CN115938480A - Long-read sequencing optimizes device and system for error correction method of genome assembly results - Google Patents
Long-read sequencing optimizes device and system for error correction method of genome assembly resultsDownload PDFInfo
- Publication number
- CN115938480A CN115938480ACN202111114295.1ACN202111114295ACN115938480ACN 115938480 ACN115938480 ACN 115938480ACN 202111114295 ACN202111114295 ACN 202111114295ACN 115938480 ACN115938480 ACN 115938480A
- Authority
- CN
- China
- Prior art keywords
- error correction
- long
- sequencing data
- results
- subsets
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images



Landscapes
- Management, Administration, Business Operations System, And Electronic Commerce (AREA)
Abstract
Translated fromChineseDescription
Translated fromChinese技术领域technical field
本发明涉及生物信息领域,具体地,涉及长读长测序对基因组组装结果纠错方法优化装置和系统, 更具体地,涉及纠错长读长组装结果的方法、纠错长读长组装结果的装置、及计算机可读存储介质。The present invention relates to the field of biological information, in particular, to a device and system for optimizing an error correction method for genome assembly results by long-read sequencing, and more specifically, to a method for error-correcting long-read assembly results, and a method for error-correcting long-read assembly results devices, and computer-readable storage media.
背景技术Background technique
随着技术发展,以Pacbio及Nanopore为代表的单分子测序数据拥有超长读长的特点可以解决基因 组复杂区域的拼接问题,因此单分子测序目前已成为基因组组装分析的重要测序技术之一。然而单分子 测序拥有长读长的同时也有高的测序错误率,一些组装软件例如WTDBG2、Miniasm、Flye等可以使用 含有高测序错误的数据进行拼接,所以对于长读长组装结果的纠错尤为重要。另一些软件先对高测序错 误率的数据纠错然后组装,例如Canu软件组装的准确性较高,但是其计算分析成本高,对于高度复杂 基因组的拼接效果不好并不能适用于所有的物种;而falcon等软件可以有效解决高度复杂的区域,但是 其拼接结果准确性较低,组装完成后对于基因组组装结果的纠错也是必不可少的一步。With the development of technology, single-molecule sequencing data represented by Pacbio and Nanopore have the characteristics of ultra-long read length, which can solve the splicing problem of complex regions of the genome. Therefore, single-molecule sequencing has become one of the important sequencing technologies for genome assembly analysis. However, single-molecule sequencing has a long read length and a high sequencing error rate. Some assembly software such as WTDBG2, Miniasm, and Flye can use data with high sequencing errors for splicing, so error correction for long-read assembly results is particularly important. . Other software first corrects the data with a high sequencing error rate and then assembles it. For example, Canu software has higher assembly accuracy, but its calculation and analysis costs are high, and the splicing effect of highly complex genomes is not good and cannot be applied to all species. ; while software such as falcon can effectively solve highly complex regions, but the accuracy of the splicing results is low, and the error correction of the genome assembly results after assembly is also an essential step.
对于组装后的纠错环节,尽管短读长数据相对于长读长数据准确性高,但是长读长数据具有超长的 读长特点可以跨过大的重复序列区域,对基因组复杂区域的矫正更为准确,基因组组装矫正首先使用长 读长数据纠错才能获得高质量高准确度的组装结果。For error correction after assembly, although short-read data is more accurate than long-read data, long-read data has the characteristics of ultra-long read length, which can span large repetitive sequence regions and correct complex regions of the genome To be more accurate, genome assembly correction first uses long-read data for error correction to obtain high-quality and high-accuracy assembly results.
尽管目前长读长的比对软件都使用Minimap2替换所有的比对软件,并且在长读长对基因组的纠错 应用中有非常好的效果。尤其是纠错软件输入文件的格式为paf格式时,比对速度非常很快。但是对于 大基因组,在数据纠错步骤依然需要很大的内存,并且消耗时间很长。如果将大基因组分割,对每个分 割的小块单独比对和纠错,虽然这种方法类似于对小基因组进行比对和纠错,在一定程度上可以降低内 存和缩短分析时间,但是由于真核生物内有大量的重复序列,序列之间存在一定的相似性,这种局部比 对并不能考虑到全基因组的比对信息,从而对整个基因组的纠错结果存在一定的偏向性,导致结果不准 确。因此目前纠错软件仅支持全局比对(即所有数据比对到全基因组上),然后基于全局比对的结果进 行纠错(即所有的数据的比对结果对基因组序列上进行纠错),这样在纠错过程中依然存在内存和时间 消耗的问题。因此,仍需进一步优化纠错方式以降低纠错过程中运行的峰值内存和执行时间。Although the current long-read alignment software uses Minimap2 to replace all alignment software, it has a very good effect in the application of long-read length to genome error correction. Especially when the input file format of the error correction software is paf format, the comparison speed is very fast. But for large genomes, the data error correction step still requires a lot of memory and takes a long time. If the large genome is divided, each divided small block is compared and error-corrected separately. Although this method is similar to the comparison and error-correction of the small genome, it can reduce the memory and shorten the analysis time to a certain extent. However, since there are a large number of repetitive sequences in eukaryotes, there is a certain similarity between the sequences. This kind of local comparison cannot take into account the comparison information of the whole genome, so there is a certain bias in the error correction results of the whole genome. , leading to inaccurate results. Therefore, the current error correction software only supports global alignment (that is, all data are compared to the whole genome), and then error correction is performed based on the results of the global alignment (that is, the comparison results of all data are corrected on the genome sequence) , so there are still memory and time consumption problems in the error correction process. Therefore, it is still necessary to further optimize the error correction method to reduce the peak memory and execution time during the error correction process.
发明内容Contents of the invention
本申请是基于发明人对以下问题的发现和认识作出的:This application is made based on the inventor's discovery and recognition of the following problems:
多项研究表明大基因组的长读长纠错过程中内存飙升,耗时很长;因此,发明人经过大量研究发现, 在长读长数据对基因组纠错过程中,将基因组模块处理、比对结果模块处理和长度长数据模块处理,有 效将一个纠错大任务切分成多个纠错小任务;保留了长读长数据与参考基因组之间的全部关联信息,保 证了纠错结果的准确性;同时,这种切分方式可以多个子任务并行执行,有效降低纠错过程耗费的运行 的峰值内存和执行时间,达到时间效率最大化,实现整个基因组纠错分析成本的降低。A number of studies have shown that the memory soars during the long-read error correction process of large genomes, which takes a long time; therefore, the inventors have found through a lot of research that in the process of long-read data for genome error correction, the genome modules are processed and compared. The result module processing and the long-length data module processing can effectively divide a large error correction task into multiple small error correction tasks; retain all the correlation information between the long-read data and the reference genome, and ensure the accuracy of the error correction results Accuracy; at the same time, this segmentation method can execute multiple subtasks in parallel, effectively reducing the peak memory and execution time consumed by the error correction process, maximizing time efficiency, and reducing the cost of error correction analysis for the entire genome.
在本发明的第一方面,本发明提出了一种用于长读段测序数据纠错的方法。根据本发明的实施例, 包括:(1)对参考序列进行分组,以便获得由所述参考序列的一部分构成的多个参考序列子集;(2) 针对所述多个参考序列子集的每一个,分别进行纠错处理,所述纠错处理是基于下列进行的:(a)所 述参考序列子集中所包含的参考序列;(b)总比对结果中与(a)对应的所述比对结果;(c)所述长 读段测序数据中与(a)对应的部分数据;(d)将步骤(2)中各所述参考序列子集中得到的纠错结果 进行整合,以便获得所述长读段测序数据纠错结果。根据本发明实施例的方法对长读段测序数据进行纠 错,所述方法通过多任务并行进行数据导入、比对、纠错操作,显著降低了数据纠错的时间,提高了测 序数据的纠错效率。In the first aspect of the present invention, the present invention proposes a method for error correction of long-read sequencing data. According to an embodiment of the present invention, it includes: (1) grouping reference sequences so as to obtain a plurality of reference sequence subsets composed of a part of the reference sequences; (2) for each of the plurality of reference sequence subsets One, performing error correction processing respectively, and the error correction processing is based on the following: (a) the reference sequences contained in the reference sequence subset; (b) the total alignment results corresponding to (a) Comparison results; (c) part of the data corresponding to (a) in the long-read sequencing data; (d) integrating the error correction results obtained in each of the reference sequence subsets in step (2), so as to obtain The error correction result of the long-read sequencing data. According to the method of the embodiment of the present invention, the long-read sequencing data is corrected. The method performs data import, comparison, and error correction operations in parallel through multiple tasks, which significantly reduces the time for data error correction and improves the accuracy of the sequencing data. error correction efficiency.
根据本发明的实施例,上述方法还可以进一步包括如下附加技术特征至少之一:According to an embodiment of the present invention, the above method may further include at least one of the following additional technical features:
根据本发明的实施例,对长读段测序数据纠错前,进一步包括以下处理:(3)将所述长读段测序 数据进行分组,以便获得由测序读段构成的多个测序数据子集;(4)将所述多个测序数据子集的每一 个分别与所述参考序列进行比对,以便获得各所述测序数据子集的比对结果;(5)将所述多个测序数 据子集的比对结果合并,以便获得所述多个测序数据子集的总比对结果。According to an embodiment of the present invention, before correcting the long-read sequencing data, the following processing is further included: (3) grouping the long-read sequencing data so as to obtain multiple sequencing data subsets composed of sequencing reads (4) comparing each of the plurality of sequencing data subsets with the reference sequence, so as to obtain the comparison results of each of the sequencing data subsets; (5) comparing the plurality of sequencing data subsets The alignment results of the data subsets are combined to obtain a total alignment result of the plurality of sequencing data subsets.
根据本发明的实施例,所述长测序读段为长度10K以上的测序读段。According to an embodiment of the present invention, the long sequencing reads are sequencing reads with a length of more than 10K.
根据本发明的实施例,所述分组是随机进行的。According to an embodiment of the present invention, the grouping is performed randomly.
根据本发明的实施例,每个所述测序数据子集中所述测序读段的数目不受特别限制。According to an embodiment of the present invention, the number of the sequencing reads in each of the sequencing data subsets is not particularly limited.
根据本发明的实施例,在步骤(3)之前,包括:对所述长读段测序数据进行组装,以便获得初步 组装结果,所述初步组装结果构成步骤(4)中的所述参考序列。According to an embodiment of the present invention, before step (3), it includes: assembling the long-read sequencing data so as to obtain a preliminary assembly result, which constitutes the reference sequence in step (4).
根据本发明的实施例,所述步骤(4)和步骤(2)的至少之一为多任务同时进行。According to an embodiment of the present invention, at least one of the step (4) and the step (2) is performed simultaneously by multiple tasks.
根据本发明的实施例,所述对所述参考序列进行分组是基于下列标准进行的:(1)对所述参考序 列中的每一条序列不进行内部切分;(2)各所述参考序列子集中含有序列的总长度差异不超过20%。According to an embodiment of the present invention, the grouping of the reference sequences is based on the following criteria: (1) each sequence in the reference sequences is not internally segmented; (2) each of the reference sequences The total length of the sequences contained in the subset of sequences differs by no more than 20%.
在本发明的第二方面,本发明提出了一种测序方法。根据本发明的实施例,包括:获取核酸样本; 对所述核酸样本进行长读段测序,以便获得长读段测序数据;对所述测序数据,按照第一方面所述的方 法进行纠错处理,以便获得经过纠错的测序结果。根据本发明实施例的方法对长读段测序数据进行纠错, 所述方法通过多任务并行进行数据导入、比对、纠错操作,显著降低了数据纠错的时间,提高了测序数 据的纠错效率。In the second aspect of the present invention, the present invention provides a sequencing method. According to an embodiment of the present invention, it includes: obtaining a nucleic acid sample; performing long-read sequencing on the nucleic acid sample to obtain long-read sequencing data; performing error correction on the sequencing data according to the method described in the first aspect processing to obtain error-corrected sequencing results. According to the method of the embodiment of the present invention, the long-read sequencing data is corrected. The method performs data import, comparison, and error correction operations in parallel through multiple tasks, which significantly reduces the time for data error correction and improves the accuracy of the sequencing data. error correction efficiency.
根据本发明的实施例,上述方法还可以进一步包括如下附加技术特征至少之一:According to an embodiment of the present invention, the above method may further include at least one of the following additional technical features:
根据本发明的实施例,所述核酸样本来源于未知基因组序列的宿主。According to an embodiment of the present invention, the nucleic acid sample is derived from a host whose genome sequence is unknown.
在本发明的第三方面,本发明提出了一种用于长读段测序数据纠错的装置。根据本发明的实施例, 包括:第一分组模块,用于将所述长读段测序数据进行分组,以便获得由测序读段构成的多个测序数据 子集;比对模块,用于将所述多个测序数据子集的每一个分别与参考序列进行比对,以便获得各所述测 序数据子集的比对结果;比对结果合并模块,用于将所述多个测序数据子集的比对结果合并,以便获得 所述多个测序数据子集的总比对结果;第二分组模块,用于对所述参考序列进行分组,以便好的由所述 参考序列的一部分构成的多个参考序列子集;纠错模块,用于针对所述多个参考序列子集的每一个,分 别进行纠错处理,所述纠错处理是基于下列进行的:(a)所述参考序列子集中所包含的参考序列;(b) 总比对结果中与(a)对应的部分比对结果;(c)所述测序数据中与(a)对应的部分测序数据;纠错 结果整合模块,用于将所述多个参考序列子集中得到的纠错结果进行整合,以便获得所述长读段测序数 据纠错结果。In the third aspect of the present invention, the present invention proposes a device for error correction of long-read sequencing data. According to an embodiment of the present invention, it includes: a first grouping module for grouping the long-read sequencing data so as to obtain multiple sequencing data subsets composed of sequencing reads; a comparison module for grouping the long-read sequencing data Each of the multiple sequencing data subsets is compared with a reference sequence, so as to obtain the comparison results of each of the sequencing data subsets; the comparison result merging module is used to combine the multiple sequencing data subsets The alignment results of the plurality of sequencing data subsets are combined so as to obtain the total alignment result of the plurality of sequencing data subsets; the second grouping module is used to group the reference sequences so that a good multiple composed of a part of the reference sequences reference sequence subsets; an error correction module, configured to perform error correction processing on each of the plurality of reference sequence subsets, and the error correction processing is based on the following: (a) the reference sequence Reference sequences included in the subset; (b) partial alignment results corresponding to (a) in the total alignment results; (c) partial sequencing data corresponding to (a) in the sequencing data; error correction result integration module , used to integrate the error correction results obtained in the plurality of reference sequence subsets, so as to obtain the error correction results of the long-read sequencing data.
在本发明的第四方面,本发明提出了一种用于长读段测序数据纠错的装置。根据本发明的实施例, 所述装置包括存储器和处理器;所述存储器,包括用于存储程序;所述处理器,包括用于通过执行所述 存储器存储的程序以实现第一方面所述的方法。根据本发明实施例的装置,对长读段测序数据进行纠错, 通过多任务并行进行数据导入、比对、纠错操作,可显著提高测序数据的纠错效率,降低时间成本。In the fourth aspect of the present invention, the present invention proposes a device for error correction of long-read sequencing data. According to an embodiment of the present invention, the device includes a memory and a processor; the memory is used to store programs; the processor is used to implement the program described in the first aspect by executing the program stored in the memory method. According to the device of the embodiment of the present invention, error correction is performed on long-read sequencing data, and data import, comparison, and error correction operations are performed in parallel through multiple tasks, which can significantly improve the error correction efficiency of sequencing data and reduce time costs.
在本发明的第五方面,本发明提出了一种计算机可读存储介质的装置。根据本发明的实施例,所述 存储介质中存储有程序,所述程序能够被处理器执行以实现第一方面所述的方法。In a fifth aspect of the present invention, the present invention provides an apparatus of a computer-readable storage medium. According to an embodiment of the present invention, a program is stored in the storage medium, and the program can be executed by a processor to implement the method described in the first aspect.
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本 发明的实践了解到。Additional aspects and advantages of the invention will be set forth in the description which follows, and in part will be obvious from the description, or may be learned by practice of the invention.
附图说明Description of drawings
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其 中:The above-mentioned and/or additional aspects and advantages of the present invention will become apparent and easily understood from the description of the embodiments in conjunction with the following drawings, wherein:
图1是根据本发明实施例的长读长纠错流程,其中,FIG. 1 is a long-read error correction process according to an embodiment of the present invention, wherein,
步骤101表示组装结果文件和长读长数据文件的准备,
步骤102表示将长读长数据比对到组装结果文件上,
步骤103表示根据比对结果的信息,基于长读长数据对基因组组装结果进行矫正,
步骤104表示纠错矫正后生成的最终的纠错结果;
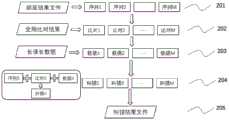
图2是根据本发明实施例的拆分比对、拆分纠错方法的流程图,其中:Fig. 2 is a flow chart of a split comparison and split error correction method according to an embodiment of the present invention, wherein:
步骤201表示基因组准备及切块,此步骤将基因组序列切分为M个模块,
步骤202表示比对文件准备及切块,此步骤最终获得与M个子参考序列对应的M个比对结果,
步骤203表示长读长数据切块,此步骤最终获得与M个比对结果对应的M个长读长数据,
步骤204表示对于M个子块纠错,最终获得M个纠错结果,Step 204 represents error correction for M sub-blocks, and finally M error correction results are obtained,
步骤205表示纠错结果合并,得到最终的纠错结果问题;以及,Step 205 represents merging the error correction results to obtain the final error correction result question; and,
图3是根据本发明实施例的获得比对结果的流程图,其中:Fig. 3 is a flowchart of obtaining comparison results according to an embodiment of the present invention, wherein:
步骤301表示将长读长数据切分获得N个数据切块,Step 301 represents segmenting the long-read data to obtain N data segments,
步骤302表示将N个子块数据比对到参考基因组,获得N个比对结果;Step 302 represents comparing N sub-block data to the reference genome to obtain N comparison results;
步骤303表示将N个比对结果合并成一个最终的长读长比对结果;以及Step 303 represents merging the N alignment results into one final long-read alignment result; and
图4是根据本发明实施例的长读段测序数据纠错的装置,包括:第一分组模块、比对模块、合并比 对模块(比对结果合并模块)、第二分组模块、纠错模块、纠错结果整合模块。Fig. 4 is a device for error correction of long-read sequencing data according to an embodiment of the present invention, including: a first grouping module, a comparison module, a merge comparison module (combination module for comparison results), a second grouping module, and an error correction module , Error correction result integration module.
具体实施方式Detailed ways
下面详细描述本发明的实施例,所述实施例的示例在附图中示出。下面通过参考附图描述的实施例 是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。Embodiments of the invention are described in detail below, examples of which are illustrated in the accompanying drawings. The embodiments described below by referring to the figures are exemplary and are intended to explain the present invention, but should not be construed as limiting the present invention.
此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所 指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。 在本发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。In addition, the terms "first" and "second" are used for descriptive purposes only, and cannot be interpreted as indicating or implying relative importance or implicitly specifying the quantity of the indicated technical features. Thus, the features defined as "first" and "second" may explicitly or implicitly include at least one of these features. In the description of the present invention, "plurality" means at least two, such as two, three, etc., unless otherwise specifically defined.
下面参考附图描述本公开实施例的长读长测序对基因组组装结果纠错方法优化装置和系统。在具体 描述本发明实施例之前,为了便于理解,首先对长读长测序对基因组组装结果纠错方法进行介绍:The device and system for optimizing the error correction method for genome assembly results by long-read sequencing according to the embodiments of the present disclosure will be described below with reference to the accompanying drawings. Before describing the embodiment of the present invention in detail, in order to facilitate understanding, firstly, the error correction method for genome assembly results by long-read sequencing is introduced:
1.参考基因组的准备及切块1. Preparation and dicing of the reference genome
基于长读长组装软件对长读长数据进行组装,得到含错误率的长读长组装结果,即如步骤201所示 组装结果文件。The long-read data is assembled based on the long-read assembly software, and a long-read assembly result including an error rate is obtained, which is an assembly result file as shown in
组装结果文件的切分过程:首先统计参考基因组的总大小及最长序列的长度,并确定子块大小。如 果切分块的长度差异较大,会影响后续分析内存的设定和时间效率。为保证切分子块大小的一致性,本 方法建议至少保证设定的子块长度大于最长的参考基因组序列。确定子块长度后,读取组装结果文件, 对长度进行累加,在超过指定子块大小的文件时,生成第一个子块,再用同样的方法构建下一个子块, 如步骤201所示,最终构建M个子块。本方法基于序列名称及其对应的序列长度来切分,并不会打断 序列。The segmentation process of the assembly result file: first count the total size of the reference genome and the length of the longest sequence, and determine the sub-block size. If there is a large difference in the length of the segmented blocks, it will affect the memory setting and time efficiency of subsequent analysis. In order to ensure the consistency of the sub-block size, this method recommends at least ensuring that the set sub-block length is greater than the longest reference genome sequence. After determining the length of the sub-block, read the assembly result file, accumulate the length, and generate the first sub-block when the file exceeds the specified sub-block size, and then use the same method to construct the next sub-block, as shown in
2.比对文件准备及切块2. Comparison file preparation and cutting
该过程包括以下几个步骤:The process consists of the following steps:
(1)数据比对(1) Data comparison
利用Minimap2比对软件将测序的长读长数据比对到参考基因组上。本方法支持全局比对结果和切 块比对结果,如附图3所示,切块比对结果是指在参考基因组不做切分的前提下,对长读长数据经过切 分、比对、排序、合并后得到的比对结果。长读长的数据格式有两种:bam格式(Pacbio的原始下机数 据文件)和Fastq格式(Pacbio原始下机数据文件转换后获得或者Nanopore原始下机数据文件),此处 示出两种不同格式数据的切分方法。The sequenced long-read data was compared to the reference genome using the Minimap2 comparison software. This method supports global comparison results and block comparison results. As shown in Figure 3, the block comparison results refer to long-read data after segmentation and comparison on the premise that the reference genome is not segmented. , sorting, and merging the comparison results. There are two long-read data formats: bam format (Pacbio’s original off-machine data file) and Fastq format (Pacbio’s original off-machine data file converted or Nanopore’s original off-machine data file), two of which are shown here Segmentation methods for data in different formats.
bam格式文件的切分:Segmentation of bam format files:
Pacbio测序是在一张含有数百万零模波导孔(简称ZMW孔,是一种用于将光线限制在小的观测体 积的纳米光学设备,是一种具有导电层的小孔)的芯片上完成的,测序时DNA模板通过ZMW孔底部 时与固定的DNA聚合酶发生聚合反应合成DNA,通过检测合成时激发的荧光信号来完成实时测序。由 于Pacbio的文库时DNA的双链两侧加上测序接头形成哑铃状,在测序时DNA双链打开,在同一个ZMW 孔内循环测序,下机数据去掉序列接头序列并标记有ZMW孔的信息,因此相同ZMW孔的测序reads 来自同一条DNA片段。如果将相同DNA片段序列放在不同的子文件,则会导致比对的冗余和计算量、 存储量的加大。因此在对Pacbiobam文件切分时需要将相同ZMW孔的序列放在同一个子文件中。这一 过程通过Pacbio的官方下载地址(https://www.pacb.com/support/software-downloads/)下SMRT Link安 装包中的软件程序bamsieve实现。首先基于软件bamsieve的参数“--show-zmws”获得Pacbio下机数据包 含芯片板的ZMW的列表,并获得ZMW的总数目M,通过设定拆分后子文件的数目N(例如设定100, 则对ZMW列表信息拆分100份),确定ZMW每个子文件中ZMW的数目,生成子ZMW列表,再根 据bamsieve参数“—whitelist”抽取与每个子ZMW列表对应的长读长的reads信息,以此获得N个bam 格式的子文件。Pacbio sequencing is performed on a chip containing millions of zero-mode waveguide holes (ZMW holes for short, a nano-optical device used to confine light to a small observation volume, a small hole with a conductive layer) When sequencing, the DNA template passes through the bottom of the ZMW hole and polymerizes with the immobilized DNA polymerase to synthesize DNA, and the real-time sequencing is completed by detecting the fluorescent signal excited during the synthesis. In Pacbio’s library, the double strands of the DNA are added with sequencing adapters to form a dumbbell shape. During sequencing, the DNA double strands are opened, and the sequence is cycled in the same ZMW hole. The off-machine data removes the sequence adapter sequence and marks the ZMW hole. information, so the sequencing reads of the same ZMW well come from the same DNA fragment. If the same DNA fragment sequence is placed in different sub-files, it will lead to the redundancy of the comparison and the increase of the amount of calculation and storage. Therefore, when dividing the Pacbiobam file, it is necessary to put the sequence of the same ZMW hole in the same sub-file. This process is realized through the software program bamsieve in the SMRT Link installation package under Pacbio's official download address (https://www.pacb.com/support/software-downloads/). First, based on the parameter "--show-zmws" of the software bamsieve, the Pacbio off-machine data contains a list of ZMWs of the chip board, and the total number M of ZMWs is obtained. By setting the number N of sub-files after splitting (for example, setting 100, split the ZMW list information into 100 copies), determine the number of ZMWs in each sub-file of the ZMW, generate the sub-ZMW list, and then extract the long reads corresponding to each sub-ZMW list according to the bamsieve parameter "-whitelist" information to obtain N subfiles in bam format.
Fastq文件的切分:Segmentation of Fastq files:
Fastq文件的切分只需计算Fastq格式文件的总reads条数S,并设定子文件数目N(例如设定为100), 确定每个子文件中reads的数目,先对reads拆分,再基于reads的序列名称列表将长读长序列切分为N 个子文件。The segmentation of Fastq files only needs to calculate the total number of reads S of the Fastq format file, and set the number of sub-files N (for example, set to 100), determine the number of reads in each sub-file, first split the reads, and then based on The sequence name list of reads divides the long read sequence into N subfiles.
如附图3中步骤301所示,数据切分后,获得N个数据切块。将各个子块数据比对到参考基因组, 获得N个比对结果,如附图步骤302所示,本方法建议的比对软件为Minimap2,然后再合并成最终的 比对结果,即附图步骤303。由于比对结果有两种格式paf和bam格式,对于paf由于是表格形式,因 此直接将所有文件合并。而对于bam格式,则使用samtoolssort先对每个子文件比对结果排序后再用 samtools merge合并成最终的bam格式的比对结果,其中samtools的下载地址 (https://github.com/samtools/samtools)。如果使用Pacbio官方纠错软件GCPP,则可以使用smartlink 的软件包pbmm2比对(包含了minimap2的比对和排序),“dataset create--type AlignmentSet”来实现比 对结果的合并。As shown in
(2)比对文件的拆分(2) Splitting of comparison files
(1)的比对结果即为附图2中202所示全局比对结果,附图2中202步骤示出基于每个序列文件 序列X及全局比对结果,获得比对结果X文件,其中X=1,2~M。The comparison result of (1) is the global comparison result shown in 202 in the accompanying drawing 2, and the 202 step shows in the accompanying drawing 2 based on each sequence file sequence X and the global comparison result, obtains the comparison result X file, wherein X=1, 2~M.
若全局比对文件为paf格式,则基于模式匹配的方式,获取与参考基因组子块对应的的子比对文件; 若比对文件为bam格式,则根据序列子块文件生成序列的bed文件,bed文件起始位置、终止位置为参 考序列中每条序列的起始位置和终止位置,再利用samtoolsview来实现bam文件的拆分。如果使用 Pacbio官方纠错软件GCPP,则可使用smartlink的软件包“dataset split–contigs”拆分。If the global comparison file is in the paf format, the sub-comparison file corresponding to the reference genome sub-block is obtained based on pattern matching; if the comparison file is in the bam format, the bed file of the sequence is generated according to the sequence sub-block file, The start position and end position of the bed file are the start position and end position of each sequence in the reference sequence, and then use samtoolsview to realize the splitting of the bam file. If you use Pacbio's official error correction software GCPP, you can use smartlink's software package "dataset split–contigs" to split.
此外,比对文件进行切分处理时,并不是直接使用子块的比对结果,须对子块的比对结果先合并, 再基于参考基因组拆分的子块抽取对应的比对结果,所述参考序列子集的比对结果,包含了所有测序长 读长数据针对该子集序列的所有比对结果,因为在纠错时需要考虑参考基因组每个位置的最优比对信 息,切块的比对结果中每一块的最优并不能反应全部数据在全基因组水平的最优,只有将整个子块比对 文件合并后,确定参考基因组每个位置上所有长读长序列的比对信息,才能获取参考基因组每个位置的 最优比对。In addition, when the comparison file is split, the comparison results of the sub-blocks are not directly used. The comparison results of the sub-blocks must be merged first, and then the corresponding comparison results are extracted based on the sub-blocks split by the reference genome. The alignment results of the subset of reference sequences mentioned above include all the alignment results of all sequenced long-read data for this subset of sequences, because the optimal alignment information for each position of the reference genome needs to be considered when correcting errors. The optimality of each block in the block comparison results does not reflect the optimality of all data at the genome-wide level. Only after merging the entire sub-block alignment files, the alignment of all long-read sequences at each position in the reference genome can be determined. In order to obtain the optimal alignment of each position in the reference genome.
Minimap2在生成paf格式文件时,所消耗的时间和内存资源均很小,在CPU资源比较充沛的情况 下,可以考虑该比对模式不进行切分处理。Minimap2 consumes very little time and memory resources when generating paf format files. In the case of relatively abundant CPU resources, it can be considered that this comparison mode does not perform segmentation processing.
3.长读长数据切块3. Long-read data dicing
bam文件本身带有原始长读长序列信息,在对比对文件拆分时,已经完成对于长读长序列信息的拆 分;而paf比对格式文件,由于paf文件并未包含碱基序列信息,因此在纠错时需要读取长读长数据的 信息,需要对每个子块匹配到与之比对结果相对应的长读长信息。The bam file itself contains the original long-read sequence information, and the long-read sequence information has been split when the alignment file is split; while the paf alignment format file, since the paf file does not contain base sequence information, Therefore, it is necessary to read the information of the long-read data during error correction, and it is necessary to match the long-read information corresponding to the comparison result for each sub-block.
如附图2中203步骤所示,将长读长数据基于paf格式的比对结果进行切块,获取每个比对子文件 中包含的长读长数据子集。具体做法是对步骤202中的每个paf格式的比对文件子块中获取对应的长读 长序列名称列表,长读长序列名称列表根据模式匹配的方法从原始长读长测序数据文件中抽取长读长数 据,即步骤203中的数据子块:数据X,其中X=1,2~M,此步骤长读长数据格式为Fasta和Fastq格 式。As shown in
4.参考序列子块纠错4. Reference sequence sub-block error correction
如附图2中204所示,序列子块X、比对子块X、长读长数据子块X基于纠错软件生成与之对应的 纠错子块X,其中X=1,2~M。这一步的实现依赖于参考基因组的子块纠错过程是独立完成的。常用 的长读长数据纠错软件Arrow、RACON软件的纠错方法中,每一条序列的纠错是基于与之对应的比对 结果来获取一致性序列,其纠错都是相对独立的,所以此方案是可行的。这种方法将大的比对信息和大 的长读长数据转化为上百个小的比对块和上百个小的长读长数据,大大降低峰值内存需求,并缩短分析 时间。As shown in 204 in accompanying drawing 2, the sequence sub-block X, the comparison sub-block X, and the long-read data sub-block X generate the corresponding error correction sub-block X based on the error correction software, wherein X=1, 2~M . The realization of this step relies on the fact that the sub-block error correction process of the reference genome is completed independently. In the error correction methods of commonly used long-read data error correction software Arrow and RACON software, the error correction of each sequence is based on the corresponding alignment results to obtain consistent sequences, and the error correction is relatively independent, so This solution is feasible. This method converts large alignment information and large long-read data into hundreds of small comparison blocks and hundreds of small long-read data, greatly reducing peak memory requirements and shortening analysis time.
5.纠错结果合并5. Combination of error correction results
最终结果如附图2中205所示,将M个纠错子块的纠错结果合并,获得最终的一个一致性序列文 件,该文件即为长读长数据对参考基因组的纠错结果。The final result is shown in 205 in Figure 2. The error correction results of the M error correction sub-blocks are combined to obtain a final consensus sequence file, which is the error correction result of the long-read data for the reference genome.
本发明将大的数据量切分成很多小数据量,但是同时保留了长读长数据与整个基因组序列的全部关 联,而基因组的纠错过程是逐条序列进行纠错,因此有效保证了基因组纠错结果的准确性。该切分方式 在前期处理时只消耗很小的资源,但是在纠错时可以实现单个子任务的运行时间和峰值内存的降低,还 可以多个子任务同时并行,有效缩短了运行时间,从而实现整个基因组纠错分析的整体成本。整个长读 长纠错优化策略,尤其适用于有很多小机器组成的集群的使用,其分析效率非常高。The present invention divides a large amount of data into many small amounts of data, but at the same time retains all the correlations between long-read data and the entire genome sequence, and the error correction process of the genome is to correct errors one by one, thus effectively ensuring genome correction. accuracy of the results. This segmentation method consumes only a small amount of resources during pre-processing, but it can reduce the running time and peak memory of a single subtask during error correction. It can also parallelize multiple subtasks at the same time, effectively shortening the running time, thus realizing The overall cost of error correction analysis across the genome. The entire long-read error correction optimization strategy is especially suitable for the use of clusters composed of many small machines, and its analysis efficiency is very high.
作为一种示例,参见图4,图4为本发明实施例中的能够实施上述长读段测序数据纠错装置,根据 本发明的实施例,该装置包括:第一分组模块,用于将所述长读段测序数据进行分组,以便获得由测序 读段构成的多个测序数据子集;比对模块,用于将所述多个测序数据子集的每一个分别与参考序列进行 比对,以便获得各所述测序数据子集的比对结果;第二分组模块,用于对所述参考序列进行分组,以便 好的由所述参考序列的一部分构成的多个参考序列子集;纠错模块,用于针对所述多个参考序列子集的 每一个,分别进行纠错处理,所述纠错处理是基于下列进行的:(a)所述参考序列子集中所包含的参考 序列;(b)总比对结果中与(a)对应的部分比对结果;(c)所述测序数据中与(a)对应的部分测序数据; 纠错结果整合模块,用于将所述多个参考序列子集中得到的纠错结果进行整合,以便获得所述长读段测 序数据纠错结果。As an example, refer to Fig. 4. Fig. 4 is an error correction device capable of implementing the above-mentioned long-read sequencing data in an embodiment of the present invention. According to an embodiment of the present invention, the device includes: a first grouping module for grouping all The long-read sequencing data is grouped so as to obtain multiple sequencing data subsets composed of sequencing reads; the comparison module is used to compare each of the multiple sequencing data subsets with a reference sequence, In order to obtain the comparison results of each of the sequencing data subsets; the second grouping module is used to group the reference sequences so as to obtain a plurality of reference sequence subsets composed of a part of the reference sequence; error correction A module, configured to perform error correction processing on each of the plurality of reference sequence subsets, the error correction processing is performed based on the following: (a) the reference sequence contained in the reference sequence subset; ( b) part of the comparison results corresponding to (a) in the total comparison results; (c) part of the sequencing data corresponding to (a) in the sequencing data; an error correction result integration module, which is used to combine the multiple references The error correction results obtained in the sequence subsets are integrated to obtain the error correction results of the long-read sequencing data.
利用该系统能够有效地对前面所述的长读段、参考基因组和比对结果进行分组处理,保留了长读段 与参考基因组之间的全部关联信息,因此,在保证了纠错结果的准确性的同时,有效的将一个纠错大任 务切分成多个纠错子任务,多个子任务并行执行,有效降低纠错过程耗费的运行的峰值内存和执行时间, 达到时间效率最大化,实现整个基因组纠错分析成本的降低。The system can effectively group the long reads, reference genome and comparison results mentioned above, and retain all the association information between the long reads and the reference genome, thus ensuring the accuracy of error correction results At the same time, it effectively divides a large error correction task into multiple error correction subtasks, and multiple subtasks are executed in parallel, effectively reducing the peak memory and execution time consumed by the error correction process, maximizing time efficiency, and realizing the entire Reduced cost of genome error correction analysis.
本发明还提供了一种计算机设备,包括存储器和处理器;所述存储器,包括用于存储程序;所述处 理器,包括用于通过执行所述存储器存储的程序以实现前面所述的长读段数据纠错的方法。The present invention also provides a computer device, including a memory and a processor; the memory includes a program for storing a program; the processor includes a program for executing the program stored in the memory to realize the aforementioned long Method for error correction of read segment data.
另外,本发明还提供了一种计算机可读存储介质,所述存储介质中存储有程序,所述程序能够被处 理器执行以实现前面所述的长读段数据纠错的方法。In addition, the present invention also provides a computer-readable storage medium, in which a program is stored, and the program can be executed by a processor to implement the above-mentioned method for correcting errors in long-read segment data.
下面参考具体实施例,对本发明进行描述,需要说明的是,这些实施例仅仅是描述性的,而不以任 何方式限制本发明。The present invention is described below with reference to specific embodiments, and it should be noted that these embodiments are only descriptive, and do not limit the present invention in any way.
实施例1将长读长初步组装结果进行模块比对、模块纠错Example 1 Perform module comparison and module error correction on the preliminary assembly results of long reads
本实施例中,所使用的某动物基因组大小约3G,用Nanopore Promethion测序深度约50X,总 数据量约150G的数据,使用RACON软件对其总长度约2.87G的初级组装结果纠错,其中,下文 所述参考基因组即为长读长初步组装结果。In this example, the genome size of a certain animal used is about 3G, the sequencing depth is about 50X with Nanopore Promethion, and the total data volume is about 150G, and the primary assembly result with a total length of about 2.87G is corrected using RACON software. Among them, The reference genome described below is the preliminary long-read assembly result.
1、比对文件准备1. Preparation of comparison files
数据比对:将50X的长读长的Fastq格式数据按500M数据量一份进行切块,共切分成299个 子块。将每个数据子块比对到2.87G参考基因组上进行全局比对,生成paf格式的比对子块结果。 待所有的子任务均比对完成,将比对得到的299个paf格式的子比对文件,经过合并最终得到一个 paf格式的总比对文件,具体流程如图3所示。Data comparison: The 50X long-read Fastq format data is divided into 500M pieces of data, and divided into 299 sub-blocks in total. Each data sub-block is compared to the 2.87G reference genome for global comparison, and the comparison sub-block result in paf format is generated. After all the sub-tasks are compared, the 299 sub-comparison files in paf format will be merged to obtain a total comparison file in paf format. The specific process is shown in Figure 3.
2、参考基因组的准备及切块2. Preparation and dicing of the reference genome
统计参考基因组的总大小为2.87G,序列格式为Fasta,组装序列最大长度为36M,子文件数 据量大小设置为50M,最终获得切分后子文件,其数目是60。The total size of the statistical reference genome is 2.87G, the sequence format is Fasta, the maximum length of the assembled sequence is 36M, the data size of the sub-file is set to 50M, and the number of sub-files obtained after segmentation is 60.
3、总比对文件的拆分。3. Compare the splitting of files.
步骤1中获得的paf格式的比对结果中,第六列是参考基因组的序列名称,因此依据步骤2 中每个参考基因组子块的参考序列名称,提取paf格式的总比对结果文件中与之对应的比对信息。 即,最终生成60个比对子文件,其中每个比对子文件的第六列包含了它所对应的参考基因组子块 的所有序列名称。In the comparison result in paf format obtained in step 1, the sixth column is the sequence name of the reference genome, so according to the reference sequence name of each reference genome sub-block in step 2, extract the total comparison result file in paf format with The corresponding comparison information. That is, 60 comparison subfiles are finally generated, and the sixth column of each comparison subfile contains all sequence names of its corresponding reference genome subblock.
4、长读长数据切块4. Long read and long data dicing
步骤1中获得的paf格式的比对结果,第一列反映参与比对的测序数据序列名称。因此对步骤 3中获得的60个子比对文件,每一个做如下处理:根据每个paf文件第一列所提供的长读长序列 名称信息,从全部的长读长数据文件中抽取对应的序列名称,并提取序列生成新的长读长数据子 文件。最终得到60个格式与长读长数据文件格式一致的长读长数据子文件。此处应注意,从长读 长数据中抽取的每个长读长数据子块的序列数目应与比对文件中使用的序列数目一致。最终参考 序列子块、比对子块与长读长数据子块有一一对应关系,此实施例中均为60个。For the alignment result in paf format obtained in step 1, the first column reflects the sequence name of the sequencing data involved in the alignment. Therefore, each of the 60 sub-comparison files obtained in step 3 is processed as follows: according to the long-read sequence name information provided in the first column of each paf file, extract the corresponding sequence from all long-read data files name, and extract the sequence to generate a new long-read data subfile. Finally, 60 long-read data subfiles with the same format as the long-read data file were obtained. It should be noted here that the sequence number of each long-read data sub-block extracted from the long-read data should be consistent with the sequence number used in the alignment file. Finally, there is a one-to-one correspondence between the reference sequence sub-block, the comparison sub-block and the long-read data sub-block, all of which are 60 in this embodiment.
5、参考基因组子块数据纠错5. Reference genome sub-block data error correction
利用Racon v1.3.3软件,以具有一一对应关系的参考基因组子块、比对子块、长读长数据子块 作为输入,对参考基因组子块数据进行纠错,得到60个格式为Fasta的纠错子文件。Using Racon v1.3.3 software, taking reference genome sub-blocks, alignment sub-blocks, and long-read data sub-blocks with one-to-one correspondence as input, correcting the reference genome sub-block data, and obtaining 60 Fasta format Correct subfiles.
6、纠错结果合并6. Combination of error correction results
将每个纠错子文件结果合并,获得Fasta格式的最终一致性序列,即长读长数据对参考基因组 的最终纠错结果。Merge the results of each error correction subfile to obtain the final consensus sequence in Fasta format, that is, the final error correction result of the long-read data for the reference genome.
7、时间和资源的统计7. Time and resource statistics
具体时间和资源统计的结果如表1所示,在比对步骤,长读长数据拆分使用线程是5,拆分时 间为1.67h,峰值内存0.2G,总CPU时为8.33;数据比对将大的比对任务拆分成299个子比对任 务,CPU设置为5,所有比对任务的峰值内存为17G,最大运行时间为0.12h,总CPU时为96.1; 比对结果合并CPU时为0.05。因此本实施例的纠错步骤总CPU时为104.48。The results of specific time and resource statistics are shown in Table 1. In the comparison step, the number of threads used for long-read long data splitting is 5, the splitting time is 1.67h, the peak memory is 0.2G, and the total CPU time is 8.33; the data ratio Split the large comparison task into 299 sub-comparison tasks, set the CPU to 5, the peak memory of all comparison tasks is 17G, the maximum running time is 0.12h, and the total CPU time is 96.1; the comparison results are combined with CPU is 0.05. Therefore, the total CPU time of the error correction step in this embodiment is 104.48.
在纠错步骤,基因组拆分CPU时为0.1;比对结果拆分CPU时为3;对测序长读长数据拆分, 每个子任务使用线程为5,峰值内存为0.3G,60个任务的运行时间峰值为1.28h,总CPU时为243; RACON纠错线程设置为5,峰值内存为29G,峰值运行时间为4.71h,消耗总CPU时为302.56; 纠错结果合并总CPU时为0.08。因此,本实施例整个纠错步骤的CPU时为548.74。In the error correction step, when the genome splits the CPU, it is 0.1; when the comparison result splits the CPU, it is 3; for the sequencing long-read data split, each subtask uses 5 threads, the peak memory is 0.3G, and 60 tasks The peak running time is 1.28h, and the total CPU time is 243; the RACON error correction thread is set to 5, the peak memory is 29G, the peak running time is 4.71h, and the total CPU consumption is 302.56; the error correction results combined with the total CPU time is 0.08. Therefore, the CPU time of the entire error correction step in this embodiment is 548.74.
经过拆分后,使用5CPU 32G的机器即可实现8h左右完成长读长数据对基因组的RACON纠 错,整个分析过程总CPU时为653.22。After splitting, using a 5CPU 32G machine can complete the RACON error correction of the long-read data to the genome in about 8 hours, and the total CPU time for the entire analysis process is 653.22.
表1:Table 1:


实施例2将长读长初步组装结果进行全局比对,模块纠错Example 2 Global comparison of long-read preliminary assembly results, module error correction
本实施例所使用的某动物基因组大小约3G,用Nanopore Promethion测序深度约50X,总数据 量约150G的数据,使用RACON软件对其总长度约2.87G的初级组装结果纠错。The genome size of an animal used in this example is about 3G, the sequencing depth is about 50X with Nanopore Promethion, and the total data volume is about 150G, and the primary assembly result with a total length of about 2.87G is corrected using RACON software.
1、全局比对1. Global comparison
将数据量约150G的Fastq格式数据比对到2.87G的初级组装结果,生成1个paf格式的比对结果, 比对软件使用minimap2。Compare the data in Fastq format with a data volume of about 150G to the primary assembly result of 2.87G, and generate a comparison result in paf format. The comparison software uses minimap2.
2、分块纠错2. Block error correction
统计参考基因组的总大小为2.87G,组装序列最大长度为36M,切分的子块大小设置为50M,基于 参考基因组的序列长度及序列名称进行切块,最终切分后基因组子文件数目是60。The total size of the statistical reference genome is 2.87G, the maximum length of the assembled sequence is 36M, the size of the segmented sub-block is set to 50M, and the segment is performed based on the sequence length and sequence name of the reference genome, and the number of genome sub-files after the final segmentation is 60 .
基于参考基因组切分后子文件中对应的参考序列名称,与(1)获得的paf格式的比对文件的第六列 名称信息进行匹配,提取与每个参考基因组子块对应的比对结果,生成与参考基因组子块完全对应的子 比对文件列表,共60个子比对结果文件。根据每一个子比对文件第一列所提供的长读长序列名称信息, 从全部的长读长数据文件中抽取对应的序列信息,生成60个与参考序列子块、比对子块对应的长读长 数据子文件。Based on the corresponding reference sequence name in the sub-file after the reference genome segmentation, match with the sixth column name information of the comparison file in paf format obtained in (1), and extract the comparison result corresponding to each reference genome sub-block, Generate a sub-comparison file list that completely corresponds to the reference genome sub-block, a total of 60 sub-comparison result files. According to the long-read sequence name information provided in the first column of each sub-comparison file, extract the corresponding sequence information from all long-read data files, and generate 60 sub-blocks corresponding to the reference sequence and comparison sub-blocks Long read long data subfile.
利用Racon v1.3.3软件,基于每个参考基因组子文件及其对应的比对子文件和长读长数据子文件对 参考基因组子文件进行纠错,得到每个参考基因组子文件的纠错结果。Using Racon v1.3.3 software, based on each reference genome subfile and its corresponding comparison subfile and long-read data subfile, the reference genome subfile was corrected to obtain the error correction result of each reference genome subfile.
合并纠错结果获得最终的纠错结果。The error correction results are combined to obtain the final error correction result.
3、时间和资源统计3. Time and resource statistics
时间和资源统计的结果如表2所示,在比对环节,该方法采取全部长读长数据比对到全基因组的策 略,使用线程为20,峰值内存为20.92G,运行时间为3.45h,比对消耗总的CPU时为69.05。The results of time and resource statistics are shown in Table 2. In the comparison process, this method adopts the strategy of comparing all long-read data to the whole genome, using 20 threads, peak memory of 20.92G, and running time of 3.45h , compared to the total CPU consumption is 69.05.
实施例2与实施例1差异仅在比对环节,但两种方法得到的比对结果完全一致,因此对于纠错环节, 具体操作及资源利用时间也一致,此处不再做详细介绍。该步骤总CPU时为548.74。The difference between Embodiment 2 and Embodiment 1 is only in the comparison link, but the comparison results obtained by the two methods are completely consistent, so for the error correction link, the specific operation and resource utilization time are also consistent, and will not be described in detail here. The total CPU hours for this step was 548.74.
本实施例整体分析所用时间为9.67h,峰值内存29G,峰值线程20,总CPU时为614.43。The overall analysis time of this embodiment is 9.67h, the peak memory is 29G, the peak thread is 20, and the total CPU time is 614.43.
表2:Table 2:

实施例3将长读长初步组装结果进行全局比对和全局纠错Example 3 Global comparison and global error correction of the preliminary assembly results of long reads
本实施例所使用的某动物基因组大小约3G,用Nanopore Promethion测序深度约50X,总数据 量约150G的数据,使用RACON软件对其总长度约2.87G的初级组装结果纠错。The genome size of an animal used in this example is about 3G, the sequencing depth is about 50X with Nanopore Promethion, and the total data volume is about 150G, and the primary assembly result with a total length of about 2.87G is corrected using RACON software.
将数据量约150G的Fastq格式数据比对到2.87G的初级组装结果,生成paf格式的比对结果,比对 软件使用minimap2;用比对结果、原始Fastq格式的测序长读长数据、初级组装结果作为输入,使用 Racon v1.3.3进行长读长数据对初级组装结果的纠错。Compare the Fastq format data with a data volume of about 150G to the primary assembly result of 2.87G, and generate the comparison result in paf format. The comparison software uses minimap2; use the comparison result, the sequencing long read data in the original Fastq format, and primary assembly The results are used as input, and Racon v1.3.3 is used for error correction of long-read data to primary assembly results.
时间和资源的使用结果如表3所述,本实施例中比对步骤的线程数设置为32,峰值内存21.16G, 总运行时间为2.09h,比对总的CPU时为66.98。纠错过程的线程设置为60,峰值内存为444.15G,运 行时间为89.68h,纠错步骤的CPU时为5380.18。因此整个纠错过程所用总时间为91.77h,峰值内存445G, 峰值线程数60,总CPU时为5646.95。The usage results of time and resources are as described in Table 3. In this embodiment, the number of threads in the comparison step is set to 32, the peak memory is 21.16G, the total running time is 2.09h, and the total CPU time is 66.98 when compared. The thread setting of the error correction process is 60, the peak memory is 444.15G, the running time is 89.68h, and the CPU time of the error correction step is 5380.18. Therefore, the total time used for the entire error correction process is 91.77h, the peak memory is 445G, the peak number of threads is 60, and the total CPU time is 5646.95.
表3:table 3:
实施例4纠错方法对比Embodiment 4 Error Correction Method Comparison
本实施例将实施例1~3所述方法进行比较,具体比较过程及结果如下所示:In this embodiment, the methods described in Examples 1 to 3 are compared, and the specific comparison process and results are as follows:
1、比对过程的比较1. Comparison of comparison process
表4中展示了实施例1(将长读段测序数据拆分后各长读段数据子集与参考基因组比对再合并), 实施例2(直接比对,线程设置20),实施例3(直接比对,线程设置32)所示的三种方法的比对过程 资源消耗情况。其中时间代表峰值运行时间;实际线程为分析时设置的线程数;峰值内存是指运行过程 中最大的内存使用;实际CPU时为经过核算整个比对过程的总CPU时,其中实施例1包括数据拆分和 比对结果合并的CPU时使用。比对条目指比对后的比对信息的行数,比对文件大小是最终生成的比对 文件大小。由表4可以看出,三种方法得到的比对文件大小和比对结果一致,将比对文件排序后,用diff 命令查看,三个结果完全一致。而从时间使用上,线程设置越高,总CPU时越少。在线程资源有限的 情况下,建议使用实施例1所述方法,即对长读段测序数据拆分后获得的多个长读段测序数据子集与参考基因组进行比对,获得各所述测序数据子集的比对结果,该方法可以多任务并行,提升分析效率,峰 值内存也略有降低,总的运行时间最短而且可控。Table 4 shows Example 1 (after the long-read sequencing data is split, each long-read data subset is compared with the reference genome and then merged), Example 2 (direct comparison, thread setting 20), Example 3 The comparison process resource consumption of the three methods shown in (direct comparison, thread setting 32). Wherein the time represents the peak running time; the actual thread is the number of threads set during the analysis; the peak memory refers to the maximum memory use in the running process; the actual CPU is the total CPU of the entire comparison process after accounting, and wherein embodiment 1 includes data Used when splitting and merging CPUs for comparison results. The comparison entry refers to the number of rows of comparison information after comparison, and the comparison file size is the size of the final generated comparison file. It can be seen from Table 4 that the size of the comparison files obtained by the three methods is consistent with the comparison results. After sorting the comparison files and viewing them with the diff command, the three results are completely consistent. In terms of time usage, the higher the thread setting, the less the total CPU time. In the case of limited thread resources, it is recommended to use the method described in Example 1, that is, to compare multiple subsets of long-read sequencing data obtained after splitting the long-read sequencing data with the reference genome to obtain each sequence According to the comparison results of data subsets, this method can multi-task in parallel, improve the analysis efficiency, and the peak memory is also slightly reduced, and the total running time is the shortest and controllable.
表4:Table 4:

2、纠错过程的比较2. Comparison of error correction process
由于不论在比对时是否对测序的长读长数据拆分,所有的比对结果都一致。因此,此处只示出实施 例2(模块纠错)和实施例3(全局纠错)两个实施方法的比较结果。All alignment results are consistent regardless of whether the sequenced long-read data is split during alignment. Therefore, only the comparison results of the two implementation methods of embodiment 2 (module error correction) and embodiment 3 (global error correction) are shown here.
表5中展示了实施例2和实施例3的纠错过程中时间和资源的使用结果,由结果可以看出,实施例 2所述方法在时间、线程使用、峰值内存和总的CPU数目都远低于实施例3所述方法。此外,发明人对 全基因组切块后,以耗时最长的一个子块(约52M)为例,对实施例2所述纠错方法的效果进行举例说 明,其中,CPU设置为5,参考序列导入时间0.395s,长读长数据导入时间为1614.617s,比对结果导入 时间为4.017s,从比对结果中确定overlap关系时间为2050.158s,界面输出时间11.790s,获得纠错后一 致性序列时间为13271.216s,总时间为16953.343s,总时间显著减少。Table 5 shows the usage results of time and resources in the error correction process of Embodiment 2 and Embodiment 3. As can be seen from the results, the method described in Embodiment 2 has the same effect on time, thread usage, peak memory and total number of CPUs. Much lower than the method described in Example 3. In addition, after the inventor cuts the entire genome into blocks, he takes the longest time-consuming sub-block (about 52M) as an example to illustrate the effect of the error correction method described in Example 2, wherein the CPU is set to 5, The reference sequence import time was 0.395s, the long-read data import time was 1614.617s, the comparison result import time was 4.017s, the overlap relationship determined from the comparison results was 2050.158s, and the interface output time was 11.790s. The consistency sequence time is 13271.216s and the total time is 16953.343s, the total time is significantly reduced.
表5:table 5:
此外,发明人对实施例1(优化后方法,即模块纠错)所述方法与实施例3(优化前方法,即全局 纠错)所述方法进行比较,其中,实施例2、实施例3各自所述方法中每个步骤消耗的时间占总时间的 百分比如表6所示,经过基因组切分后纠错,长读长数据的导入时间占总耗时的百分比从原来的93.86% 降低到9.2%,将耗时最长的环节转移到取一致性序列步骤,这一步骤占总耗时的百分比从原来3.6%提 升到78.28%,而由于基因组取一致性序列是基于与该序列对应的比对结果完成,而每一个参考基因组 子块的比对结果均是完整的,因此取一致性序列不论序列是否切分,其时间效率变化不大。因此实施例 1所述方法通过降低数据导入时间有效优化了整体的纠错效率。In addition, the inventor compares the method described in Embodiment 1 (the method after optimization, that is, module error correction) with the method described in Embodiment 3 (the method before optimization, that is, global error correction), wherein, Embodiment 2, Embodiment 3 The percentage of time consumed by each step in the respective methods as a percentage of the total time is shown in Table 6. After genome segmentation and error correction, the percentage of import time for long-read data in the total time was reduced from 93.86% to 9.2%, the longest time-consuming link is transferred to the step of obtaining the consensus sequence, and the percentage of this step in the total time consumption has increased from 3.6% to 78.28%, and since the consensus sequence of the genome is based on the corresponding The comparison result of each reference genome sub-block is completed, and the comparison result of each reference genome sub-block is complete. Therefore, no matter whether the sequence is segmented or not, the time efficiency of taking the consensus sequence does not change much. Therefore, the method described in Embodiment 1 effectively optimizes the overall error correction efficiency by reducing the data import time.
表6:Table 6:
实施例1所述方法与实施例3所述方法纠错结果比较如表7所示,其中,第一列Ctg_N50_len为组 装的N50长度(将基因组组装结果按照从大到小排序,总长度大于50%的序列长度,可以反应基因组整 体的长度水平,从而判断基因组序列的完整性),第二列Ctg_length为组装的序列总长度,第三列 Busco_comp与基准通用单拷贝直系同源基因比较的完整基因数目所占的比例。理论上单拷贝的直系同 源基因集基准在物种中是保守的,单拷贝的,对于组装结果鉴定出单拷贝的直系同源基因占基准数据集 的比例可以评估基因组的完整性,完整的单拷贝基因占比越高,组装越完整,最大值为100。第四列和 第五列是Merqury的评估结果,它是将来自高精度测序reads的一组k-mer与基因组组装结果进行了比 较评估。第四列QV(Pred)指Merqury评估的QV值,反应基因组的精确度。第五列Completeness为Merqury 评估的Completeness的值。从表7数据可以看出,纠错后相对于原始数据,质量值从22提升到28,Kmer 覆盖度完整性从82%提升到92%以上,BUSCO评估完整性从63.3%提升到84%以上。实施例2所述方 法和实施例3所述方法的结果略有差异,但是由于分析软件存在误差,所以整体基因组的准确性和完整 性相当。实施例2所述方法在保证分析结果不变的前提下,有效解决长长数据纠错过程中内存消耗大, 分析时间长的问题。The error correction results of the method described in Example 1 and the method described in Example 3 are compared as shown in Table 7, wherein the first column Ctg_N50_len is the N50 length of the assembly (the genome assembly results are sorted from large to small, and the total length is greater than 50% of the sequence length, which can reflect the overall length of the genome, thereby judging the integrity of the genome sequence), the second column Ctg_length is the total length of the assembled sequence, the third column Busco_comp is compared with the benchmark universal single-copy orthologous gene The proportion of the number of complete genes. Theoretically, the single-copy orthologous gene set benchmark is conserved in the species, single-copy, and the proportion of single-copy orthologous genes identified in the assembly results to the benchmark data set can evaluate the integrity of the genome, and the complete single-copy The higher the proportion of copy genes, the more complete the assembly, and the maximum value is 100. The fourth and fifth columns are the evaluation results of Merqury, which compares a set of k-mers from high-precision sequencing reads with the genome assembly results. The fourth column QV(Pred) refers to the QV value evaluated by Merqury, which reflects the accuracy of the genome. The fifth column Completeness is the value of Completeness evaluated by Merqury. As can be seen from the data in Table 7, compared with the original data after error correction, the quality value has increased from 22 to 28, the Kmer coverage integrity has increased from 82% to over 92%, and the BUSCO evaluation integrity has increased from 63.3% to over 84%. . The results of the method described in Example 2 and the method described in Example 3 are slightly different, but due to errors in the analysis software, the accuracy and completeness of the overall genome are comparable. The method described in Embodiment 2 effectively solves the problems of large memory consumption and long analysis time in the error correction process of long data under the premise of ensuring that the analysis results remain unchanged.
表7:Table 7:

3、结论:3. Conclusion:
综上所述,当机器由大量小CPU资源组成,单台机器CPU限制的情况下,建议使用模块比对、模 块纠错的方法,其中,模块纠错步骤的优化效果明显,本实施例中CPU时降为原来的1/7,整体分析时 间降为原来的1/10,因此,采用实施例1所述的模块比对、模块纠错的方法可有效提升纠错过程的时间 效率和CPU使用率。To sum up, when the machine is composed of a large number of small CPU resources and the CPU of a single machine is limited, it is recommended to use the method of module comparison and module error correction. Among them, the optimization effect of the module error correction step is obvious. This embodiment The CPU time is reduced to 1/7 of the original, and the overall analysis time is reduced to 1/10 of the original. Therefore, the method of module comparison and module error correction described in Embodiment 1 can effectively improve the time efficiency of the error correction process and CPU usage.
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示 例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个 实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且, 描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外, 在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例 或示例的特征进行结合和组合。In the description of this specification, descriptions referring to the terms "one embodiment", "some embodiments", "examples", "specific examples", or "some examples" mean that the specific embodiments described in conjunction with the embodiments or examples A feature, structure, material, or characteristic is included in at least one embodiment or example of the invention. In this specification, the schematic representations of the above terms are not necessarily directed to the same embodiment or example. Furthermore, the described specific features, structures, materials or characteristics may be combined in any suitable manner in any one or more embodiments or examples. In addition, those skilled in the art can combine and combine different embodiments or examples and features of different embodiments or examples described in this specification without conflicting with each other.
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为 对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和 变型。Although the embodiments of the present invention have been shown and described above, it can be understood that the above embodiments are exemplary and should not be construed as limiting the present invention, and those skilled in the art can make the above-mentioned The embodiments are subject to variations, modifications, substitutions and variations.
Claims (12)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111114295.1ACN115938480A (en) | 2021-09-23 | 2021-09-23 | Long-read sequencing optimizes device and system for error correction method of genome assembly results |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111114295.1ACN115938480A (en) | 2021-09-23 | 2021-09-23 | Long-read sequencing optimizes device and system for error correction method of genome assembly results |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN115938480Atrue CN115938480A (en) | 2023-04-07 |
Family
ID=86549455
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111114295.1APendingCN115938480A (en) | 2021-09-23 | 2021-09-23 | Long-read sequencing optimizes device and system for error correction method of genome assembly results |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN115938480A (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116343919A (en)* | 2023-04-11 | 2023-06-27 | 天津大学四川创新研究院 | Whole genome map drawing and sequencing method |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103797486A (en)* | 2011-06-06 | 2014-05-14 | 皇家飞利浦有限公司 | Methods for assembling nucleic acid sequence data |
| CN106795568A (en)* | 2014-10-10 | 2017-05-31 | 因维蒂公司 | Method, system and the process of the DE NOVO assemblings of read is sequenced |
| CN108460245A (en)* | 2017-02-21 | 2018-08-28 | 深圳华大基因科技服务有限公司 | The method and apparatus for assembling result using two generation of three generations's sequence optimisation |
| CN108629156A (en)* | 2017-03-21 | 2018-10-09 | 深圳华大基因科技服务有限公司 | The method, apparatus and computer readable storage medium of three generations's sequencing data error correction |
| CN111326212A (en)* | 2020-02-18 | 2020-06-23 | 福建和瑞基因科技有限公司 | A method for detecting structural variation |
| US20200364371A1 (en)* | 2017-05-11 | 2020-11-19 | Ethan Huang | Methods and systems for anonymizing genome segments and sequences and associated information |
| CN113012758A (en)* | 2021-04-26 | 2021-06-22 | 武汉华大基因技术服务有限公司 | Method and device for correcting long read length assembly result by using short read length sequence |
| CN113345521A (en)* | 2021-05-31 | 2021-09-03 | 天津大学 | Coding and recovering method using large fragment DNA storage |
- 2021
- 2021-09-23CNCN202111114295.1Apatent/CN115938480A/enactivePending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103797486A (en)* | 2011-06-06 | 2014-05-14 | 皇家飞利浦有限公司 | Methods for assembling nucleic acid sequence data |
| CN106795568A (en)* | 2014-10-10 | 2017-05-31 | 因维蒂公司 | Method, system and the process of the DE NOVO assemblings of read is sequenced |
| CN108460245A (en)* | 2017-02-21 | 2018-08-28 | 深圳华大基因科技服务有限公司 | The method and apparatus for assembling result using two generation of three generations's sequence optimisation |
| CN108629156A (en)* | 2017-03-21 | 2018-10-09 | 深圳华大基因科技服务有限公司 | The method, apparatus and computer readable storage medium of three generations's sequencing data error correction |
| US20200364371A1 (en)* | 2017-05-11 | 2020-11-19 | Ethan Huang | Methods and systems for anonymizing genome segments and sequences and associated information |
| CN111326212A (en)* | 2020-02-18 | 2020-06-23 | 福建和瑞基因科技有限公司 | A method for detecting structural variation |
| CN113012758A (en)* | 2021-04-26 | 2021-06-22 | 武汉华大基因技术服务有限公司 | Method and device for correcting long read length assembly result by using short read length sequence |
| CN113345521A (en)* | 2021-05-31 | 2021-09-03 | 天津大学 | Coding and recovering method using large fragment DNA storage |
Non-Patent Citations (1)
| Title |
|---|
| 马东娜;张兴坦;魏柳锋;李仪莹;钟伟民;赵茜;尤民生;: "基因组二代测序数据与三代测序数据的混合校正和组装", 基因组学与应用生物学, no. 04, 25 April 2018 (2018-04-25)* |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116343919A (en)* | 2023-04-11 | 2023-06-27 | 天津大学四川创新研究院 | Whole genome map drawing and sequencing method |
| CN116343919B (en)* | 2023-04-11 | 2023-12-08 | 天津大学四川创新研究院 | Whole genome map drawing and sequencing method |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Laehnemann et al. | Denoising DNA deep sequencing data—high-throughput sequencing errors and their correction | |
| Nikolayeva et al. | edgeR for differential RNA-seq and ChIP-seq analysis: an application to stem cell biology | |
| Fonseca et al. | Tools for mapping high-throughput sequencing data | |
| AU2022298428B2 (en) | Gene sequencing analysis method and apparatus, and storage medium and computer device | |
| Wright et al. | Preprocessing and quality control for whole-genome sequences from the Illumina HiSeq X platform | |
| CN116564415B (en) | Flow sequencing analysis methods, devices, storage media and computer equipment | |
| Utturkar et al. | Bacterial differential expression analysis methods | |
| Pireddu et al. | MapReducing a genomic sequencing workflow | |
| CN112397142A (en) | Gene variation detection method and system for multi-core processor | |
| CN115938480A (en) | Long-read sequencing optimizes device and system for error correction method of genome assembly results | |
| CN111767256B (en) | Method for separating sample read data from fastq file | |
| Marco‐Sola et al. | Efficient Alignment of Illumina‐Like High‐Throughput Sequencing Reads with the GEnomic Multi‐tool (GEM) Mapper | |
| CN114237911A (en) | CUDA-based gene data processing method and device and CUDA framework | |
| CN117893512B (en) | Nucleic acid detection and data analysis methods, equipment, systems and storage media | |
| CN118571324A (en) | Data processing method and device, storage medium and electronic device | |
| US20200243162A1 (en) | Method, system, and computing device for optimizing computing operations of gene sequencing system | |
| CN117789820A (en) | Methods and systems for detecting structural variations in third-generation whole-exome sequencing transcript data | |
| Clement et al. | Parallel mapping approaches for GNUMAP | |
| HK40085281A (en) | Long-read sequencing optimizes device and system for error correction method of genome assembly results | |
| WO2024187428A1 (en) | Assembly process for constructing high-quality microbial genomes on basis of stlfr metagenomic sequencing data | |
| Harrath et al. | Comparative evaluation of short read alignment tools for next generation DNA sequencing | |
| CN119673284A (en) | Third-generation sequencing read analysis methods, applications and devices | |
| CN107403076B (en) | DNA sequence processing method and equipment | |
| Jo | Multi-threading the generation of Burrows-Wheeler Alignment | |
| CN107688727A (en) | Biological sequence clusters and the recognition methods of transcript hypotype and device in total length transcript profile |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code | Ref country code:HK Ref legal event code:DE Ref document number:40085281 Country of ref document:HK |