According to the present invention, when the sustained-release preparation of the present invention is administered to a living body, a film composed of a water-insoluble polymer and a water-soluble polymer (hereinafter referred to as a water-soluble pore-forming film), or water A film comprising an insoluble polymer and an enteric polymer (hereinafter referred to as an enteric pore-forming film), or a film comprising a water-insoluble polymer, a water-soluble polymer and an enteric polymer (hereinafter referred to as such The water-soluble polymer and / or enteric polymer in the water-soluble enteric pore-forming film are dissolved in the gastrointestinal fluid, and pores are formed in the film. The drug on and near the uncoated tablet is gradually released. On the other hand, the polyvinyl alcohol in the uncoated tablet gradually swells to generate a swelling force, and the film ruptures due to the swelling force, and the drug in the uncoated tablet is released freely. However, the matrix of the uncoated tablet becomes a gel matrix with the swelling of polyvinyl alcohol, and the drug contained therein is gradually released.
[0010]
In the sustained-release preparation of the present invention, if necessary, a film made of an enteric polymer (hereinafter referred to as enteric film) is formed on the surface of the sustained-release preparation in order to further maintain the drug plasma concentration. (See FIGS. 2 and 6). In order to ensure the plasma concentration of the drug in the initial stage of administration, if necessary, a mixture of the drug and a water-soluble polymer on the surface of the continuous preparation or the continuous preparation coated with the enteric coating, Alternatively, a solid dispersion made of them can be coated (see FIGS. 3, 4, 7 and 8).
[0011]
DETAILED DESCRIPTION OF THE INVENTION
Examples of the water-soluble polymer used in the present invention include water-soluble cellulose ether and polyvinyl pyrrolidone.
[0012]
The water-soluble cellulose ether is obtained by substituting a part of an alcohol group which is a glucose residue in cellulose with a form such as methyl ether, hydroxypropyl ether and / or hydroxyethyl ether, and is soluble in water. is there.
[0013]
Examples of the water-soluble cellulose ether include methyl cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose and the like.
[0014]
The methyl cellulose preferably has a methoxyl group content of 26.0 to 33.0%. 1070 of the 12th revised Japanese Pharmacopoeia (published by Daiichi Hogaku Publishing Co., Ltd., 1991, hereinafter abbreviated as Pharmacopeia). The methylcellulose described on the page is preferably used. This can be obtained, for example, under the trade name Metrows SM (manufactured by Shin-Etsu Chemical Co., Ltd.). Hydroxypropylcellulose (hereinafter also referred to as HPC) preferably has a hydroxypropoxyl group content of 53.4 to 77.5%, and hydroxypropylcellulose described on page 1023 of the Japanese Pharmacopoeia is suitably used. This can be obtained, for example, under the trade name Nisso HPC (manufactured by Nippon Soda Co., Ltd.). Hydroxypropyl methylcellulose (hereinafter also referred to as HPMC) preferably has a methoxyl group and hydroxypropoxyl group content of 19.0 to 30.0% and 4.0 to 12.0%, respectively. Hydroxypropyl methylcellulose 2208, hydroxypropylmethylcellulose 2906 and hydroxypropylmethylcellulose 2910 described on page 1027 are preferably used. These can be obtained, for example, under the trade names Metroze 90SH, Metroze 65SH and TC-5 (all manufactured by Shin-Etsu Chemical Co., Ltd.).
[0015]
The polyvinyl pyrrolidone having a molecular weight of about 25,000 to about 1200,000 can be used, but a molecular weight of about 40,000 is preferable, and the 12th revised Japanese Pharmacopoeia Second Supplement (published by Daiichi Hogaku Publishing Co., Ltd., 1995) Polyvinylpyrrolidone K30 described on page 75 is preferably used.
[0016]
The water-soluble polymer can be used both in the uncoated tablet and in the film, and these water-soluble polymers can be appropriately mixed and used. For example, one type of water-soluble cellulose ether can be used alone, or as a mixture of two or more types, or as a mixture of one or more types of water-soluble cellulose ethers and polyvinyl pyrrolidone. It is preferable to use only 1 type, or 2 or more types of water-soluble cellulose ether. In the film, these water-soluble polymers and enteric polymers described later can be appropriately mixed and used.
[0017]
Among the above water-soluble polymers, hydroxypropyl methylcellulose having a methoxyl group and hydroxypropoxyl group content of 28.0 to 30.0% and 7.0 to 12.0%, respectively, is preferable. Of hydroxypropylmethylcellulose 2910 is particularly preferably used.
[0018]
The water-soluble polymer used in the uncoated tablet is blended to control the absorption of the drug, and the blending form may be changed according to the degree of drug absorbability. For example, in the case of a drug that is well absorbed from the gastrointestinal tract, the drug and the water-soluble polymer are simply mixed and used as a mixture, or a mixture of the drug and the other (b) and (c) components When granulating, an aqueous solution of a water-soluble polymer may be used as a binder. Further, a mixture of a drug and a water-soluble polymer may be used in the above granulation instead of the drug alone. On the other hand, if the drug is poorly absorbed from the gastrointestinal tract, such as a poorly water-soluble drug, use it as a solid dispersion obtained by removing the solvent from the organic solvent solution of the drug and water-soluble polymer, or use part of it. In the same manner as described above, a mixture with a water-soluble polymer or a mixture thereof can be used as a granulated product. By making the poorly water-soluble drug into a solid dispersion, the drug absorbability from the digestive tract can be improved.
[0019]
As the polyvinyl alcohol used in the present invention, for example, any of the completely saponified products or partially saponified products described on pages 304 and 307 of the Pharmaceutical Additives Standard (published by Yakuji Nippo Co., Ltd., 1993) can be used. Those having a large swelling power when contacted with water are preferably used. Examples thereof include polyvinyl alcohol having a saponification degree of 93.5 to 97.5%. For example, Kuraray Poval PVA-613, Kuraray Poval PVA-CST, Kuraray Poval PVA-CSTS, and Kuraray Poval PVA-CS are commercially available (all manufactured by Kuraray Co., Ltd.).
[0020]
As the trisodium citrate, sodium sulfate or sodium chloride used in the present invention, their anhydrides and hydrates thereof can be used, respectively. Moreover, these can also be mixed and used suitably.
[0021]
Examples of the water-insoluble polymer used in the present invention include ethyl cellulose, ethyl acrylate-methyl methacrylate-methacrylate-modified trimethylammonium ethyl copolymer, and the like.
[0022]
The ethyl cellulose preferably has an ethoxyl group content of 46.5 to 51.0%, and ethyl cellulose described on page 74 of the pharmaceutical additive standard is suitably used. This can be obtained, for example, under the trade name Etosel (manufactured by Dow Chemical). As the ethyl acrylate-methyl methacrylate-methacrylate trimethylammonium ethyl copolymer, the aminoalkyl methacrylate copolymer RS described on page 45 of the standard for pharmaceutical additives is preferably used. This can be obtained, for example, under the trade name Eudragit RS (Rame Pharma).
[0023]
Among the above water-insoluble polymers, ethyl cellulose having an ethoxyl group content of 46.5 to 51.0% is particularly preferable.
[0024]
Examples of the enteric polymer used in the present invention include hydroxypropylmethylcellulose phthalate (hereinafter also referred to as HPMCP), hydroxypropylmethylcellulose acetate succinate, carboxymethylethylcellulose, and the like.
[0025]
The hydroxypropyl methylcellulose phthalate has a methoxyl group, hydroxypropoxyl group and carboxybenzoyl group content of 18.0 to 24.0%, 5.0 to 10.0%, and 21.0 to 35.0, respectively. %, And hydroxypropylmethylcellulose phthalate 200731 and hydroxypropylmethylcellulose phthalate 220824 described in pp. 1028 and 1029 are preferably used. These are available under the trade names HPMCP HP-55 and HPMCP HP-50 (both manufactured by Shin-Etsu Chemical Co., Ltd.), respectively. Hydroxypropyl methylcellulose acetate succinate is a mixed ester of acetic acid and monosuccinic acid of hydroxypropyl methylcellulose, and methoxyl group, hydroxypropoxyl group, acetyl group and succinoyl group are 12.0 to 28.0%, 4.0 to 23 respectively. 0.0%, 2.0-16.0%, 4.0-28.0% are preferable, and hydroxypropyl methylcellulose acetate succinate described on page 257 of the Pharmaceutical Additives Standard is preferably used. These can be obtained, for example, under the trade name Shin-Etsu Coat (AQOAT) (manufactured by Shin-Etsu Chemical Co., Ltd.). As the carboxymethyl ethyl cellulose, a mixed ether of carboxymethyl and ethyl of cellulose, preferably having carboxymethyl group and ethoxyl group of 8.9 to 14.9% and 32.5 to 43.0%, respectively, is a pharmaceutical additive. Carboxymethyl ethyl cellulose described on page 105 of the standard is preferably used. These can be obtained, for example, under the trade name CMEC (manufactured by Freund Corporation). In addition, these enteric polymers can be used by appropriately mixing them.
[0026]
Among the enteric polymers, the contents of methoxyl group, hydroxypropoxyl group and carboxybenzoyl group are 18.0 to 22.0%, 5.0 to 9.0%, and 27.0 to 35.0%, respectively. The hydroxypropyl methylcellulose phthalate described in page 1028 of the Japanese Pharmacopoeia is preferably used.
[0027]
The sustained-release preparation of the present invention can be produced by the following production method. First, (1) selected from the group consisting of a drug or a drug and a water-soluble polymer in component (1), polyvinyl alcohol as component (2), and trisodium citrate, sodium sulfate and sodium chloride as component (1) After mixing seeds or two or more salts, the mixture of these components (a), (b) and (c) is granulated using an aqueous solution of a water-soluble polymer as a binder, and drug-containing granulation is performed. Or a mixture of the components (b) and (c) is sprayed and dried with a solution of the drug and water-soluble polymer dissolved in an organic solvent (this solution is called a solid dispersion solution), or The solid dispersion solution is added to the mixture of components (b) and (c) and kneaded, and then the solvent is distilled off and pulverized to produce a drug-containing granulated product.
[0028]
Next, the drug-containing granulated product is tableted to prepare an uncoated tablet.
[0029]
Further, a liquid in which a water-insoluble polymer and a water-soluble polymer are dispersed or dissolved in water and / or an organic solvent (hereinafter, this liquid is referred to as a water-soluble pore-forming film solution) is sprayed on the surface of the uncoated tablet. Prepare a tablet that is dried and coated with a water-soluble pore-forming film (hereinafter referred to as a water-soluble pore-forming film-coated tablet), or combine a water-insoluble polymer and an enteric polymer with water and / or an organic solvent. A tablet (hereinafter referred to as enteric pore-forming film-coated tablet) coated with an enteric pore-forming film by spraying and drying a liquid dispersed or dissolved in the liquid (hereinafter referred to as enteric pore-forming film liquid). Or a water-insoluble polymer, a water-soluble polymer and an enteric polymer dispersed or dissolved in water and / or an organic solvent (hereinafter, this solution is referred to as a water-soluble enteric pore-forming film). Sprayed and dried) Tablets were coated with a water-soluble enteric pore-forming membrane Te (hereinafter referred to as water-soluble enteric pore-forming membrane coated tablets) by preparing, it is possible to produce a depot preparation of the present invention.
[0030]
In the sustained-release preparation of the present invention, if necessary, an enteric polymer is dispersed or dissolved in water and / or an organic solvent on the surface of the sustained-release preparation by the same method as described above. By spraying a solution (hereinafter referred to as enteric coating solution), drying and coating to prepare a tablet (hereinafter referred to as enteric coating coated tablet), the plasma concentration of the drug is reduced. Furthermore, it can also be set as the formulation which can be sustained. In addition, if necessary, the surface of the water-soluble pore-forming film-coated tablet, enteric pore-forming film-coated tablet, water-soluble enteric pore-forming film-coated tablet, or enteric film-coated tablet may be combined with a drug and a water-soluble polymer. By coating the mixture or a solid dispersion comprising them in the same manner as described above, the plasma concentration of the drug at the initial administration can be ensured.
[0031]
Polyvinyl alcohol and the mixture consisting of one or more salts selected from the group consisting of trisodium citrate, sodium sulfate and sodium chloride may contain usual excipients used in pharmaceuticals (for example, starch , White sugar, lactose, mannitol, crystalline cellulose, etc.) can be appropriately mixed. Further, the granulated product may be one or more of a lubricant (eg, magnesium stearate, talc, etc.) and a fluidizing agent (eg, light anhydrous silicic acid, hydrous silicon dioxide, synthetic aluminum silicate, etc.). Are appropriately mixed to prepare a plain tablet.
[0032]
The diameter of the uncoated tablet is usually 2 to 10 mm, preferably 2 to 6 mm, more preferably 5 mm.
[0033]
When the water-soluble polymer is used as a binder, the amount of the water-soluble polymer added to the uncoated tablet is usually 0.01 to 20 parts by weight, preferably 0. 05 to 5 parts by weight. In the case of using a mixture of the above-mentioned polyvinyl alcohol and one or two or more salts selected from the group consisting of trisodium citrate, sodium sulfate and sodium chloride in combination with a water-soluble polymer, The amount is usually 0.01 to 150 parts by weight, preferably 0.1 to 30 parts by weight, based on 1 part by weight of the drug.
[0034]
On the other hand, when the water-soluble polymer is used for the preparation of the solid dispersion, the amount of the water-soluble polymer is usually 1 to 7 parts by weight, preferably 1.5 to 3 parts by weight with respect to 1 part by weight of the drug. Part.
[0035]
The organic solvent used for the solid dispersion solution is not particularly limited as long as it can dissolve both the drug and the water-soluble polymer. For example, one kind of methanol, ethanol, isopropanol, acetone, chloroform, dichloromethane, or Two or more kinds can be mixed and used. The amount of the organic solvent used is suitably 1.2 to 3 times the amount required to dissolve the drug and the water-soluble polymer at room temperature.
[0036]
The amount of polyvinyl alcohol in the uncoated tablet is usually 0.1 to 400 parts by weight, preferably 1 to 50 parts by weight, per 1 part by weight of the drug. The amount of one or more salts selected from the group consisting of trisodium citrate, sodium sulfate and sodium chloride is usually 0.01 to 300 parts by weight, preferably 0, per 1 part by weight of the drug. .1 to 50 parts by weight.
[0037]
The use amount of the water-insoluble polymer for film formation and the water-soluble polymer and / or enteric polymer is usually 5 to 20% by weight based on the weight of the uncoated tablet. The mixing ratio of the water-insoluble polymer and the water-soluble polymer and / or the enteric polymer is 1 part by weight of the water-insoluble polymer, when the water-soluble polymer and the enteric polymer are used alone or in combination. In any case, the total amount is usually 0.4 to 0.8 parts by weight.
[0038]
In addition to water-insoluble polymers, water-soluble polymers and / or enteric polymers, water-soluble pore-forming film liquids, enteric pore-forming film liquids, water-soluble enteric pore-forming film liquids and enteric film liquids In addition, one or more additives such as a plasticizer (for example, triacetin, triethyl citrate, macrogol 6000, etc.) can be appropriately mixed. The blending amount is 5 to 40% by weight based on the total amount of the water-insoluble polymer and the water-soluble polymer and / or enteric polymer.
[0039]
The organic solvent used for the water-soluble pore-forming film solution, the enteric pore-forming film solution, or the water-soluble enteric pore-forming film solution is not particularly limited. For example, one kind of methanol, ethanol, isopropanol, acetone, chloroform, and dichloromethane is used. Alternatively, two or more types can be mixed and used, but when using a mixed solvent of water and an organic solvent, it is necessary to select an organic solvent in which both solvents are mixed.
[0040]
In the case of an enteric coating-coated tablet, the amount of enteric polymer used is based on the weight of the water-soluble pore-forming coating-coated tablet, the enteric pore-forming coating-coated tablet, or the water-soluble enteric pore-forming coating-coated tablet. 4 to 12% by weight is suitable.
[0041]
When the drug is coated on the tablet surface using a binder or a solid dispersion solution in order to ensure the plasma concentration of the drug at the beginning of administration, the drug-containing film or solid dispersion film has a drug amount. As 1 to 30% by weight, preferably 5 to 25% by weight of the total amount of the drug used in the sustained-release preparation of the present invention.
[0042]
When the drug is unstable with respect to light, in order to maintain the stability of the drug, for example, hydroxypropylmethylcellulose and titanium oxide, iron sesquioxide, yellow iron sesquioxide, edible
[0043]
Although the above-described method has been described with respect to a method for producing a tablet, the sustained-release preparation of the present invention is not limited to a tablet, and other dosage forms can also be used. For example, it is produced using uncoated tablets having a diameter of 2 to 6 mm. These can also be filled into capsules by a conventional method to form capsules.
[0044]
The drug contained in the sustained-release preparation of the present invention is not particularly limited as long as it can increase the therapeutic effect by maintaining the plasma concentration over a long period of time. Circulatory drugs (eg, nifedipine, romeridine hydrochloride, nicardipine hydrochloride, nitroglycerin, isosorbide nitrate, captopril, delapril hydrochloride, ifenprodil tartrate), digestive drugs (eg, domperidone, metoclopramide), antiallergic agents (eg, Emedastine fumarate), antidepressants (for example, trazodone hydrochloride), analgesics (for example, indomethacin, ibuprofen), anticancer agents (for example, altretamine, ftorafur, fluorouracil, etoposide) and the like. The compounding quantity of these drugs should just be a unit dose which shows the medicinal effect known about each of these drugs, for example, for a once-daily administration, a daily dose is mix | blended.
[0045]
Among the above drugs, the sustained-release preparation of the present invention containing nifedipine, which is a poorly water-soluble drug, is excellent in oral absorption and plasma concentration, and is useful as a once-daily preparation of the drug.
[0046]
The sustained-release preparation of the present invention is usually orally administered once or twice a day for the daily dose of an adult for the purpose of preventing and treating various diseases.
[0047]
【The invention's effect】
Release of the drug from the sustained release formulation of the present invention occurs in at least the following two stages.
[0048]
First, as a first step, a water-soluble pore-forming film composed of a water-insoluble polymer and a water-soluble polymer and / or an enteric polymer, an enteric pore-forming film or a water-soluble enteric pore-forming film, As the soluble polymer and / or enteric polymer dissolves in the digestive tract fluid, pores are formed in the film. The gastrointestinal fluid permeates into the uncoated tablet from the pores generated in this way, and the drug in the uncoated tablet is dissolved and gradually released. At this time, it is also possible to suppress the release of the drug by adding a water-soluble polymer to the uncoated tablet. Moreover, at the same time as the drug is gradually released, the polyvinyl alcohol as the component (b) swells by the digestive tract fluid that has penetrated into the uncoated tablet, which facilitates the release of the drug. The blended (c) ingredients trisodium citrate, sodium sulfate and sodium chloride show the swelling suppression action of polyvinyl alcohol, their salts dissolve in the digestive tract fluid, and the solution gradually goes out of the film. By diffusing, the concentration of salt in the uncoated tablet is lowered, and then the polyvinyl alcohol begins to swell by the digestive tract fluid.
[0049]
As a second step, when the swelling power of polyvinyl alcohol increases, the film ruptures and the drug in the uncoated tablet is released freely. In this case, the polyvinyl alcohol in the uncoated tablet forms a matrix, and the drug is contained therein, so that the drug is gradually released as the matrix gels and gradually disintegrates from the surface. Is done.
[0050]
Based on the above, the drug release pattern of the whole formulation is a biphasic pattern with a slow initial release rate and a fast late phase, a sigmoid pattern with a slow release rate in the early and late stages, and a fast intermediate release rate, or drug release. A zero-order pattern with a constant speed is shown.
[0051]
In the sustained-release preparation of the present invention, the blended amount of polyvinyl alcohol in the uncoated tablet, the blended amount of the water-soluble polymer, and the trisodium citrate, sodium sulfate, and Adjust the blending amount of one or more salts selected from the group consisting of sodium chloride, the blending ratio of the water-insoluble polymer and the water-soluble polymer and / or enteric polymer, the coating amount of the film, etc. The first stage release rate at which the drug is released through the pores of the film, the burst time of the film, and the second stage release rate at which the drug is released after the film ruptures can be arbitrarily adjusted. Thereby, formulations having various release patterns as described above can be prepared. Therefore, in the sustained-release preparation of the present invention, the release pattern suitable for the drug can be appropriately selected according to the drug. Therefore, the sustained-release preparation of the present invention is suitable for an oral dosage form once or twice a day. Can be a long-acting formulation.
[0052]
Coating the surface of the above-mentioned sustained-release preparation with an enteric coating, or coating the above-mentioned sustained-release preparation or enteric coating-coated tablet with a mixture of a drug and a water-soluble polymer, or a solid dispersion comprising them. Thus, the plasma concentration of the drug can be further sustained, and the plasma concentration of the drug in the initial administration can be ensured.
[0053]
From these facts, the sustained-release preparation of the present invention can be made into a preparation taking into account the circadian rhythm of the living body.
[0054]
Furthermore, since one embodiment of the preparation of the present invention is in the form of a small tablet, it is easy not only to adjust the dose according to symptoms and age, but also as a capsule by filling them in a capsule. (See Test Examples 1 to 3 below).
[0055]
The effects of the present invention will be described in detail below by giving test examples. The drug was evaluated using emedastine fumarate, lomelidine hydrochloride, trazodone hydrochloride and nifedipine.
[0056]
[Test Example 1] (Dissolution test)
(1) Sample
Tablet of Example 1 to Tablet of Example 18, Tablet of Example 20, Tablet of Example 22 and Tablet of Example 24 (see Tables 1 to 4; unit is mg)
[0057]
[Table 1]
[Table 2]
[Table 3]
[Table 4]
The test was conducted according to the pharmacopeia dissolution method No. 2 (paddle method). The test solution used was one of the collateral disintegration test method, the first solution, the second solution, or an acetic acid / sodium acetate buffer solution (pH 4.0). The temperature of the test solution was 37 ° C., and the rotational speed of the paddle was 50 rpm. The elution rates of emedastine fumarate, trazodone hydrochloride or nifedipine were determined by measuring the absorbance at 280 nm, 312 nm and 325 nm, respectively. The elution rate of romeridine hydrochloride was determined by a liquid chromatographic method (hereinafter referred to as HPLC method) under the following conditions.
HPLC method conditions:
Column: L-columnTM ODS [150 mm x 4.6 mm, manufactured by Chemicals Inspection Association]
Eluent: A mixed solution of 3 volumes of methanol and 1 volume of 0.5 W / V% aqueous sodium lauryl sulfate adjusted to pH 2.5 with phosphoric acid
-Column temperature: 50 ° C
・ Flow rate: 1.4ml / min
Detection method: Absorbance measurement at UV225nm
(3) Test results
The results (average values of 3 to 6 cases) are shown in FIGS.
FIG. 9 is a diagram in which the effect of a test solution (pH) was examined using emedastine fumarate as a drug. As is apparent from FIG. 9, in any test solution, emedastine fumarate elution starts from the first hour, and after about 5 hours the film has ruptured, the elution rate increases, and the dissolution rate is determined by the test solution. Was almost unaffected.
[0061]
FIG. 10 is a diagram in which the effect on the dissolution rate when the amount of polyvinyl alcohol in the uncoated tablet is changed using emedastine fumarate as a drug is examined. As is clear from FIG. 10, as the amount of polyvinyl alcohol was increased, the rupture time of the film became faster, and consequently the elution rate became faster.
[0062]
FIG. 11 is a diagram in which the effect on the dissolution rate when the amount of salt in an uncoated tablet, that is, trisodium citrate, is changed using emedastine fumarate as a drug is examined. As is apparent from FIG. 11, as the amount of trisodium citrate was decreased, the rupture time of the film was increased, and as a result, the dissolution rate was increased.
[0063]
FIG. 12 and FIG. 13 are diagrams in which the influence on the dissolution rate when the amount of the water-soluble pore-forming film is changed using emedastine fumarate as a drug is examined. As apparent from FIGS. 12 and 13, as the coating amount of the water-soluble pore-forming film was increased, the rupture time of the film was delayed, and consequently the dissolution rate was decreased.
[0064]
FIG. 14 is a diagram in which the effect of the presence or absence of the addition of a water-soluble polymer in the uncoated tablet on the dissolution rate was examined using emedastine fumarate as a drug. As is apparent from FIG. 14, the elution rate was slowed by adding the water-soluble polymer.
[0065]
FIG. 15 and FIG. 16 are diagrams in which the influence on the dissolution rate when the coating amount of the water-soluble pore-forming film was changed using lomerizine hydrochloride as a drug was examined. As apparent from FIGS. 15 and 16, as the coating amount of the water-soluble pore-forming film was increased, the rupture time of the film was delayed, and the elution rate was slowed.
[0066]
FIG. 17 is a diagram in which the effect on dissolution rate is examined when trazodone hydrochloride is used as a drug and the uncoated tablet diameter is 8 mm and the coating amount of the water-soluble pore-forming film is changed. As is clear from FIG. 17, even when the diameter of the uncoated tablet was increased, as the coating amount of the water-soluble pore-forming film was increased, the rupture time of the film was delayed, and consequently the dissolution rate was decreased.
[0067]
FIG. 18 shows the dissolution test results of a water-soluble pore-forming film-coated tablet (Example 18) when nifedipine is used as a drug. As is apparent from FIG. 18, the elution of nifedipine from the sustained-release preparation of Example 18 started from the third hour, and after the film ruptured in 3.5 hours, the nifedipine was rapidly eluted.
[0068]
FIG. 19 shows a tablet in which an enteric coating is applied to the surface of a water-soluble pore-forming film-coated tablet when nifedipine is used as the drug (Example 20), and an enteric coating is applied to the surface of the water-soluble pore-forming film-coated tablet. Further, dissolution test results of a tablet (Example 22) coated with a solid dispersion film on its surface and a tablet (Example 24) coated with a solid dispersion film on the surface of an enteric pore-forming film-coated tablet [Test solution: Disintegration test method described in pharmacopoeia, second liquid (pH 6.8)]. The elution of nifedipine in the second liquid (pH 6.8) from the long-acting preparation of Example 22 started from the first hour, and after 2 hours, the nifedipine was rapidly eluted. In the first solution (pH 1.2), no nifedipine was eluted until 10 hours. The dissolution of nifedipine from the long-acting formulation of Example 22 started immediately after the start of the test, and after 2 hours, the nifedipine was rapidly eluted. The dissolution of nifedipine from the long-acting formulation of Example 24 started immediately after the start of the test, and the drug was rapidly eluted after the film ruptured after 3 hours. In both cases of the sustained-release preparations of Example 22 and Example 24, in the first liquid (pH 1.2), only the nifedipine in the solid dispersion film coated on the outermost layer was gradually eluted. The film did not rupture until 10 hours.
[0069]
[Test Example 2] (Measurement of urinary excretion rate)
The absorption test of the preparation of the present invention was conducted using nifedipine as a drug, and 2,6-dimethyl-4- (2-nitrophenyl) -3,5-pyridinedicarboxylic acid monomethyl ester (hereinafter, the main metabolite of nifedipine). The change in the urinary excretion rate of metabolite) was used as an index. The urine concentration of the main metabolite of nifedipine was quantified by the HPLC method under the following conditions.
HPLC method conditions:
Column: Nova-PakTM C18 [150 mm × 3.9 mm, manufactured by Waters]
-Eluent: mixed solution of 5 volumes of 0.05M acetic acid and 1 volume of acetonitrile
-Column temperature: 25 ° C
・ Flow rate: 1.0 ml / min
・ Detection method: Absorbance measurement at UV290nm
(1) Sample
Capsule of Example 19, Capsule of Example 21, Capsule of Example 23, Capsule of Example 25 and Tablet of Comparative Example 1
(2) Test method
Each sample (equivalent amount of
(3) Test results
Using nifedipine as a drug, the capsule of Example 19 and 4 tablets of Comparative Example 1 filled with 4 water-soluble pore-forming film-coated tablets of Example 18 were excreted from urine when administered to humans. The results of comparing the urinary excretion rates of the main metabolites are shown in FIG. As is clear from FIG. 20, the urinary excretion rate when the sustained-release preparation (capsule) of the present invention is administered is compared with the urinary excretion rate when the tablet (sustained preparation) of Comparative Example 1 is administered. For a long time.
[0070]
Furthermore, the capsule of Example 21 in which four tablets of Example 20 in which the surface of the water-soluble pore-forming film-coated tablet was coated with an enteric film were filled in capsules, and the enteric film on the surface of the water-soluble pore-forming film-coated tablet And the capsule of Example 23 in which four tablets of Example 22 having a solid dispersion film coated on the surface thereof were filled in capsules, and the solid dispersion film on the surface of the enteric pore-forming film-coated tablet The results of measuring the urinary excretion rate of the main metabolite excreted from urine when the capsule of Example 25 filled with 4 coated tablets of Example 24 in capsules were administered to humans are shown in FIGS. It shows in FIG. As is apparent from FIGS. 21 to 23, the urinary excretion rate is long when the capsule is filled with the sustained-release preparation of the present invention in which the enteric film is coated on the surface of the water-soluble pore-forming film-coated tablet. Sustained. In addition, when the sustained-release preparation of the present invention in which a solid dispersion film is coated on the surface of a water-soluble pore-forming film-coated tablet or an enteric film-coated tablet is filled in a capsule and administered, the initial urinary excretion rate is secured. And it lasted for a long time.
[0071]
[Test Example 3] (Measurement of plasma concentration)
The plasma concentration of nifedipine was quantified by gas chromatography (hereinafter referred to as GC method) under the following conditions.
Conditions for the GC method:
・ Detector: Electron capture detector
Column: 3% OV-17 (3 mm × 3.1 m, manufactured by GL Sciences Inc.)
Column temperature: 270 ° C
(1) Sample
Capsule of Example 19
(2) Test method
Six healthy adult volunteers were orally administered a sample (equivalent to 20 mg of nifedipine) 30 minutes after breakfast, and after administration for a certain period of time (2, 4, 6, 8, 10, 12, 15, 24, 28, 32, 36 48 hours) Blood was collected every time, and the obtained blood was centrifuged to obtain plasma. The concentration of nifedipine in plasma obtained every hour was quantified by the GC method.
(3) Test results
FIG. 24 shows the time course of the plasma concentration (average value) of nifedipine when the water-soluble pore-forming film-coated tablet of the present invention was administered to humans.
As is apparent from FIG. 24, when the sustained-release preparation of the present invention was orally administered to humans, the plasma concentration of nifedipine was sustained for a long time.
Therefore, the preparation of the present invention is excellent in oral absorption of poorly water-soluble drugs and sustainability in plasma concentration, and is useful as a preparation for drug administration once or twice a day.
[0072]
【Example】
Hereinafter, the present invention will be described with reference to examples and comparative examples.
Example 1
98 g of emedastine fumarate (Japanese Patent Publication No. 2-24821), polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.) 734.7 g, trisodium citrate dihydrate (100-mesh sieve product) ) A mixture of 244.9 g and lactose 98 g was fluidized in a fluidized bed granulator, and a solution obtained by dissolving 24.4 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 463 g of water was used as a binder. After spraying, it was dried and sized with an 18-mesh sieve to obtain a drug-containing granulated product. 1114 g of this granulated product was mixed with 11.3 g of magnesium stearate and 11.3 g of light anhydrous silicic acid, and this mixture was tableted with a rotary tableting machine to 50 mg per tablet using a 5 mm diameter punch. 753 g (about 15100) tablets were obtained.
Next, 0.43 parts by weight of hydroxypropyl methylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) with respect to 1 part by weight of ethyl cellulose (trade name: Etocel standard-10, manufactured by Dow Chemical Co., Ltd.) Triethyl acid (trade name:
[0073]
Example 2
Fluidized bed construction of 72 g of emedastine fumarate, 495 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 180 g of trisodium citrate dihydrate (100 mesh sieve passed product) and lactose 117 g. Fluidized in a granulator, sprayed with a solution of 18 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 342 g of water as a binder, dried, and sized with an 18 mesh sieve. A drug-containing granulated product was obtained. 855 g of this granulated product was mixed with 10.5 g of magnesium stearate and 7 g of light anhydrous silicic acid, and this mixture was compressed to 50 mg per tablet with a rotary tableting machine using a punch with a diameter of 5 mm, and 737 g of plain tablets (About 14700 pieces) were obtained.
Next, 350 g (about 7000) of the uncoated tablets were sprayed with a water-soluble pore-forming film solution in the same manner as in Example 1 and dried to obtain tablets having a weight per tablet of 56 mg. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0074]
Example 3
Fluidized bed construction of a mixture of 152 g emedastine fumarate, 950 g polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 380 g trisodium citrate dihydrate (100 mesh sieve passed product) and lactose 342 g. Fluidized in a granulator, sprayed as a binder with a solution of 38 g of hydroxypropyl cellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 722 g of water, dried, and sized with an 18 mesh sieve. A drug-containing granulated product was obtained. 1800 g of this granulated product was mixed with 18.4 g of magnesium stearate and 18.4 g of light anhydrous silicic acid, and this mixture was compressed to 50 mg per tablet with a rotary tableting machine using a 5 mm diameter punch. 1680 g (about 33600) of tablets were obtained.
Next, 350 g (about 7000) of the uncoated tablets were sprayed with a water-soluble pore-forming film solution onto the uncoated tablets in the same manner as in Example 1 to obtain tablets with a weight of 56 mg per tablet. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0075]
Example 4
A fluidized bed mixture of 72 g emedastine fumarate, 450 g polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 36 g trisodium citrate dihydrate (100 mesh sieve product) and lactose 306 g Fluidized in a granulator, sprayed with a solution of 18 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 342 g of water as a binder, dried, and sized with an 18 mesh sieve. A granulated product was obtained. 855 g of this granulated product was mixed with 10.5 g of magnesium stearate and 7 g of light anhydrous silicic acid, and this mixture was compressed to 50 mg per tablet with a rotary tableting machine using a punch with a diameter of 5 mm. (About 14400) were obtained.
Next, 350 g (about 7000) of the uncoated tablets were sprayed with a water-soluble pore-forming film solution onto the uncoated tablets in the same manner as in Example 1 to obtain tablets with a weight of 56 mg per tablet. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0076]
Example 5
Fluidized bed construction of 72 g of emedastine fumarate, 450 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 54 g of trisodium citrate dihydrate (100-mesh sieve product) and 288 g of lactose Fluidized in a granulator, sprayed with a solution of 18 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 342 g of water as a binder, dried, and sized with an 18 mesh sieve. A granulated product was obtained. 855 g of this granulated product was mixed with 10.5 g of magnesium stearate and 7 g of light anhydrous silicic acid, and this mixture was compressed to 50 mg per tablet with a rotary tableting machine using a punch with a diameter of 5 mm. (About 14800 pieces) were obtained.
Next, 350 g (about 7000) of the uncoated tablets were sprayed with a water-soluble pore-forming film solution onto the uncoated tablets in the same manner as in Example 1 to obtain tablets with a weight of 56 mg per tablet. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0077]
Example 6
Fluidized bed mixture of 72 g of emedastine fumarate, 540 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 180 g of trisodium citrate dihydrate (100 mesh sieve product) and lactose 72 g Fluidized in a granulator, sprayed with a solution of 18 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 342 g of water as a binder, dried, and sized with an 18 mesh sieve. A granulated product was obtained. 860 g of this granulated product was mixed with 10.5 g of magnesium stearate and 7 g of light anhydrous silicic acid, and the mixture was compressed to 50 mg per tablet with a rotary tableting machine using a punch with a diameter of 5 mm. About 14200).
Next, 0.43 parts by weight of hydroxypropyl cellulose (trade name: Nisso HPC, manufactured by Nippon Soda Co., Ltd.) is added to 1 part by weight of ethyl cellulose (trade name: Etcelle standard-10, manufactured by Dow Chemical Company), citric acid 0.29 parts by weight of triethyl (trade name:
[0078]
Example 7
In the same manner as in Example 6, the mixture containing emedastine fumarate was tableted into 50 mg per tablet with a 5 mm diameter punch to obtain 708 g (about 14,200) of plain tablets.
Next, a water-soluble pore-forming film solution was sprayed onto the plain tablets and dried in the same manner as in Example 6 except that 350 g (about 7000) of these uncoated tablets was increased by 1 mg in the amount of coating per tablet. A tablet with a weight of 57 mg was obtained. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0079]
Example 8
Fluidized bed granulation of a mixture of 72 g emedastine fumarate, 540 g polyvinyl alcohol (trade name: Kuraray Poval PVA-CS, manufactured by Kuraray Co., Ltd., 100-mesh sieve product), 180 g sodium chloride (100-mesh sieve product) and lactose 72 g Fluidized in the machine, sprayed as a binder a solution obtained by dissolving 18 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 342 g of water, dried, and sized with an 18 mesh sieve. Grains were obtained. 850 g of this granulated product was mixed with 10.4 g of magnesium stearate and 7 g of light anhydrous silicic acid, and this mixture was compressed to 50 mg per tablet with a rotary tableting machine using a punch having a diameter of 5 mm. (About 15100 pieces) were obtained.
Next, 350 g (about 7000) of the uncoated tablets were sprayed with a water-soluble pore-forming film solution onto the uncoated tablets in the same manner as in Example 1 to obtain tablets with a weight of 56 mg per tablet. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0080]
Example 9
In the same manner as in Example 8, the mixture containing emedastine fumarate was tableted to 50 mg per tablet with a rotary tableting machine using a punch with a diameter of 5 mm to obtain 755 g (about 15100) of uncoated tablets.
Next, a water-soluble pore-forming film solution was sprayed and dried on the uncoated tablets in the same manner as in Example 1 except that 350 mg (about 7000) of the uncoated tablets was increased by 1 mg. Tablets with a weight per tablet of 57 mg were obtained. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0081]
Example 10
In a fluid bed granulator, 72 g of emedastine fumarate, 540 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 180 g of sodium sulfate / anhydride (100 mesh sieve passed product) and 72 g of lactose were mixed. It is made to flow, and after spraying as a binder a solution obtained by dissolving 18 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 342 g of ethanol, the mixture is dried and granulated with an 18 mesh sieve. Obtained. This granulated product (850 g) was mixed with magnesium stearate (10.4 g) and light anhydrous silicic acid (7 g). (About 15400 pieces) were obtained.
Next, 350 g (about 7000) of the uncoated tablets were sprayed with a water-soluble pore-forming film solution onto the uncoated tablets in the same manner as in Example 1 to obtain tablets with a weight of 56 mg per tablet. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0082]
Example 11
72 g of emedastine fumarate, 540 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 180 g of sodium sulfate / anhydride (100 mesh sieve product) and hydroxypropyl cellulose (trade name: Nisso HPC, Japan (Soda Co., Ltd.) 72 g of the mixture was fluidized in a fluidized bed granulator, a solution of 18 g of hydroxypropylcellulose dissolved in 342 g of ethanol was sprayed as a binder, dried, granulated with an 18 mesh sieve and granulated I got a thing. This granulated product (850 g) was mixed with magnesium stearate (10.4 g) and light anhydrous silicic acid (7 g), and this mixture was tableted into 50 mg per tablet with a rotary tableting machine using a 5 mm diameter punch. (About 15200) was obtained.
Next, 350 g (about 7000) of the uncoated tablets were sprayed with a water-soluble pore-forming film solution onto the uncoated tablets in the same manner as in Example 1 to obtain tablets with a weight of 56 mg per tablet. The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0083]
Example 12
A mixture of 90 g of lomelidine hydrochloride (Japanese Patent Publication No. 3-13232), 360 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 360 g of sodium sulfate / anhydride (100 mesh sieve-passed product) and 54 g of lactose In a fluidized bed granulator, sprayed with a solution of 18 g of hydroxypropylcellulose (trade name: Nisso HPC, manufactured by Nippon Soda Co., Ltd.) in 342 g of ethanol as a binder, dried, and dried to a 18 mesh sieve. To obtain a granulated product. 850 g of this granulated product is mixed with 12.1 g of magnesium stearate and 5.2 g of light anhydrous silicic acid, and the mixture is compressed to 50 mg per tablet with a rotary tableting machine using a punch with a diameter of 5 mm. 750 g (about 15000 pieces) was obtained.
Next, 0.49 parts by weight of hydroxypropyl methylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) is added to 1 part by weight of ethyl cellulose (trade name: Etocel standard-10, manufactured by Dow Chemical Co., Ltd.), Triethyl acid (trade name:
[0084]
Example 13
In the same manner as in Example 12, the mixture containing romeridine hydrochloride was tableted into 50 mg per tablet with a rotary tableting machine using a 5 mm diameter punch to obtain 750 g of uncoated tablets (about 15,000).
Next, water-soluble pore-forming film solution was sprayed onto the uncoated tablets and dried in the same manner as in Example 12 except that 350 mg (about 7000) of the uncoated tablets was increased by 1 mg in the coating amount per tablet. A tablet having a weight per unit of 57 mg was obtained. The long-acting preparation of the present invention thus produced contains 5 mg of lomelidine hydrochloride per tablet.
[0085]
Example 14
A fluidized bed of a mixture of 90 g of romeridine hydrochloride, 450 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 270 g of trisodium citrate dihydrate (100 mesh sieve product) and lactose 57.6 g Fluidized in a granulator, sprayed with a solution of 18 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 342 g of water as a binder, dried, and sized with an 18 mesh sieve A shig granulated product was obtained. 855 g of this granulated product was mixed with 7 g of magnesium stearate and 7 g of light anhydrous silicic acid, and this mixture was compressed into 50 mg per tablet using a punch with a diameter of 5 mm, and 735 g of plain tablets (about 14700).
Next, 0.47 parts by weight of hydroxypropyl methylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) is added to 1 part by weight of ethyl cellulose (trade name: Etcelle standard-10, manufactured by Dow Chemical Company), Triethyl acid (trade name:
[0086]
Example 15
In the same manner as in Example 14, the mixture containing romeridine hydrochloride was tableted into 50 mg per tablet with a rotary tableting machine using a 5 mm diameter punch to obtain 750 g of uncoated tablets (about 15,000).
Next, water-soluble pore-forming film solution was sprayed onto the uncoated tablets and dried in the same manner as in Example 16 except that 350 mg (about 7000) of the uncoated tablets was increased by 1 mg. A tablet having a weight per unit of 57 mg was obtained. The long-acting preparation of the present invention thus produced contains 5 mg of lomelidine hydrochloride per tablet.
[0087]
Example 16
337.5 g of trazodone hydrochloride (Japanese Patent Publication No. 44-7341), 360 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.) and trisodium citrate dihydrate (product passed through 100 mesh sieve) 180 g of the mixture was fluidized in a fluidized bed granulator, sprayed with a solution of 18 g of hydroxypropylcellulose (trade name: Nisso HPC, Nippon Soda Co., Ltd.) in 342 g of water as a binder, dried, and 18 The granulated product was obtained by sizing with a mesh sieve. 866 g of this granulated product is mixed with 8.7 g of magnesium stearate and 4.4 g of light anhydrous silicic acid, and this mixture is compressed to 202 mg per tablet with a rotary tablet using a punch with a diameter of 8 mm. 750 g (about 3700) tablets were obtained.
Next, 0.47 parts by weight of hydroxypropyl methylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) is added to 1 part by weight of ethyl cellulose (trade name: Etcelle standard-10, manufactured by Dow Chemical Company), Triethyl acid (trade name:
[0088]
Example 17
In the same manner as in Example 16, the mixture containing trazodone hydrochloride was tableted into 202 mg per tablet with a rotary tableting machine using a punch with a diameter of 8 mm, to obtain 750 g (about 3700) of plain tablets.
Next, a water-soluble pore-forming film solution was sprayed onto the plain tablets and dried in the same manner as in Example 16 except that 350 mg (about 1700) of these uncoated tablets were increased by 7 mg in the amount of coating per tablet. A tablet with a weight per weight of 220 mg was obtained. The long-acting preparation of the present invention thus produced contains 75 mg of trazodone hydrochloride per tablet.
[0089]
Example 18
101.4 g of nifedipine and 152 g of hydroxypropylmethylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) are dissolved in a mixed solvent of 821 g of dichloromethane and 547 g of ethanol to obtain a solid dispersion solution. In a fluid bed granulator, a mixture of 810.8 g of polyvinyl alcohol (trade name: Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.) and 135.8 g of trisodium citrate dihydrate (100 mesh sieve product) After flowing and spraying the solid dispersion solution, it was dried and sized with a 30-mesh sieve to obtain a granulated product. 592 g of this granulated product was mixed with 3 g of magnesium stearate and 5 g of light anhydrous silicic acid, and the mixture was compressed to 60 mg per tablet with a rotary tableting machine using a 5 mm diameter punch, and 460 g (about 7600 g) Obtained).
Next, 0.6 parts by weight of hydroxypropyl methylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) is added to 1 part by weight of ethyl cellulose (trade name: Etcelle standard-10, manufactured by Dow Chemical Company). It dissolved in the mixed solvent of dichloromethane and ethanol [1: 1 (weight ratio)] so that the density | concentration of the sum total of both might be 5 weight%, and it was set as the water-soluble pore formation film liquid. This water-soluble pore-forming film solution was sprayed and dried on 350 g (about 5800) of the above-mentioned uncoated tablets, and the water-soluble pore-forming film was coated to obtain tablets having a weight per tablet of 65 mg. The long-acting preparation of the present invention thus produced contains 5 mg of nifedipine per tablet.
[0090]
Example 19
Four long-acting preparations of Example 18 were filled into No. 2 hard capsules to form capsules.
[0091]
Example 20
338 g of nifedipine and 507 g of hydroxypropylmethylcellulose (trade name: TC-5E: manufactured by Shin-Etsu Chemical Co., Ltd.) are dissolved in a mixed solvent of 2740 g of dichloromethane and 1824 g of ethanol to obtain a solid dispersion solution. A mixture of 2703 g of polyvinyl alcohol (Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.) and 453 g of trisodium citrate dihydrate (100-mesh passed product) was fluidized in a fluid bed granulator, and a solid dispersion solution was obtained. After spraying, it was dried and sized with a 30-mesh sieve to obtain a granulated product. 1852 g of magnesium stearate and 30 g of light anhydrous silicic acid were mixed with 3552 g of this granulated product, and the mixture was compressed to 60 mg per tablet with a rotary tableting machine using a 5 mm diameter punch, and 3420 g (about 57000) of uncoated tablets. Obtained).
Next, 0.7 parts by weight of hydroxypropyl methylcellulose (trade name: TC-5E: manufactured by Shin-Etsu Chemical Co., Ltd.) is added to 1 part by weight of ethyl cellulose (trade name: Etocel standard-10, manufactured by Dow Chemical Company). It dissolved in the mixed solvent of dichloromethane and ethanol [1: 1 (weight ratio)] so that the density | concentration of the sum total of both might be 5 weight%, and it was set as the water-soluble pore formation film liquid. After spraying and drying the water-soluble pore-forming film liquid on 350 g (about 5800) of the above-mentioned uncoated tablets, the water-soluble pore-forming film was coated to obtain tablets having a weight of 65 mg per tablet, and further hydroxypropylmethylcellulose Enteric coating solution dissolved in a mixed solvent of dichloromethane and ethanol [1: 1 (weight ratio)] so that the concentration of phthalate [trade name: HPMCP (HP-50), manufactured by Shin-Etsu Chemical Co., Ltd.] is 5% by weight. Was sprayed and dried, and an enteric film was coated to obtain tablets each having a weight of 68 mg. The long-acting preparation of the present invention thus produced contains 5 mg of nifedipine per tablet.
[0092]
Example 21
Four long-acting preparations of Example 20 were filled into No. 2 hard capsules to form capsules.
[0093]
Example 22
300 g of nifedipine and 450 g of hydroxypropyl methylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) are dissolved in a mixed solvent of 2432 g of dichloromethane and 1624 g of ethanol to obtain a solid dispersion solution. A mixture of 3200 g of polyvinyl alcohol (Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.) and 536 g of trisodium citrate / anhydride (100-mesh passed product) is fluidized in a fluid bed granulator, and the solid dispersion solution is sprayed. After drying, the mixture was sized with a 30-mesh sieve to obtain a granulated product. This granulated product (4190 g) was mixed with magnesium stearate (22.4 g) and light anhydrous silicic acid (37.4 g), and this mixture was tableted with a rotary tableting machine using a 5 mm diameter punch, and the weight per tablet was 56 g (88.88 mg) of uncoated tablets (containing 3.75 mg of nifedipine per tablet) were obtained.
Next, 0.7 parts by weight of hydroxypropyl methylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) with respect to 1 part by weight of ethyl cellulose (trade name: Etocel standard-10, manufactured by Dow Chemical Company) It dissolved in the mixed solvent of dichloromethane and ethanol [1: 1 (weight ratio)] so that the density | concentration of the sum total of both might be 5 weight%, and it was set as the water-soluble pore formation film liquid. 350 g (about 6200) of the above-mentioned uncoated tablets are put in a coating pan, and this water-soluble pore-forming film solution is sprayed on the uncoated tablets, dried and coated to form a water-soluble pore having a weight of 61.38 mg per tablet. After obtaining the film-coated tablet, further, dichloromethane and ethanol [1: 1 (weight ratio) so that the concentration of hydroxypropylmethylcellulose phthalate (trade name: HPMCP (HP-50), manufactured by Shin-Etsu Chemical Co., Ltd.) is 5% by weight. The enteric coating solution dissolved in the mixed solvent of)] was sprayed and dried on the above tablets, and the enteric coating was coated to obtain an enteric coating-coated tablet having a weight per tablet of 64.38 mg.
Next, 1.5 parts by weight of hydroxypropylmethylcellulose (trade name: TC-5RW, manufactured by Shin-Etsu Chemical Co., Ltd.) is added to 1 part by weight of nifedipine, and dichloromethane and ethanol so that the concentration of hydroxypropylmethylcellulose is 5% by weight. It was dissolved in a mixed solvent of [6: 4 (weight ratio)] to obtain a solid dispersion solution. The enteric film-coated tablet is sprayed with this solid dispersion solution and dried to coat the solid dispersion film (corresponding to 1.25 mg of nifedipine per tablet), and the weight per tablet is 67.5 mg. A sustained formulation was obtained. The long-acting preparation of the present invention thus produced contains 5 mg of nifedipine per tablet.
[0094]
Example 23
Four long-acting preparations of Example 22 were filled into No. 2 hard capsules to form capsules.
[0095]
Example 24
90 g of nifedipine and 135.6 g of hydroxypropylmethylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) are dissolved in a mixed solvent of 732 g of dichloromethane and 488 g of ethanol to obtain a solid dispersion solution. A mixture of 600 g of polyvinyl alcohol (Kuraray Poval PVA-CSTS, manufactured by Kuraray Co., Ltd.), 160.8 g of trisodium citrate / anhydride (100 mesh passed product) and crystalline cellulose (trade name: Avicel PH301, manufactured by Asahi Kasei Kogyo Co., Ltd.) After flowing in a fluidized bed granulator and spraying and drying the solid dispersion solution, it was sized with a 30 mesh sieve to obtain a granulated product. 6.4 g of magnesium stearate and 10.7 g of light anhydrous silicic acid were mixed with 1088 g of this granulated product, and this mixture was compressed with a rotary tableting machine using a 5 mm diameter punch, and the weight per tablet was 800 g (about 15400) of 51.9 mg uncoated tablets (containing 3.75 mg of nifedipine per tablet) were obtained. Next, hydroxypropyl methylcellulose phthalate [trade name: HPMCP (HP-55), manufactured by Shin-Etsu Chemical Co., Ltd.] 0.67 with respect to 1 part by weight of ethyl cellulose (trade name: Etocel standard-10, manufactured by Dow Chemical Company) Parts by weight, 0.33 parts by weight of triethyl citrate (trade name:
Next, 1.5 parts by weight of hydroxypropylmethylcellulose (trade name: TC-5RW, manufactured by Shin-Etsu Chemical Co., Ltd.) is added to 1 part by weight of nifedipine, and dichloromethane and ethanol so that the concentration of hydroxypropylmethylcellulose is 5% by weight. It was dissolved in a mixed solvent of [6: 4 (weight ratio)] to obtain a solid dispersion solution. This enteric pore-forming film-coated tablet is sprayed with this solid dispersion solution and dried to coat the solid dispersion film (corresponding to 1.25 mg of nifedipine per tablet), and the weight per tablet is 59.85 mg. A sustained formulation of the invention was obtained. The long-acting preparation of the present invention thus produced contains 5 mg of nifedipine per tablet.
[0096]
Example 25
Four long-acting preparations of Example 24 were filled into No. 2 hard capsules to form capsules.
[0097]
Example 26
In the same manner as in Example 8, the mixture containing emedastine fumarate was tableted to 50 mg per tablet with a rotary tableting machine using a punch with a diameter of 5 mm to obtain 755 g (about 15100) of uncoated tablets.
Next, 0.21 part by weight of hydroxypropyl methylcellulose (trade name: TC-5E, manufactured by Shin-Etsu Chemical Co., Ltd.) is added to 1 part by weight of ethyl cellulose (trade name: Etcelle standard-10, manufactured by Dow Chemical Co.). 0.21 part by weight of propylmethylcellulose phthalate [trade name: HPMCP (HP-55), manufactured by Shin-Etsu Chemical Co., Ltd.], 0.29 part by weight of triethyl citrate (trade name:
[0098]
Example 27
In the same manner as in Example 1, a mixture containing emedastine fumarate was tableted to prepare an uncoated tablet, and the surface of the uncoated tablet was sprayed with a water-soluble pore-forming film solution and dried to obtain 56 mg per tablet. Ethanol so that the concentration of hydroxypropylmethylcellulose phthalate (trade name: HPMCP (HP-50), manufactured by Shin-Etsu Chemical Co., Ltd.) is 5% by weight on the surface of 200 g (about 3560) of water-soluble pore-forming film-coated tablets. An enteric coating solution dissolved in a mixed solvent of water and water [4: 1 (weight ratio)] was sprayed and dried, and the enteric coating was coated to obtain an enteric coating coated tablet having a weight per tablet of 59 mg. . The long-acting preparation of the present invention thus produced contains 4 mg emedastine fumarate per tablet.
[0099]
Example 28
In the same manner as in Example 1, a mixture containing emedastine fumarate was tableted to prepare an uncoated tablet, and the surface of the uncoated tablet was sprayed with a water-soluble pore-forming film solution and dried to obtain 56 mg per tablet. On the surface of 100 g (about 1780) of water-soluble pore-forming film-coated tablets, 2 parts by weight of hydroxypropyl methylcellulose (trade name: TC-5MW, manufactured by Shin-Etsu Chemical Co., Ltd.) is further added to 1 part by weight of emedastine fumarate. A solid dispersion solution dissolved in a mixed solvent of ethanol and water [1: 1 (weight ratio)] is sprayed and dried so that the concentration of hydroxypropylmethylcellulose is 5% by weight, and the solid dispersion film is coated (per tablet) The sustained-release preparation of the present invention having a weight per tablet of 59 mg was obtained. The long-acting preparation of the present invention thus produced contains 5 mg of emedastine fumarate per tablet.
[0100]
Example 29
In the same manner as in Example 27, a mixture containing emedastine fumarate was tableted to prepare an uncoated tablet, and a water-soluble pore-forming film solution and an enteric film solution were sequentially sprayed and dried on the surface of the uncoated tablet. Hydroxypropyl methylcellulose (trade name: TC-5MW, manufactured by Shin-Etsu Chemical Co., Ltd.) 2 with respect to 1 part by weight of emedastine fumarate on the surface of 100 g (about 1690) of enteric-coated coated tablets of 59 mg per tablet A solid dispersion solution in which parts by weight are dissolved in a mixed solvent of ethanol and water [1: 1 (weight ratio)] so that the concentration of hydroxypropylmethylcellulose is 5% by weight is sprayed and dried to coat the solid dispersion film (A corresponding amount of 1 mg emedastine fumarate per tablet) to obtain a sustained-release preparation of the present invention having a weight per tablet of 62 mg. The long-acting preparation of the present invention thus produced contains 5 mg of emedastine fumarate per tablet.
[0101]
Comparative Example 1
Prepared according to Example 1 described in JP-B-6-11699.
A) Core part
16.5 g of crystalline nifedipine [average particle size 3.3 μm (measured by air permeation method)] is mixed with 194.0 g of lactose and 75.0 g of corn starch, and this mixture is mixed with 5.0 g of corn starch and 70.0 g of hot water. Granulated in paste and then dried. The granulate was sieved and mixed with 25.0 g microcrystalline cellulose and 1.0 g magnesium stearate. This mixture was compressed into 63.3 mg uncoated tablets having a diameter of 6 mm. A gastric juice-resistant coating was applied to the core by using an organic solvent solution of hydroxypropylmethylcellulose phthalate [trade name: HPMCP (HP-55), manufactured by Shin-Etsu Chemical Co., Ltd.]. The coated tablet weighed 70.3 mg.
B) Granules for coating
8.4 g of nifedipine 20.0 g of lactose, 0.8 g of colloidal silica, M-type hydroxypropylcellulose [trade name: Nisso HPC (HPC-M), manufactured by Nippon Soda Co., Ltd.] 35.0 g, L-type hydroxypropylcellulose [ Product name: Nisso HPC (HPC-L), manufactured by Nippon Soda Co., Ltd.] 87.4 g and 16.0 g of citric acid were mixed with the solution of 1.0 g of L-type hydroxypropylcellulose in a fluid bed granulator. Granulated. The dried and sieved granulate was then mixed with 1.4 g of magnesium stearate.
[0102]
These granular materials and the core described in A) were compressed into a compression-molded tablet having a diameter of 10 mm and a weight of 410 mg by a compression molding machine.
[Brief description of the drawings]
FIG. 1 shows cross-sectional views of the sustained-release preparations of the present invention (Examples 1 to 17 and Example 26).
FIG. 2 shows a cross-sectional view of the long-acting formulation of the present invention (Example 27).
FIG. 3 shows a cross-sectional view of the sustained release formulation (Example 28) of the present invention.
FIG. 4 shows a cross-sectional view of the long-acting formulation of the present invention (Example 29).
FIG. 5 shows a cross-sectional view of the long-acting formulation of the present invention (Example 18).
FIG. 6 shows a cross-sectional view of the long-acting formulation of the present invention (Example 20).
FIG. 7 shows a cross-sectional view of the long-acting preparation of the present invention (Example 24).
FIG. 8 shows a cross-sectional view of the long-acting preparation of the present invention (Example 22).
FIG. 9 is a graph showing the dissolution test results of emedastine fumarate from the tablet of Example 1, where ◇ indicates the disintegration test method described in the Japanese Pharmacopoeia, first solution (pH 1.2) as the test solution In this case, the ♦ mark indicates that the acetic acid / sodium acetate buffer solution (pH 4.0) described in the pharmacopoeia is used as a test solution, and the △ mark indicates the disintegration test method / second solution (
FIG. 10 is a graph showing the results of dissolution test of emedastine fumarate from the tablets of Examples 1 to 3 [test solution: disintegration test method described in pharmacopoeia, first solution (pH 1.2)]. Indicates the tablet of Example 1, o indicates the tablet of Example 2, and Δ indicates the tablet of Example 3.
FIG. 11 is a graph showing the results of dissolution test of emedastine fumarate from the tablets of Examples 3 to 5 [test solution: disintegration test method described in pharmacopoeia, first solution (pH 1.2)], Δ mark Indicates the tablet of Example 3, o indicates the tablet of Example 4, and ◇ indicates the tablet of Example 5.
FIG. 12 is a graph showing the results of dissolution test of emedastine fumarate from the tablets of Examples 6 to 7 [test solution: disintegration test method described in pharmacopoeia, first solution (pH 1.2)], ○ mark Indicates the tablet of Example 6, and Δ indicates the tablet of Example 7.
FIG. 13 is a graph showing the results of the dissolution test of emedastine fumarate from the tablets of Examples 8 to 9 [test solution: disintegration test method described in the Japanese Pharmacopoeia, first solution (pH 1.2)]. Indicates the tablet of Example 8, and Δ indicates the tablet of Example 9.
FIG. 14 is a graph showing the results of dissolution test of emedastine fumarate from the tablets of Examples 10 to 11 [test solution: disintegration test method described in pharmacopoeia, first solution (pH 1.2)], and Δ mark Indicates the tablet of Example 10, and ● indicates the tablet of Example 11.
FIG. 15 is a graph showing the results of a dissolution test of lomerizine hydrochloride from the tablets of Examples 12 to 13 [test solution: disintegration test method described in pharmacopoeia, first solution (pH 1.2)]. The tablet of Example 12 and the Δ mark indicate the tablet of Example 13.
FIG. 16 is a graph showing the results of dissolution test of lomerizine hydrochloride from the tablets of Examples 14 to 15 [test solution: disintegration test method described in pharmacopoeia, first solution (pH 1.2)], Tablets of Example 14 and Δ marks indicate tablets of Example 15.
FIG. 17 is a graph showing the results of dissolution test of trazodone hydrochloride from the tablets of Examples 16 to 17 [test solution: disintegration test method described in pharmacopoeia, first solution (pH 1.2)]. The tablet of Example 16 and the Δ mark indicate the tablet of Example 17.
18 is a graph showing the results of a dissolution test of nifedipine from the tablet of Example 18 [test solution: disintegration test method described in pharmacopoeia, second solution (pH 6.8)]. FIG.
FIG. 19 is a result of dissolution test of nifedipine from the tablet of Example 20, the tablet of Example 22 and the tablet of Example 24 [test solution: disintegration test method described in pharmacopoeia, second solution (pH 6.8)] result. △ mark indicates the tablet of Example 20, ◇ mark indicates the tablet of Example 22, and ♦ mark indicates the tablet of Example 24.
FIG. 20 is a graph showing the time course of nifedipine urinary excretion rate after oral administration of the capsule of Example 19 and the tablet of Comparative Example 1 to humans. The mark indicates the tablet of Comparative Example 1.
FIG. 21 is a graph showing the time course of nifedipine urinary excretion rate after oral administration of the capsule of Example 21 to humans.
FIG. 22 is a graph showing the time course of the nifedipine urinary excretion rate after oral administration of the capsule of Example 23 to humans.
FIG. 23 is a graph showing the time course of the nifedipine urinary excretion rate after oral administration of the capsule of Example 25 to humans.
FIG. 24 is a graph showing the time course of nifedipine plasma concentration after oral administration of the capsule of Example 19 to humans.
[Explanation of symbols]
1: (A) a mixture of a drug and a water-soluble polymer, or a solid dispersion comprising them,
(B) Uncoated tablets containing polyvinyl alcohol and (c) one or more salts selected from the group consisting of trisodium citrate, sodium sulfate and sodium chloride
In the uncoated tablets of FIGS. 1 to 4, the drug is indicated by a black discontinuous phase, and components other than the drug are indicated by a white continuous phase. Moreover, in the uncoated tablet of FIGS. 5-8, the solid dispersion which consists of a drug and water-soluble polymer is shown by a black continuous phase, and components other than a solid dispersion are shown by a white discontinuous phase.
2: A film comprising a water-insoluble polymer and a water-soluble polymer and / or an enteric polymer
3: Film made of enteric polymer
4: Mixture of drug and water-soluble polymer, or solid dispersion film made of them
Claims (16)
Translated fromJapanesePriority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP35197295AJP4072597B2 (en) | 1994-12-27 | 1995-12-26 | Sustained formulation |
Applications Claiming Priority (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP6-340045 | 1994-12-27 | ||
| JP34004594 | 1994-12-27 | ||
| JP34040794 | 1994-12-28 | ||
| JP6-340407 | 1994-12-28 | ||
| JP3615695 | 1995-01-31 | ||
| JP5353495 | 1995-02-17 | ||
| JP7-36156 | 1995-08-07 | ||
| JP7-222566 | 1995-08-07 | ||
| JP7-53534 | 1995-08-07 | ||
| JP22256695 | 1995-08-07 | ||
| JP35197295AJP4072597B2 (en) | 1994-12-27 | 1995-12-26 | Sustained formulation |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH09104620A JPH09104620A (en) | 1997-04-22 |
| JP4072597B2true JP4072597B2 (en) | 2008-04-09 |
Family
ID=27549798
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP35197295AExpired - Fee RelatedJP4072597B2 (en) | 1994-12-27 | 1995-12-26 | Sustained formulation |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4072597B2 (en) |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000128779A (en)* | 1998-10-20 | 2000-05-09 | Mitsui Chemicals Inc | Controlled release medicine type preparation |
| WO2000071099A1 (en)* | 1999-05-20 | 2000-11-30 | Elan Corporation, Plc | Multiparticulate controlled release selective serotonin reuptake inhibitor formulations |
| GB0025208D0 (en) | 2000-10-13 | 2000-11-29 | Euro Celtique Sa | Delayed release pharmaceutical formulations |
| US20030175349A1 (en)* | 2001-01-30 | 2003-09-18 | Council Of Scientific And Industrial Research | Pharmaceutical compostion for extended/sustained release of a therapeutically active ingredient |
| DK1377276T3 (en)* | 2001-04-10 | 2012-01-02 | Sun Pharma Advanced Res Co Ltd | Calculation of pulse-release composition |
| JP2005104914A (en)* | 2003-09-30 | 2005-04-21 | Kyowa Yakuhin Kogyo Kk | Basic agent-containing pharmaceutical preparation |
| CA2491665A1 (en)* | 2004-12-24 | 2006-06-24 | Louis Cartilier | Tablet formulation for the sustained release of active substances |
| PT1931346E (en)* | 2005-09-09 | 2012-08-14 | Angelini Labopharm Llc | COMPOSITION OF TRAZODONE FOR ADMINISTRATION ONCE A DAY |
| KR101434316B1 (en)* | 2006-04-19 | 2014-08-28 | 아지노모토 가부시키가이샤 | Film coating method of solid dispersion |
| KR100758592B1 (en)* | 2006-08-29 | 2007-09-13 | 주식회사유한양행 | Film coating composition |
| CA2661683C (en)* | 2006-08-31 | 2015-11-24 | Eurand, Inc | Drug delivery systems comprising solid solutions of weakly basic drugs |
| US8778398B2 (en) | 2008-11-04 | 2014-07-15 | Jazz Pharmaceuticals, Inc. | Immediate release formulations and dosage forms of gamma-hydroxybutyrate |
| US20120076865A1 (en) | 2010-03-24 | 2012-03-29 | Jazz Pharmaceuticals, Inc. | Controlled release dosage forms for high dose, water soluble and hygroscopic drug substances |
| US10398662B1 (en) | 2015-02-18 | 2019-09-03 | Jazz Pharma Ireland Limited | GHB formulation and method for its manufacture |
| US11602513B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11986451B1 (en) | 2016-07-22 | 2024-05-21 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12186296B1 (en) | 2016-07-22 | 2025-01-07 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11504347B1 (en) | 2016-07-22 | 2022-11-22 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11602512B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| UY37341A (en) | 2016-07-22 | 2017-11-30 | Flamel Ireland Ltd | FORMULATIONS OF GAMMA-MODIFIED RELEASE HYDROXIBUTIRATE WITH IMPROVED PHARMACOCINETICS |
| US20180263936A1 (en) | 2017-03-17 | 2018-09-20 | Jazz Pharmaceuticals Ireland Limited | Gamma-hydroxybutyrate compositions and their use for the treatment of disorders |
| JP2018184360A (en)* | 2017-04-25 | 2018-11-22 | ニプロ株式会社 | Enteric-coated sustained release formulation for oral administration |
| PL3616696T3 (en)* | 2017-04-28 | 2025-05-05 | Astellas Pharma Inc. | Orally administrable enzalutamide-containing pharmaceutical composition |
| JP7180592B2 (en) | 2017-04-28 | 2022-11-30 | アステラス製薬株式会社 | Pharmaceutical composition |
| WO2019172420A1 (en)* | 2018-03-09 | 2019-09-12 | 協和発酵キリン株式会社 | Pharmaceutical composition |
| EP3883549A1 (en) | 2018-11-19 | 2021-09-29 | Jazz Pharmaceuticals Ireland Limited | Alcohol-resistant drug formulations |
| CA3127871A1 (en) | 2019-03-01 | 2020-09-10 | Flamel Ireland Limited | Gamma-hydroxybutyrate compositions having improved pharmacokinetics in the fed state |
| TW202139986A (en) | 2020-02-21 | 2021-11-01 | 愛爾蘭商爵士製藥愛爾蘭有限責任公司 | Methods of treating idiopathic hypersomnia |
| US11779557B1 (en) | 2022-02-07 | 2023-10-10 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11583510B1 (en) | 2022-02-07 | 2023-02-21 | Flamel Ireland Limited | Methods of administering gamma hydroxybutyrate formulations after a high-fat meal |
- 1995
- 1995-12-26JPJP35197295Apatent/JP4072597B2/ennot_activeExpired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JPH09104620A (en) | 1997-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4072597B2 (en) | Sustained formulation | |
| US6156343A (en) | Controlled release preparation | |
| JP5808670B2 (en) | Composition containing weakly basic drug and sustained release dosage form | |
| CA2342340C (en) | New sustained release oral formulations | |
| KR101752014B1 (en) | Orally disintegrating tablet compositions comprising combinations of high and low-dose drugs | |
| US20040052844A1 (en) | Time-controlled, sustained release, pharmaceutical composition containing water-soluble resins | |
| US20130259906A1 (en) | Pharmaceutical composition comprising one or more fumaric acid esters | |
| JP2002527468A (en) | Pulsating dose oral drug delivery system | |
| JPH061716A (en) | Dosage form with long-term release of active ingredient | |
| MX2008014455A (en) | Controlled dose drug delivery system. | |
| JP2001507359A (en) | Sustained release cisapride mini tablet formulation | |
| JP2009504795A (en) | Solid pharmaceutical composition comprising 1- (4-chloroanilino) -4- (4-pyridylmethyl) phthalazine and a pH adjuster | |
| JP4457003B2 (en) | Controlled release pharmaceutical composition | |
| TWI883162B (en) | A febuxostat tablet | |
| JPH09504779A (en) | Drug transfer composition for α-adrenoreceptor blocker | |
| EP3796908B1 (en) | Controlled release propiverine formulations | |
| KR20180024505A (en) | Formulation of fenofibric acid with improved bioavailability | |
| JP2002068964A (en) | Sustained release tablet for oral administration | |
| JP2003267889A (en) | Sustainable pharmaceutical preparation | |
| JP3122478B2 (en) | Lower gastrointestinal release oral formulation | |
| AU2021345210A9 (en) | Multiparticulate dosage forms comprising deutetrabenazine | |
| WO2025215658A1 (en) | Pharmaceutical compositions of ketotifen | |
| CN104997749A (en) | Controlled release preparation, preparation method and application thereof | |
| JP2008115083A (en) | Coated granule containing tramadol hydrochloride | |
| JP2005510449A (en) | Improved controlled release oral dosage form |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal | Free format text:JAPANESE INTERMEDIATE CODE: A131 Effective date:20061128 | |
| A521 | Written amendment | Free format text:JAPANESE INTERMEDIATE CODE: A523 Effective date:20070226 | |
| A131 | Notification of reasons for refusal | Free format text:JAPANESE INTERMEDIATE CODE: A131 Effective date:20070807 | |
| A521 | Written amendment | Free format text:JAPANESE INTERMEDIATE CODE: A523 Effective date:20071105 | |
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) | Free format text:JAPANESE INTERMEDIATE CODE: A01 Effective date:20071204 | |
| A711 | Notification of change in applicant | Free format text:JAPANESE INTERMEDIATE CODE: A711 Effective date:20071207 | |
| A61 | First payment of annual fees (during grant procedure) | Free format text:JAPANESE INTERMEDIATE CODE: A61 Effective date:20071221 | |
| A521 | Written amendment | Free format text:JAPANESE INTERMEDIATE CODE: A821 Effective date:20071207 | |
| FPAY | Renewal fee payment (event date is renewal date of database) | Free format text:PAYMENT UNTIL: 20110201 Year of fee payment:3 | |
| R150 | Certificate of patent or registration of utility model | Free format text:JAPANESE INTERMEDIATE CODE: R150 | |
| LAPS | Cancellation because of no payment of annual fees |