CN112125916A - Novel small molecule inhibitors of insulin-like growth factor-1 receptor and their uses - Google Patents
Novel small molecule inhibitors of insulin-like growth factor-1 receptor and their usesDownload PDFInfo
- Publication number
- CN112125916A CN112125916ACN202011040572.4ACN202011040572ACN112125916ACN 112125916 ACN112125916 ACN 112125916ACN 202011040572 ACN202011040572 ACN 202011040572ACN 112125916 ACN112125916 ACN 112125916A
- Authority
- CN
- China
- Prior art keywords
- compound
- small molecule
- molecule inhibitor
- mice
- halogenated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000003112inhibitorSubstances0.000titleclaimsabstractdescription20
- 150000003384small moleculesChemical class0.000titleclaimsabstractdescription19
- 102100039688Insulin-like growth factor 1 receptorHuman genes0.000titleclaimsabstractdescription17
- 101710184277Insulin-like growth factor 1 receptorProteins0.000titleclaimsabstractdescription16
- 206010028980NeoplasmDiseases0.000claimsabstractdescription31
- 239000003814drugSubstances0.000claimsabstractdescription25
- 229940079593drugDrugs0.000claimsabstractdescription21
- 239000002246antineoplastic agentSubstances0.000claimsabstractdescription8
- 201000011510cancerDiseases0.000claimsabstractdescription7
- 229940041181antineoplastic drugDrugs0.000claimsabstractdescription5
- 125000004431deuterium atomChemical group0.000claimsabstractdescription3
- 150000001875compoundsChemical class0.000claimsdescription61
- 206010018338GliomaDiseases0.000claimsdescription17
- 208000032612Glial tumorDiseases0.000claimsdescription14
- -1nitro, aminoChemical group0.000claimsdescription12
- 125000003545alkoxy groupChemical group0.000claimsdescription11
- 150000003839saltsChemical class0.000claimsdescription11
- 125000000217alkyl groupChemical group0.000claimsdescription9
- 229910052736halogenInorganic materials0.000claimsdescription8
- 150000002367halogensChemical class0.000claimsdescription8
- 206010058467Lung neoplasm malignantDiseases0.000claimsdescription6
- 125000003118aryl groupChemical group0.000claimsdescription6
- 229910052799carbonInorganic materials0.000claimsdescription6
- 201000005202lung cancerDiseases0.000claimsdescription6
- 208000020816lung neoplasmDiseases0.000claimsdescription6
- 229910052757nitrogenInorganic materials0.000claimsdescription6
- 238000006467substitution reactionMethods0.000claimsdescription5
- NOIXNOMHHWGUTG-UHFFFAOYSA-N2-[[4-[4-pyridin-4-yl-1-(2,2,2-trifluoroethyl)pyrazol-3-yl]phenoxy]methyl]quinolineChemical groupC=1C=C(OCC=2N=C3C=CC=CC3=CC=2)C=CC=1C1=NN(CC(F)(F)F)C=C1C1=CC=NC=C1NOIXNOMHHWGUTG-UHFFFAOYSA-N0.000claimsdescription4
- 206010006187Breast cancerDiseases0.000claimsdescription4
- 208000026310Breast neoplasmDiseases0.000claimsdescription4
- 230000001419dependent effectEffects0.000claimsdescription4
- 229910052805deuteriumInorganic materials0.000claimsdescription4
- 201000010099diseaseDiseases0.000claimsdescription4
- 208000037265diseases, disorders, signs and symptomsDiseases0.000claimsdescription4
- 230000003287optical effectEffects0.000claimsdescription4
- 238000002360preparation methodMethods0.000claimsdescription4
- 208000030289Lymphoproliferative diseaseDiseases0.000claimsdescription3
- 208000014767Myeloproliferative diseaseDiseases0.000claimsdescription3
- 230000000259anti-tumor effectEffects0.000claimsdescription3
- 125000004432carbon atomChemical groupC*0.000claimsdescription3
- 210000001035gastrointestinal tractAnatomy0.000claimsdescription3
- 230000002071myeloproliferative effectEffects0.000claimsdescription3
- 230000002195synergetic effectEffects0.000claimsdescription3
- 230000008685targetingEffects0.000claimsdescription3
- 206010003571AstrocytomaDiseases0.000claimsdescription2
- 206010009944Colon cancerDiseases0.000claimsdescription2
- 208000001333Colorectal NeoplasmsDiseases0.000claimsdescription2
- 206010025323LymphomasDiseases0.000claimsdescription2
- 208000009277Neuroectodermal TumorsDiseases0.000claimsdescription2
- 206010060862Prostate cancerDiseases0.000claimsdescription2
- 208000000236Prostatic NeoplasmsDiseases0.000claimsdescription2
- 208000005718Stomach NeoplasmsDiseases0.000claimsdescription2
- 239000002253acidSubstances0.000claimsdescription2
- 229910052783alkali metalInorganic materials0.000claimsdescription2
- 229910052784alkaline earth metalInorganic materials0.000claimsdescription2
- 229940125644antibody drugDrugs0.000claimsdescription2
- 239000002585baseSubstances0.000claimsdescription2
- 229940044683chemotherapy drugDrugs0.000claimsdescription2
- 208000035250cutaneous malignant susceptibility to 1 melanomaDiseases0.000claimsdescription2
- 125000004093cyano groupChemical group*C#N0.000claimsdescription2
- 206010017758gastric cancerDiseases0.000claimsdescription2
- 208000032839leukemiaDiseases0.000claimsdescription2
- 201000001441melanomaDiseases0.000claimsdescription2
- 102000039446nucleic acidsHuman genes0.000claimsdescription2
- 108020004707nucleic acidsProteins0.000claimsdescription2
- 150000007523nucleic acidsChemical class0.000claimsdescription2
- 229910052760oxygenInorganic materials0.000claimsdescription2
- 125000003226pyrazolyl groupChemical group0.000claimsdescription2
- 201000011549stomach cancerDiseases0.000claimsdescription2
- 229910052717sulfurInorganic materials0.000claimsdescription2
- 230000000118anti-neoplastic effectEffects0.000claims1
- 208000005017glioblastomaDiseases0.000claims1
- 230000002265preventionEffects0.000claims1
- 230000002797proteolythic effectEffects0.000claims1
- 230000035755proliferationEffects0.000abstractdescription22
- YJGVMLPVUAXIQN-UHFFFAOYSA-NepipodophyllotoxinNatural productsCOC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1YJGVMLPVUAXIQN-UHFFFAOYSA-N0.000abstractdescription21
- 230000008499blood brain barrier functionEffects0.000abstractdescription17
- 210000001218blood-brain barrierAnatomy0.000abstractdescription17
- 210000004556brainAnatomy0.000abstractdescription15
- 230000004083survival effectEffects0.000abstractdescription13
- YJGVMLPVUAXIQN-XVVDYKMHSA-NpodophyllotoxinChemical groupCOC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1YJGVMLPVUAXIQN-XVVDYKMHSA-N0.000abstractdescription9
- 238000001727in vivoMethods0.000abstractdescription8
- 229960001237podophyllotoxinDrugs0.000abstractdescription7
- YVCVYCSAAZQOJI-UHFFFAOYSA-NpodophyllotoxinNatural productsCOC1=C(O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1YVCVYCSAAZQOJI-UHFFFAOYSA-N0.000abstractdescription7
- 230000035699permeabilityEffects0.000abstractdescription6
- 206010027476MetastasesDiseases0.000abstractdescription5
- 229910052731fluorineInorganic materials0.000abstractdescription5
- 230000009401metastasisEffects0.000abstractdescription5
- 125000001153fluoro groupChemical groupF*0.000abstractdescription4
- 125000004435hydrogen atomChemical group[H]*0.000abstractdescription4
- 230000010534mechanism of actionEffects0.000abstractdescription3
- 210000004881tumor cellAnatomy0.000abstractdescription3
- 210000004027cellAnatomy0.000description70
- 241000699670Mus sp.Species0.000description61
- QFLWZFQWSBQYPS-AWRAUJHKSA-N(3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acidChemical compoundCCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1QFLWZFQWSBQYPS-AWRAUJHKSA-N0.000description49
- YMWUJEATGCHHMB-UHFFFAOYSA-NDichloromethaneChemical compoundClCClYMWUJEATGCHHMB-UHFFFAOYSA-N0.000description48
- 238000012360testing methodMethods0.000description37
- IAZDPXIOMUYVGZ-UHFFFAOYSA-NDimethylsulphoxideChemical compoundCS(C)=OIAZDPXIOMUYVGZ-UHFFFAOYSA-N0.000description36
- XEKOWRVHYACXOJ-UHFFFAOYSA-NEthyl acetateChemical compoundCCOC(C)=OXEKOWRVHYACXOJ-UHFFFAOYSA-N0.000description30
- OKKJLVBELUTLKV-UHFFFAOYSA-NMethanolChemical compoundOCOKKJLVBELUTLKV-UHFFFAOYSA-N0.000description21
- YJGVMLPVUAXIQN-HAEOHBJNSA-NpicropodophyllotoxinChemical classCOC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@H]2C(OC3)=O)=C1YJGVMLPVUAXIQN-HAEOHBJNSA-N0.000description19
- 241000699666Mus <mouse, genus>Species0.000description17
- RTZKZFJDLAIYFH-UHFFFAOYSA-NDiethyl etherChemical compoundCCOCCRTZKZFJDLAIYFH-UHFFFAOYSA-N0.000description16
- BPEGJWRSRHCHSN-UHFFFAOYSA-NTemozolomideChemical compoundO=C1N(C)N=NC2=C(C(N)=O)N=CN21BPEGJWRSRHCHSN-UHFFFAOYSA-N0.000description13
- 230000000694effectsEffects0.000description13
- 238000001962electrophoresisMethods0.000description12
- 102000004169proteins and genesHuman genes0.000description12
- 108090000623proteins and genesProteins0.000description12
- 210000005013brain tissueAnatomy0.000description11
- 230000002401inhibitory effectEffects0.000description11
- 239000000203mixtureSubstances0.000description11
- 239000012074organic phaseSubstances0.000description11
- 239000007787solidSubstances0.000description11
- 238000003756stirringMethods0.000description11
- 229960004964temozolomideDrugs0.000description11
- WYURNTSHIVDZCO-UHFFFAOYSA-NTetrahydrofuranChemical compoundC1CCOC1WYURNTSHIVDZCO-UHFFFAOYSA-N0.000description10
- 238000006243chemical reactionMethods0.000description10
- 239000006228supernatantSubstances0.000description10
- WEVYAHXRMPXWCK-UHFFFAOYSA-NAcetonitrileChemical compoundCC#NWEVYAHXRMPXWCK-UHFFFAOYSA-N0.000description9
- PMZURENOXWZQFD-UHFFFAOYSA-LSodium SulfateChemical compound[Na+].[Na+].[O-]S([O-])(=O)=OPMZURENOXWZQFD-UHFFFAOYSA-L0.000description9
- 238000001514detection methodMethods0.000description9
- 238000002474experimental methodMethods0.000description9
- 230000003285pharmacodynamic effectEffects0.000description9
- 238000000926separation methodMethods0.000description9
- HPALAKNZSZLMCH-UHFFFAOYSA-Msodium;chloride;hydrateChemical classO.[Na+].[Cl-]HPALAKNZSZLMCH-UHFFFAOYSA-M0.000description9
- 241000699660Mus musculusSpecies0.000description8
- 229940125904compound 1Drugs0.000description8
- 239000001963growth mediumSubstances0.000description8
- 239000012528membraneSubstances0.000description8
- 238000011580nude mouse modelMethods0.000description8
- 239000002953phosphate buffered salineSubstances0.000description8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-NSilicium dioxideChemical compoundO=[Si]=OVYPSYNLAJGMNEJ-UHFFFAOYSA-N0.000description7
- 239000008346aqueous phaseSubstances0.000description7
- 230000015572biosynthetic processEffects0.000description7
- 239000002285corn oilSubstances0.000description7
- 235000005687corn oilNutrition0.000description7
- 238000000338in vitroMethods0.000description7
- 239000003208petroleumSubstances0.000description7
- 239000012071phaseSubstances0.000description7
- 239000000741silica gelSubstances0.000description7
- 229910002027silica gelInorganic materials0.000description7
- 238000010898silica gel chromatographyMethods0.000description7
- KFZMGEQAYNKOFK-UHFFFAOYSA-NIsopropanolChemical groupCC(C)OKFZMGEQAYNKOFK-UHFFFAOYSA-N0.000description6
- 239000002033PVDF binderSubstances0.000description6
- ZMANZCXQSJIPKH-UHFFFAOYSA-NTriethylamineChemical compoundCCN(CC)CCZMANZCXQSJIPKH-UHFFFAOYSA-N0.000description6
- 239000000090biomarkerSubstances0.000description6
- 238000010568chiral column chromatographyMethods0.000description6
- 229940125773compound 10Drugs0.000description6
- 230000003203everyday effectEffects0.000description6
- 238000003304gavageMethods0.000description6
- ZLVXBBHTMQJRSX-VMGNSXQWSA-NjdticChemical compoundC1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1ZLVXBBHTMQJRSX-VMGNSXQWSA-N0.000description6
- 239000004005microsphereSubstances0.000description6
- 229920002981polyvinylidene fluoridePolymers0.000description6
- 238000003786synthesis reactionMethods0.000description6
- 238000012546transferMethods0.000description6
- 10210002399060S ribosomal protein L17Human genes0.000description5
- 101710089372Programmed cell death protein 1Proteins0.000description5
- 239000013592cell lysateSubstances0.000description5
- 230000019491signal transductionEffects0.000description5
- IJGRMHOSHXDMSA-UHFFFAOYSA-NAtomic nitrogenChemical compoundN#NIJGRMHOSHXDMSA-UHFFFAOYSA-N0.000description4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-NEthanolChemical compoundCCOLFQSCWFLJHTTHZ-UHFFFAOYSA-N0.000description4
- 239000006180TBST bufferSubstances0.000description4
- HEDRZPFGACZZDS-MICDWDOJSA-NTrichloro(2H)methaneChemical compound[2H]C(Cl)(Cl)ClHEDRZPFGACZZDS-MICDWDOJSA-N0.000description4
- 238000010171animal modelMethods0.000description4
- 230000003542behavioural effectEffects0.000description4
- 229940125898compound 5Drugs0.000description4
- 125000000753cycloalkyl groupChemical group0.000description4
- 238000011156evaluationMethods0.000description4
- 239000006260foamSubstances0.000description4
- 239000000499gelSubstances0.000description4
- 230000005764inhibitory processEffects0.000description4
- 238000011081inoculationMethods0.000description4
- 239000002609mediumSubstances0.000description4
- 238000000034methodMethods0.000description4
- 239000002985plastic filmSubstances0.000description4
- 229920006255plastic filmPolymers0.000description4
- 239000011541reaction mixtureSubstances0.000description4
- 239000000243solutionSubstances0.000description4
- 239000003981vehicleSubstances0.000description4
- 238000001262western blotMethods0.000description4
- NHBKXEKEPDILRR-UHFFFAOYSA-N2,3-bis(butanoylsulfanyl)propyl butanoateChemical compoundCCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCCNHBKXEKEPDILRR-UHFFFAOYSA-N0.000description3
- UHOVQNZJYSORNB-UHFFFAOYSA-NBenzeneChemical compoundC1=CC=CC=C1UHOVQNZJYSORNB-UHFFFAOYSA-N0.000description3
- 239000006144Dulbecco’s modified Eagle's mediumSubstances0.000description3
- 238000005481NMR spectroscopyMethods0.000description3
- UIIMBOGNXHQVGW-UHFFFAOYSA-MSodium bicarbonateChemical class[Na+].OC([O-])=OUIIMBOGNXHQVGW-UHFFFAOYSA-M0.000description3
- 230000001464adherent effectEffects0.000description3
- 230000004071biological effectEffects0.000description3
- 230000037396body weightEffects0.000description3
- 201000007983brain gliomaDiseases0.000description3
- 239000006285cell suspensionSubstances0.000description3
- 238000004587chromatography analysisMethods0.000description3
- 230000003247decreasing effectEffects0.000description3
- 238000001647drug administrationMethods0.000description3
- 238000011503in vivo imagingMethods0.000description3
- 238000002347injectionMethods0.000description3
- 239000007924injectionSubstances0.000description3
- 230000004048modificationEffects0.000description3
- 238000012986modificationMethods0.000description3
- 230000000877morphologic effectEffects0.000description3
- 239000000047productSubstances0.000description3
- 229920006395saturated elastomerPolymers0.000description3
- 239000007858starting materialSubstances0.000description3
- 239000011550stock solutionSubstances0.000description3
- YLQBMQCUIZJEEH-UHFFFAOYSA-NtetrahydrofuranNatural productsC=1C=COC=1YLQBMQCUIZJEEH-UHFFFAOYSA-N0.000description3
- 239000003643water by typeSubstances0.000description3
- 125000005913(C3-C6) cycloalkyl groupChemical group0.000description2
- 2380000051601H NMR spectroscopyMethods0.000description2
- OPHQOIGEOHXOGX-UHFFFAOYSA-N3,4,5-trimethoxybenzaldehydeChemical compoundCOC1=CC(C=O)=CC(OC)=C1OCOPHQOIGEOHXOGX-UHFFFAOYSA-N0.000description2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N4-DimethylaminopyridineChemical compoundCN(C)C1=CC=NC=C1VHYFNPMBLIVWCW-UHFFFAOYSA-N0.000description2
- NLXLAEXVIDQMFP-UHFFFAOYSA-NAmmonia chlorideChemical class[NH4+].[Cl-]NLXLAEXVIDQMFP-UHFFFAOYSA-N0.000description2
- 238000011729BALB/c nude mouseMethods0.000description2
- 208000003174Brain NeoplasmsDiseases0.000description2
- 238000011740C57BL/6 mouseMethods0.000description2
- OKTJSMMVPCPJKN-UHFFFAOYSA-NCarbonChemical compound[C]OKTJSMMVPCPJKN-UHFFFAOYSA-N0.000description2
- ZAMOUSCENKQFHK-UHFFFAOYSA-NChlorine atomChemical compound[Cl]ZAMOUSCENKQFHK-UHFFFAOYSA-N0.000description2
- PXGOKWXKJXAPGV-UHFFFAOYSA-NFluorineChemical compoundFFPXGOKWXKJXAPGV-UHFFFAOYSA-N0.000description2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-NHydroquinoneChemical compoundOC1=CC=C(O)C=C1QIGBRXMKCJKVMJ-UHFFFAOYSA-N0.000description2
- 229910010199LiAlInorganic materials0.000description2
- 241001465754MetazoaSpecies0.000description2
- UFWIBTONFRDIAS-UHFFFAOYSA-NNaphthaleneChemical compoundC1=CC=CC2=CC=CC=C21UFWIBTONFRDIAS-UHFFFAOYSA-N0.000description2
- 239000007983Tris bufferSubstances0.000description2
- 238000004458analytical methodMethods0.000description2
- MWPLVEDNUUSJAV-UHFFFAOYSA-NanthraceneChemical compoundC1=CC=CC2=CC3=CC=CC=C3C=C21MWPLVEDNUUSJAV-UHFFFAOYSA-N0.000description2
- 239000007640basal mediumSubstances0.000description2
- 208000030224brain astrocytomaDiseases0.000description2
- UDSAIICHUKSCKT-UHFFFAOYSA-Nbromophenol blueChemical compoundC1=C(Br)C(O)=C(Br)C=C1C1(C=2C=C(Br)C(O)=C(Br)C=2)C2=CC=CC=C2S(=O)(=O)O1UDSAIICHUKSCKT-UHFFFAOYSA-N0.000description2
- 239000000872bufferSubstances0.000description2
- 238000004364calculation methodMethods0.000description2
- 238000004113cell cultureMethods0.000description2
- 239000006143cell culture mediumSubstances0.000description2
- 229910052801chlorineInorganic materials0.000description2
- 239000000460chlorineSubstances0.000description2
- 229940125782compound 2Drugs0.000description2
- 229940126214compound 3Drugs0.000description2
- 230000034994deathEffects0.000description2
- 238000013461designMethods0.000description2
- CSJLBAMHHLJAAS-UHFFFAOYSA-Ndiethylaminosulfur trifluorideChemical compoundCCN(CC)S(F)(F)FCSJLBAMHHLJAAS-UHFFFAOYSA-N0.000description2
- 230000009977dual effectEffects0.000description2
- 235000013861fat-freeNutrition0.000description2
- 238000002073fluorescence micrographMethods0.000description2
- 239000011737fluorineSubstances0.000description2
- 239000008103glucoseSubstances0.000description2
- 239000011544gradient gelSubstances0.000description2
- 208000029824high grade gliomaDiseases0.000description2
- 238000004128high performance liquid chromatographyMethods0.000description2
- 238000011534incubationMethods0.000description2
- 239000007788liquidSubstances0.000description2
- ZCSHNCUQKCANBX-UHFFFAOYSA-Nlithium diisopropylamideChemical compound[Li+].CC(C)[N-]C(C)CZCSHNCUQKCANBX-UHFFFAOYSA-N0.000description2
- 239000012160loading bufferSubstances0.000description2
- 208000030883malignant astrocytomaDiseases0.000description2
- 201000011614malignant gliomaDiseases0.000description2
- 229910052751metalInorganic materials0.000description2
- 239000002184metalSubstances0.000description2
- 235000013336milkNutrition0.000description2
- 239000008267milkSubstances0.000description2
- 210000004080milkAnatomy0.000description2
- 239000012146running bufferSubstances0.000description2
- 238000002415sodium dodecyl sulfate polyacrylamide gel electrophoresisMethods0.000description2
- 239000002904solventSubstances0.000description2
- 210000000130stem cellAnatomy0.000description2
- 229960005322streptomycinDrugs0.000description2
- 125000001424substituent groupChemical group0.000description2
- 239000000725suspensionSubstances0.000description2
- 125000004213tert-butoxy groupChemical group[H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H]0.000description2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-NtriphenylphosphineChemical compoundC1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1RIOQSEWOXXDEQQ-UHFFFAOYSA-N0.000description2
- XLYOFNOQVPJJNP-UHFFFAOYSA-NwaterSubstancesOXLYOFNOQVPJJNP-UHFFFAOYSA-N0.000description2
- 125000004191(C1-C6) alkoxy groupChemical group0.000description1
- ORTVZLZNOYNASJ-UPHRSURJSA-N(z)-but-2-ene-1,4-diolChemical compoundOC\C=C/COORTVZLZNOYNASJ-UPHRSURJSA-N0.000description1
- NDQXKKFRNOPRDW-UHFFFAOYSA-N1,1,1-triethoxyethaneChemical compoundCCOC(C)(OCC)OCCNDQXKKFRNOPRDW-UHFFFAOYSA-N0.000description1
- 1250000058093,4,5-trimethoxyphenyl groupChemical group[H]C1=C(OC([H])([H])[H])C(OC([H])([H])[H])=C(OC([H])([H])[H])C([H])=C1*0.000description1
- OPHQOIGEOHXOGX-GQALSZNTSA-N3,4,5-tris(trideuteriomethoxy)benzaldehydeChemical compound[2H]C([2H])([2H])OC1=CC(C=O)=CC(OC([2H])([2H])[2H])=C1OC([2H])([2H])[2H]OPHQOIGEOHXOGX-GQALSZNTSA-N0.000description1
- WDBQJSCPCGTAFG-QHCPKHFHSA-N4,4-difluoro-N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclohexane-1-carboxamideChemical compoundFC1(CCC(CC1)C(=O)N[C@@H](CCN1CCC(CC1)N1C(=NN=C1C)C(C)C)C=1C=NC=CC=1)FWDBQJSCPCGTAFG-QHCPKHFHSA-N0.000description1
- 2299600005494-dimethylaminophenolDrugs0.000description1
- AUJAAMXYEHZSLP-UHFFFAOYSA-N4-ethenyloxolan-2-oneChemical compoundC=CC1COC(=O)C1AUJAAMXYEHZSLP-UHFFFAOYSA-N0.000description1
- HBAQYPYDRFILMT-UHFFFAOYSA-N8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-oneChemical classC1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=OHBAQYPYDRFILMT-UHFFFAOYSA-N0.000description1
- 208000024827Alzheimer diseaseDiseases0.000description1
- 206010003210ArteriosclerosisDiseases0.000description1
- 208000031648Body Weight ChangesDiseases0.000description1
- WKBOTKDWSSQWDR-UHFFFAOYSA-NBromine atomChemical compound[Br]WKBOTKDWSSQWDR-UHFFFAOYSA-N0.000description1
- 206010008342Cervix carcinomaDiseases0.000description1
- IGXWBGJHJZYPQS-SSDOTTSWSA-ND-LuciferinChemical compoundOC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1IGXWBGJHJZYPQS-SSDOTTSWSA-N0.000description1
- 201000004624DermatitisDiseases0.000description1
- YZCKVEUIGOORGS-OUBTZVSYSA-NDeuteriumChemical compound[2H]YZCKVEUIGOORGS-OUBTZVSYSA-N0.000description1
- KCXVZYZYPLLWCC-UHFFFAOYSA-NEDTAChemical compoundOC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=OKCXVZYZYPLLWCC-UHFFFAOYSA-N0.000description1
- 102100024785Fibroblast growth factor 2Human genes0.000description1
- 108090000379Fibroblast growth factor 2Proteins0.000description1
- 208000010412GlaucomaDiseases0.000description1
- WQZGKKKJIJFFOK-GASJEMHNSA-NGlucoseNatural productsOC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1OWQZGKKKJIJFFOK-GASJEMHNSA-N0.000description1
- 208000003084Graves OphthalmopathyDiseases0.000description1
- HTTJABKRGRZYRN-UHFFFAOYSA-NHeparinChemical compoundOC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1HTTJABKRGRZYRN-UHFFFAOYSA-N0.000description1
- 101001034652Homo sapiens Insulin-like growth factor 1 receptorProteins0.000description1
- UFHFLCQGNIYNRP-UHFFFAOYSA-NHydrogenChemical compound[H][H]UFHFLCQGNIYNRP-UHFFFAOYSA-N0.000description1
- 206010059282Metastases to central nervous systemDiseases0.000description1
- 241001529936MurinaeSpecies0.000description1
- 241000872931Myoporum sandwicenseSpecies0.000description1
- LFTLOKWAGJYHHR-UHFFFAOYSA-NN-methylmorpholine N-oxideChemical compoundCN1(=O)CCOCC1LFTLOKWAGJYHHR-UHFFFAOYSA-N0.000description1
- 208000012247Oligodendroglial tumorDiseases0.000description1
- 201000010133OligodendrogliomaDiseases0.000description1
- 206010033128Ovarian cancerDiseases0.000description1
- 206010061535Ovarian neoplasmDiseases0.000description1
- 206010061902Pancreatic neoplasmDiseases0.000description1
- 108091005804PeptidasesProteins0.000description1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-NPhenolChemical compoundOC1=CC=CC=C1ISWSIDIOOBJBQZ-UHFFFAOYSA-N0.000description1
- 229940122907Phosphatase inhibitorDrugs0.000description1
- 239000004365ProteaseSubstances0.000description1
- 229940124158Protease/peptidase inhibitorDrugs0.000description1
- 201000004681PsoriasisDiseases0.000description1
- 208000017442Retinal diseaseDiseases0.000description1
- 206010038923RetinopathyDiseases0.000description1
- 102100037486Reverse transcriptase/ribonuclease HHuman genes0.000description1
- 208000034189SclerosisDiseases0.000description1
- 108010087230SincalideProteins0.000description1
- 206010052779Transplant rejectionsDiseases0.000description1
- GLNADSQYFUSGOU-GPTZEZBUSA-JTrypan blueChemical compound[Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1NGLNADSQYFUSGOU-GPTZEZBUSA-J0.000description1
- 102000004142TrypsinHuman genes0.000description1
- 108090000631TrypsinProteins0.000description1
- 208000025865UlcerDiseases0.000description1
- 208000006105Uterine Cervical NeoplasmsDiseases0.000description1
- 230000005856abnormalityEffects0.000description1
- 238000002835absorbanceMethods0.000description1
- WETWJCDKMRHUPV-UHFFFAOYSA-Nacetyl chlorideChemical compoundCC(Cl)=OWETWJCDKMRHUPV-UHFFFAOYSA-N0.000description1
- 206010002224anaplastic astrocytomaDiseases0.000description1
- 230000001348anti-gliomaEffects0.000description1
- 229940034982antineoplastic agentDrugs0.000description1
- 230000006907apoptotic processEffects0.000description1
- 239000007864aqueous solutionSubstances0.000description1
- 208000011775arteriosclerosis diseaseDiseases0.000description1
- 208000006673asthmaDiseases0.000description1
- 125000004429atomChemical group0.000description1
- 208000010668atopic eczemaDiseases0.000description1
- 230000004888barrier functionEffects0.000description1
- 230000008901benefitEffects0.000description1
- 230000000903blocking effectEffects0.000description1
- 239000008280bloodSubstances0.000description1
- 210000004369bloodAnatomy0.000description1
- 230000004579body weight changeEffects0.000description1
- GDTBXPJZTBHREO-UHFFFAOYSA-NbromineSubstancesBrBrGDTBXPJZTBHREO-UHFFFAOYSA-N0.000description1
- 229910052794bromiumInorganic materials0.000description1
- 210000001715carotid arteryAnatomy0.000description1
- 230000015556catabolic processEffects0.000description1
- 230000003197catalytic effectEffects0.000description1
- 238000010609cell counting kit-8 assayMethods0.000description1
- 230000010261cell growthEffects0.000description1
- 230000006037cell lysisEffects0.000description1
- 230000004663cell proliferationEffects0.000description1
- 238000005119centrifugationMethods0.000description1
- 201000010881cervical cancerDiseases0.000description1
- 239000003153chemical reaction reagentSubstances0.000description1
- 238000002512chemotherapyMethods0.000description1
- RNFNDJAIBTYOQL-UHFFFAOYSA-Nchloral hydrateChemical compoundOC(O)C(Cl)(Cl)ClRNFNDJAIBTYOQL-UHFFFAOYSA-N0.000description1
- 229960002327chloral hydrateDrugs0.000description1
- 239000012230colorless oilSubstances0.000description1
- 238000007887coronary angioplastyMethods0.000description1
- 125000004122cyclic groupChemical group0.000description1
- 125000001995cyclobutyl groupChemical group[H]C1([H])C([H])([H])C([H])(*)C1([H])[H]0.000description1
- 125000000582cycloheptyl groupChemical group[H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H]0.000description1
- 125000000113cyclohexyl groupChemical group[H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H]0.000description1
- 125000000640cyclooctyl groupChemical group[H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H]0.000description1
- 125000001511cyclopentyl groupChemical group[H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H]0.000description1
- 125000001559cyclopropyl groupChemical group[H]C1([H])C([H])([H])C1([H])*0.000description1
- 238000006731degradation reactionMethods0.000description1
- 150000001975deuteriumChemical group0.000description1
- 238000010790dilutionMethods0.000description1
- 239000012895dilutionSubstances0.000description1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-Idipotassium trisodium dihydrogen phosphate hydrogen phosphate dichlorideChemical compoundP(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+]LOKCTEFSRHRXRJ-UHFFFAOYSA-I0.000description1
- 231100000673dose–response relationshipToxicity0.000description1
- 230000007783downstream signalingEffects0.000description1
- 239000003596drug targetSubstances0.000description1
- 238000002651drug therapyMethods0.000description1
- 239000012636effectorSubstances0.000description1
- 230000012202endocytosisEffects0.000description1
- 208000023437ependymal tumorDiseases0.000description1
- 230000002496gastric effectEffects0.000description1
- PCHJSUWPFVWCPO-UHFFFAOYSA-NgoldChemical compound[Au]PCHJSUWPFVWCPO-UHFFFAOYSA-N0.000description1
- 230000012010growthEffects0.000description1
- 230000009036growth inhibitionEffects0.000description1
- 125000001188haloalkyl groupChemical group0.000description1
- 125000005843halogen groupChemical group0.000description1
- 229960002897heparinDrugs0.000description1
- 229920000669heparinPolymers0.000description1
- 208000027671high grade ependymomaDiseases0.000description1
- 229910052739hydrogenInorganic materials0.000description1
- 239000001257hydrogenSubstances0.000description1
- 238000003384imaging methodMethods0.000description1
- 230000031146intracellular signal transductionEffects0.000description1
- PNDPGZBMCMUPRI-UHFFFAOYSA-NiodineChemical compoundIIPNDPGZBMCMUPRI-UHFFFAOYSA-N0.000description1
- 208000017169kidney diseaseDiseases0.000description1
- 210000005240left ventricleAnatomy0.000description1
- 231100001231less toxicToxicity0.000description1
- 231100000225lethalityToxicity0.000description1
- 229920005610ligninPolymers0.000description1
- 238000010859live-cell imagingMethods0.000description1
- 210000004185liverAnatomy0.000description1
- 201000007270liver cancerDiseases0.000description1
- 208000014018liver neoplasmDiseases0.000description1
- 239000006166lysateSubstances0.000description1
- 230000036210malignancyEffects0.000description1
- 208000015486malignant pancreatic neoplasmDiseases0.000description1
- 230000001404mediated effectEffects0.000description1
- 230000002503metabolic effectEffects0.000description1
- 230000004060metabolic processEffects0.000description1
- IZDROVVXIHRYMH-UHFFFAOYSA-Nmethanesulfonic anhydrideChemical compoundCS(=O)(=O)OS(C)(=O)=OIZDROVVXIHRYMH-UHFFFAOYSA-N0.000description1
- 239000011259mixed solutionSubstances0.000description1
- 238000002156mixingMethods0.000description1
- 210000000653nervous systemAnatomy0.000description1
- 210000004498neuroglial cellAnatomy0.000description1
- 229910000489osmium tetroxideInorganic materials0.000description1
- 201000002528pancreatic cancerDiseases0.000description1
- 208000008443pancreatic carcinomaDiseases0.000description1
- 239000008188pelletSubstances0.000description1
- 125000005010perfluoroalkyl groupChemical group0.000description1
- 125000003367polycyclic groupChemical group0.000description1
- 239000002244precipitateSubstances0.000description1
- 208000029340primitive neuroectodermal tumorDiseases0.000description1
- 230000017854proteolysisEffects0.000description1
- 238000001959radiotherapyMethods0.000description1
- 230000001105regulatory effectEffects0.000description1
- 238000002271resectionMethods0.000description1
- 208000037803restenosisDiseases0.000description1
- 206010039073rheumatoid arthritisDiseases0.000description1
- 238000013207serial dilutionMethods0.000description1
- 230000001568sexual effectEffects0.000description1
- 231100001251short-term toxicityToxicity0.000description1
- 102000034285signal transducing proteinsHuman genes0.000description1
- 108091006024signal transducing proteinsProteins0.000description1
- IZTQOLKUZKXIRV-YRVFCXMDSA-NsincalideChemical compoundC([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1IZTQOLKUZKXIRV-YRVFCXMDSA-N0.000description1
- 239000012354sodium borodeuterideSubstances0.000description1
- AKHNMLFCWUSKQB-UHFFFAOYSA-Lsodium thiosulfateChemical class[Na+].[Na+].[O-]S([O-])(=O)=SAKHNMLFCWUSKQB-UHFFFAOYSA-L0.000description1
- 238000010186stainingMethods0.000description1
- 239000013589supplementSubstances0.000description1
- 238000001356surgical procedureMethods0.000description1
- 210000001519tissueAnatomy0.000description1
- YFDSDPIBEUFTMI-UHFFFAOYSA-NtribromoethanolChemical compoundOCC(Br)(Br)BrYFDSDPIBEUFTMI-UHFFFAOYSA-N0.000description1
- 229950004616tribromoethanolDrugs0.000description1
- ITMCEJHCFYSIIV-UHFFFAOYSA-MtriflateChemical compound[O-]S(=O)(=O)C(F)(F)FITMCEJHCFYSIIV-UHFFFAOYSA-M0.000description1
- 125000000876trifluoromethoxy groupChemical groupFC(F)(F)O*0.000description1
- 239000012588trypsinSubstances0.000description1
- 230000004565tumor cell growthEffects0.000description1
- 208000001072type 2 diabetes mellitusDiseases0.000description1
- 229940121358tyrosine kinase inhibitorDrugs0.000description1
- 239000005483tyrosine kinase inhibitorSubstances0.000description1
- 150000004917tyrosine kinase inhibitor derivativesChemical class0.000description1
- 231100000397ulcerToxicity0.000description1
- 238000005292vacuum distillationMethods0.000description1
- 238000010200validation analysisMethods0.000description1
- 238000005406washingMethods0.000description1
- 230000004580weight lossEffects0.000description1
Images







Classifications
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Translated fromChineseDescription
Translated fromChinese技术领域technical field
本发明涉及新化合物,以及一种新型取代苦鬼臼毒素衍生物抑制胰岛素样生长因子-1受体IGF-1R,用于治疗IGF-1R依赖性疾病尤其是癌症的用途。The present invention relates to novel compounds and the use of a novel substituted picropodophyllotoxin derivative for inhibiting insulin-like growth factor-1 receptor IGF-1R for treating IGF-1R-dependent diseases, especially cancer.
背景技术Background technique
原发性神经系统恶性肿瘤的发生呈逐年递增趋势,胶质瘤是指起源神经胶质细胞的脑肿瘤,包括星形细胞肿瘤,少突胶质细胞肿瘤和室管膜肿瘤(Muir CS,Storm et al.,Cancer Surv,19-20:369-392,1994;Louis DN et al.,Acta Neuropathol,131:803-820,2016),是最常见的原发性肿瘤类型。以替莫唑胺为代表的化学药物疗法是当前治疗高级别胶质瘤的金标准,但仅能很有限地延长患者生存期。疗效更好的靶向药物治疗迄今尚未在胶质瘤中获得成功,因为绝大部分靶向药无法透过血脑屏障。有文献报道苦鬼臼毒素(Picropodophyllin,PPP)可通过抑制胰岛素样生长因子-1受体IGF-1R来抑制接种至大鼠脑部的人源胶质瘤细胞的增殖,提示它可能具有透过血脑屏障的特性(Yin S,et al.,Neuro-Oncology,12:19-27,2010)。The incidence of primary nervous system malignancies is increasing year by year. Glioma refers to brain tumors originating from glial cells, including astrocytic tumors, oligodendroglial tumors and ependymal tumors (Muir CS, Storm et al. al., Cancer Surv, 19-20:369-392, 1994; Louis DN et al., Acta Neuropathol, 131:803-820, 2016), is the most common primary tumor type. Chemotherapy, represented by temozolomide, is the current gold standard for the treatment of high-grade gliomas, but it can only prolong patient survival to a very limited extent. Better targeted drug therapy has so far not been successful in glioma because most targeted drugs cannot penetrate the blood-brain barrier. It has been reported in the literature that Picropodophyllin (PPP) can inhibit the proliferation of human glioma cells inoculated into the rat brain by inhibiting the insulin-like growth factor-1 receptor IGF-1R, suggesting that it may have the ability to penetrate Properties of the blood-brain barrier (Yin S, et al., Neuro-Oncology, 12:19-27, 2010).
苦鬼臼毒素是一种环木酯素类化合物,具有下述结构:Picropodophyllotoxin is a cyclic lignin compound with the following structure:

苦鬼臼毒素被认为是一种特异性的胰岛素样生长因子-1受体(insulin-likegrowth factor-1receptor,IGF-1R)酪氨酸激酶抑制剂,被用来治疗IGF-1R导致的疾病包括多种癌症,动脉硬化,牛皮癣,冠状血管成形术后再狭窄(WO02/102804),2型糖尿病,肾病,视网膜病,青光眼,甲状腺眼病(WO 2007/097707),风湿性关节炎,溃疡,多发性硬化,阿尔茨海默症,哮喘,湿疹,移植后排斥(WO 2009/157858)。苦鬼臼毒素通过阻断IGF-1R介导的细胞内信号传导通路达到抑制肿瘤细胞生长并促进其凋亡的目的(Girnita A,et al.,Cancer Res,64:236-242,2004)。多种肿瘤细胞中IGF-1R表达量显著升高,因而苦鬼臼毒素可以靶向抑制肿瘤细胞增殖,而对正常细胞毒副作用较小。Picropodophyllotoxin is considered to be a specific insulin-like growth factor-1 receptor (IGF-1R) tyrosine kinase inhibitor and is used to treat diseases caused by IGF-1R including Various cancers, arteriosclerosis, psoriasis, restenosis after coronary angioplasty (WO02/102804),
尽管苦鬼臼毒素特异性高,毒副作用小,但我们在小鼠中的研究表明它在体内代谢很快,并且不易透过血脑屏障,也许是其人体临床试验中效果不佳的重要原因。因此,需要寻找体内代谢更慢、体内药效更高、并更易于透过血脑屏障的化合物,用于治疗IGF-1R依赖性疾病尤其是胶质瘤和易发生脑转移的癌症。Although podophyllotoxin is highly specific and has few side effects, our studies in mice show that it is rapidly metabolized in the body and does not easily penetrate the blood-brain barrier, which may be an important reason for its poor effect in human clinical trials. . Therefore, it is necessary to find compounds with slower metabolism in vivo, higher drug efficacy in vivo, and easier to cross the blood-brain barrier for the treatment of IGF-1R-dependent diseases, especially gliomas and cancers prone to brain metastases.
发明内容SUMMARY OF THE INVENTION
本发明要解决的技术问题是通过设计合成新颖取代苦鬼臼毒素衍生物,延长IGF-1R抑制剂在体内的半衰期,提高其透过血脑屏障的能力,增强其抗肿瘤药效。The technical problem to be solved by the present invention is to design and synthesize novel derivatives of podophyllotoxin to prolong the half-life of the IGF-1R inhibitor in vivo, improve its ability to pass through the blood-brain barrier, and enhance its anti-tumor efficacy.
为了解决上述技术问题,本发明提供如下的技术方案:In order to solve the above-mentioned technical problems, the present invention provides the following technical solutions:
本发明提供的取代苦鬼臼毒素衍生物具有通式Ⅰ所示结构:The substituted picropodophyllotoxin derivative provided by the present invention has the structure shown in general formula I:

及其光学异构体或其作为抗肿瘤药可接受的盐(包括碱金属盐、碱土金属盐、酸加成盐、碱加成盐和烷基化盐),其中:and their optical isomers or their salts (including alkali metal salts, alkaline earth metal salts, acid addition salts, base addition salts and alkylated salts) acceptable as antineoplastic agents, wherein:
R选自F、-OH、CH3CO-、CH3COO-、CH3CH2COO-、CH3CH2CH2COO-或取代吡唑环类衍生物如通式Ⅱ所示:R is selected from F, -OH, CH3 CO-, CH3 COO-, CH3 CH2 COO-, CH3 CH2 CH2 COO- or substituted pyrazole ring derivatives as shown in general formula II:

式中:where:
B为
R3选自H、C1-C4烷基中的任一种、卤代的C1-C4烷基中的任一种、C1-C4烷氧基中的任一种、卤代的C1-C4烷氧基中的任一种、无取代或取代的五元或六元芳基;R3 is selected from H, any one of C1 -C4 alkyl, any one of halogenated C1 -C4 alkyl, any one of C1 -C4 alkoxy, halogen Any one of substituted C1 -C4 alkoxy, unsubstituted or substituted five- or six-membered aryl;
R4选自H、卤素、C1-C3烷基中的任一种、卤代的C1-C3烷基中的任一种,取代在吡唑环的3或4位;R4 is selected from H, halogen, any one of C1 -C3 alkyl, any one of halogenated C1 -C3 alkyl, and is substituted at the 3 or 4 position of the pyrazole ring;
R5、R6选自H、卤素、硝基、氨基、氰基、C1-C4烷基中的任一种、卤代的C1-C4烷基中的任一种、C1-C4烷氧基中的任一种、卤代的C1-C4烷氧基中的任一种,R5与R6协同或独立取代在通式Ⅱ中间五元环的不同位置;R5 , R6 are selected from H, halogen, nitro, amino, cyano, any one of C1 -C4 alkyl, any one of halogenated C1 -C4 alkyl, C1 Any one of -C4 alkoxy, any one of halogenated C1 -C4 alkoxy, R5 and R6 are substituted synergistically or independently at different positions in the middle five-membered ring of general formula II;
X选自O、S、N(Rh),其中Rh选自H、C1-C5烷基中的任一种、卤代的C1-C5烷基中的任一种、C1-C5烷氧基中的任一种、卤代的C1-C5烷氧基中的任一种;X is selected from O, S, N(Rh ), wherein Rh is selected from H, any of C1 -C5 alkyl, any of halogenated C1 -C5 alkyl, C Any of1 -C5 alkoxy, any of halogenated C1 -C5 alkoxy;
L选自C和N。L is selected from C and N.
其中,通式Ⅱ表示的取代吡唑环类衍生物基团与的连接位置可为R3、R4、R5、R6及B中任意可连接的碳。Wherein, the linking position of the substituted pyrazole ring derivative group represented by the general formula II can be any connectable carbon among R3 , R4 , R5 , R6 and B.
作为优选方案,通式Ⅰ所示结构化合物中至少存在一个氢被氘取代。As a preferred solution, at least one hydrogen in the compound of formula I is replaced by deuterium.
作为优选方案,该小分子抑制剂为通式Ш所示结构化合物及其光学异构体或其作为抗肿瘤药可接受的盐:As a preferred solution, the small molecule inhibitor is the structural compound represented by the general formula Ш and its optical isomer or its salt acceptable as an antitumor drug:

本发明还提供上述小分子抑制剂用于制备药物的用途,该药物用于预防和治疗不同类型癌症,例如恶性黑色素瘤;原发性神经外胚层瘤;神经胶质瘤,如恶性胶质瘤及星形胶质细胞瘤;肺癌;前列腺癌;乳腺癌;骨髓增生性及淋巴组织增生性疾病,如白血病、淋巴瘤;消化道肿瘤,如胃癌、结直肠癌、肝癌及胰腺癌;妇科癌症,如卵巢癌及宫颈癌。鉴于本发明所述的化合物具有良好的血脑屏障通透性,尤其适合治疗由上述原发肿瘤向脑部转移后形成的肿瘤。The present invention also provides the use of the above-mentioned small molecule inhibitor for preparing a medicament for preventing and treating different types of cancer, such as malignant melanoma; primary neuroectodermal tumor; glioma, such as malignant glioma and astrocytoma; lung cancer; prostate cancer; breast cancer; myeloproliferative and lymphoproliferative diseases such as leukemia, lymphoma; digestive tract tumors such as gastric, colorectal, liver and pancreatic cancer; gynecological cancers , such as ovarian cancer and cervical cancer. In view of the good permeability of the blood-brain barrier, the compounds described in the present invention are especially suitable for the treatment of tumors formed after the above-mentioned primary tumors metastasize to the brain.
至于不完全依赖IGF-1R的肿瘤,本发明化合物可以增强其它抗癌药物的效果。因此,本发明也涉及小分子抑制剂与其它抗癌药物(包括但不限于化疗药物、靶向药物、抗体药物、纳米药物、蛋白降解靶向嵌合体、核酸药物等)或治疗方法(如手术切除、放疗等)的联合治疗。As for tumors that are not completely dependent on IGF-1R, the compounds of the present invention can enhance the effect of other anticancer drugs. Therefore, the present invention also relates to small molecule inhibitors and other anticancer drugs (including but not limited to chemotherapeutic drugs, targeted drugs, antibody drugs, nano-drugs, protein degradation targeting chimeras, nucleic acid drugs, etc.) or treatment methods (such as surgery resection, radiotherapy, etc.)
本发明特别涉及小分子抑制剂用于治疗脑部胶质瘤的药物制备中的用途。The present invention particularly relates to the use of small molecule inhibitors in the preparation of medicaments for treating brain gliomas.
本发明涉及的术语说明如下:The terms involved in the present invention are explained as follows:
本发明所用术语“芳基”是指5到12个碳原子的全碳单环或稠合多环基团,具有完全共轭的π电子系统。芳环的非限制性实例有:苯环、萘环和蒽环。芳环可以是无取代或取代的。芳环的取代基选自卤素、硝基、氨基、C1-C6烷基、C1-C6烷氧基、卤代C1-C6烷基、卤代C1-C6烷氧基、C3-C6环烷基、卤代C3-C6环烷基。The term "aryl" as used herein refers to an all-carbon monocyclic or fused polycyclic group of 5 to 12 carbon atoms, with a fully conjugated pi electron system. Non-limiting examples of aromatic rings are: benzene, naphthalene, and anthracene. Aromatic rings can be unsubstituted or substituted. The substituent of the aromatic ring is selected from halogen, nitro, amino, C1 -C6 alkyl, C1 -C6 alkoxy, halogenated C1 -C6 alkyl, halogenated C1 -C6 alkoxy group, C3 -C6 cycloalkyl, halogenated C3 -C6 cycloalkyl.
本发明所用术语“环烷基”是指具有3-6个碳原子的饱和单环碳环,除非指明不同数目的原子。“环烷基”包括例如环丙基、环丁基、环戊基、环己基、环庚基和环辛基。环烷基可以是无取代或取代的。环烷基的取代可任选在任何可利用碳上被一个或多个选自烷氧基、卤素和卤代烷基,如全氟烷基的取代基取代。The term "cycloalkyl" as used herein refers to a saturated monocyclic carbocyclic ring having 3 to 6 carbon atoms, unless a different number of atoms is indicated. "Cycloalkyl" includes, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl. Cycloalkyl groups can be unsubstituted or substituted. Substitution of cycloalkyl groups can be optionally substituted on any available carbon with one or more substituents selected from alkoxy, halo, and haloalkyl, such as perfluoroalkyl.
本发明所用术语“烷氧基”是指-O-烷基基团,其中烷基如上所定义。本发明所用“烷氧基”的实例包括但不限于甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基和叔丁氧基。烷氧基可以是无取代或取代的。烷氧基可任选被卤素取代一次或多次,如三氟甲氧基。The term "alkoxy" as used herein refers to an -O-alkyl group, wherein alkyl is as defined above. Examples of "alkoxy" as used herein include, but are not limited to, methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, and t-butoxy. Alkoxy groups can be unsubstituted or substituted. The alkoxy group can be optionally substituted one or more times with halogen, such as trifluoromethoxy.
本发明所用术语“卤素”表示氟、氯、溴或碘,优选为氟或氯。The term "halogen" as used herein means fluorine, chlorine, bromine or iodine, preferably fluorine or chlorine.
本发明具有如下技术优势:The present invention has the following technical advantages:
1、新颖的取代苦鬼臼毒素衍生物结构;1. Novel structure of substituted picropodophyllotoxin derivatives;
2、本发明中涉及的小分子抑制剂具有良好的血脑屏障通透性。2. The small molecule inhibitor involved in the present invention has good blood-brain barrier permeability.
3、本发明中涉及的小分子抑制剂在机体内具有较长的半衰期。3. The small molecule inhibitor involved in the present invention has a longer half-life in the body.
附图说明Description of drawings
图1是化合物10及其手性分离化合物体外药效检测结果图;Fig. 1 is a graph showing the results of in vitro pharmacodynamic testing of
图2为化合物7和化合物11对U87肿瘤微球处理24小时后收样Western blot检测结果图,检测化合物7和化合物11对其靶标蛋白IGF1R与下游信号蛋白AKT的作用;Figure 2 shows the results of Western blot detection of U87 tumor microspheres treated with
图3为给药3小时后小鼠脑组织样的Western blot检测图,其中,给药方式为以100mg/kg剂量灌胃,检测药物对小鼠血脑屏障的透过性;Fig. 3 is the Western blot detection chart of mouse
图4为给药后不同时间段药物在小鼠血浆(上)和脑组织(下)中浓度的动态变化实验结果图,其中,给药方式为以50mg/kg剂量灌胃,检测药物在小鼠血浆和脑组织中的稳定性;Figure 4 is a graph showing the experimental results of the dynamic changes of drug concentrations in mouse plasma (top) and brain tissue (bottom) at different time periods after administration. Stability in murine plasma and brain tissue;
图5为不同剂量给药后小鼠的体重变化图,展示药物对小鼠的短期毒性试验;Figure 5 is a graph of the body weight change of mice after administration of different doses, showing the short-term toxicity test of the drug on mice;
图6为化合物11单药及其与替莫唑胺联用对原位接种于裸鼠脑部的U87-Luc胶质瘤细胞增殖的抑制作用(左)和对小鼠生存期的影响(右)结果图;Figure 6 shows the results of the inhibitory effect of compound 11 and its combination with temozolomide on the proliferation of U87-Luc glioma cells orthotopically inoculated into the brain of nude mice (left) and the effect on mouse survival (right). ;
图7为化合物11对接种于裸鼠的MDA-MB-231-Luc乳腺癌细胞脑转移的抑制作用结果图;Figure 7 is a graph showing the results of the inhibitory effect of compound 11 on brain metastasis of MDA-MB-231-Luc breast cancer cells inoculated in nude mice;
图8为化合物11单药及其与PD-1抗体联用对免疫正常小鼠中LLC肺癌细胞增殖的抑制作用(左)和对小鼠生存期的影响(右)结果图。Figure 8 is a graph showing the inhibitory effect of compound 11 alone and its combination with PD-1 antibody on the proliferation of LLC lung cancer cells in immunized normal mice (left) and the effect on mouse survival time (right).
具体实施方式Detailed ways
下面通过实施例来说明本发明的可实施性,本领域的技术人员应当理解,根据现有技术的教导,对相应的技术特征进行修改或替换,仍然属于本发明要求保护的范围。The practicability of the present invention will be described below through examples. Those skilled in the art should understand that, according to the teachings of the prior art, modification or replacement of corresponding technical features still belongs to the scope of protection of the present invention.
实施例1、相对-(5R,5aS,8aR,9R)-2,2-二氟-9-羟基-5-(3,4,5-三甲氧基苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮(化合物1)Example 1. Relative-(5R,5aS,8aR,9R)-2,2-difluoro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9 -Tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one (Compound 1)

步骤1)、4-乙烯基二氢呋喃-2(3H)-酮(中间体1.1)的合成:Step 1), the synthesis of 4-vinyldihydrofuran-2(3H)-one (intermediate 1.1):
在2-丁烯-1,4-二醇(206.4g,2.34mol,1.0eq)和原乙酸三乙酯(546.7g,3.4mol,1.4eq)的混合物中加入催化对苯二酚(54.00g,0.49mol,0.2eq),混合物加热至120℃。乙醇被持续蒸馏出来,直到无更多乙醇产生时,将反应温度升高至150℃,搅拌反应混合物48小时。通过真空蒸馏(70-75℃,2-3mmHg)收集得到中间体1.1,为无色油状物(170.0g,收率65%)。1HNMR(400MHz,CDCl3)δ(ppm)2.36(dd,J=17.4Hz,8.7Hz,1H),2.69(dd,J=17.7Hz,8.4Hz,1H),3.19-3.29(m,1H),4.01-4.14(m,1H),4.43-4.47(m,1H),5.17-5.23(m,2H),5.75-5.84(m,1H)。To a mixture of 2-butene-1,4-diol (206.4g, 2.34mol, 1.0eq) and triethyl orthoacetate (546.7g, 3.4mol, 1.4eq) was added catalytic hydroquinone (54.00g) , 0.49mol, 0.2eq), the mixture was heated to 120 °C. Ethanol was continuously distilled out until no more ethanol was produced, the reaction temperature was raised to 150°C and the reaction mixture was stirred for 48 hours. Intermediate 1.1 was collected by vacuum distillation (70-75°C, 2-3 mmHg) as a colorless oil (170.0 g, 65% yield).1 HNMR (400MHz, CDCl3 ) δ (ppm) 2.36 (dd, J=17.4Hz, 8.7Hz, 1H), 2.69 (dd, J=17.7Hz, 8.4Hz, 1H), 3.19-3.29 (m, 1H) , 4.01-4.14 (m, 1H), 4.43-4.47 (m, 1H), 5.17-5.23 (m, 2H), 5.75-5.84 (m, 1H).
步骤2)、相对-(3S,4R)-3-(羟基(3,4,5-三甲氧基苯基)甲基)-4-乙烯基二氢呋喃-2(3H)-酮(中间体1.2)的合成Step 2), relative-(3S,4R)-3-(hydroxy(3,4,5-trimethoxyphenyl)methyl)-4-vinyldihydrofuran-2(3H)-one (intermediate 1.2) Synthesis
将中间体1.1(170.0g,1.52mol,1.0eq)和四氢呋喃(1500ml)混合,氮气保护,在-78℃下搅拌,滴加二异丙基氨基锂(2.0M,834ml,1.67mol,1.1eq),并搅拌反应混合物30分钟。在相同条件下,滴加3,4,5-三甲氧基苯甲醛(327.6g,1.67mol,1.1eq)与四氢呋喃(1500ml)的混合溶液,再搅拌3小时,然后逐渐加热到环境温度过夜。反应混合物冷却至-78℃,用饱和NH4Cl淬灭。由此产生的混合物用乙酸乙酯萃取,用饱和食盐水洗涤,无水硫酸钠干燥,硅胶柱层析(200-300硅胶,石油醚:乙酸乙酯=5/1-1/1)纯化得到中间体1.2(190.1g,收率41%),为浅黄色固体。1HNMR(400MHz,CDCl3)δ(ppm)2.75-2.80(m,1H),2.91-2.96(m,0.5H),3.26-3.31(m,0.5H),3.84(s,3H),3.89(s,6H),3.92(t,1H),4.30-4.40(m,1H),4.86-4.93(m,2H),6.59(s,1H),6.61(s,1H)。Intermediate 1.1 (170.0g, 1.52mol, 1.0eq) and tetrahydrofuran (1500ml) were mixed, under nitrogen protection, stirred at -78°C, and added dropwise lithium diisopropylamide (2.0M, 834ml, 1.67mol, 1.1eq) ) and stirred the reaction mixture for 30 minutes. Under the same conditions, a mixed solution of 3,4,5-trimethoxybenzaldehyde (327.6g, 1.67mol, 1.1eq) and tetrahydrofuran (1500ml) was added dropwise, stirred for another 3 hours, and then gradually heated to ambient temperature overnight. The reaction mixture was cooled to -78°C and quenched with saturatedNH4Cl . The resulting mixture was extracted with ethyl acetate, washed with saturated brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (200-300 silica gel, petroleum ether:ethyl acetate=5/1-1/1) to obtain Intermediate 1.2 (190.1 g, 41% yield) as a pale yellow solid.1 HNMR (400MHz, CDCl3 )δ(ppm) 2.75-2.80(m, 1H), 2.91-2.96(m, 0.5H), 3.26-3.31(m, 0.5H), 3.84(s, 3H), 3.89( s, 6H), 3.92 (t, 1H), 4.30-4.40 (m, 1H), 4.86-4.93 (m, 2H), 6.59 (s, 1H), 6.61 (s, 1H).
步骤3)、相对-(3R,4R)-3-((R)-(2,2-二氟-6-羟基苯并[d][1,3]二氧杂-5-基)(3,4,5-三甲氧基苯基)甲基)-4-乙烯基二氢呋喃-2(3H)-酮(中间体1.3)的合成:Step 3), relative-(3R,4R)-3-((R)-(2,2-difluoro-6-hydroxybenzo[d][1,3]dioxan-5-yl)(3 Synthesis of ,4,5-trimethoxyphenyl)methyl)-4-vinyldihydrofuran-2(3H)-one (intermediate 1.3):
将中间体1.2(106.7g,0.41mol,1.0eq),5-酚羟基-2,2-二氟-1,3-苯并二噁唑(107.6g,0.61mol,1.5eq)和二氯甲烷(2000ml)混合搅拌,加入FeCl3(34.06g,0.20mol,0.5eq),反应混合物加热至40℃2-3小时,用饱和NaHCO3淬灭,水相进一步以二氯甲烷萃取。合并后的有机相用饱和食盐水洗涤,无水硫酸钠干燥,硅胶柱层析(200-300硅胶,石油醚:乙酸乙酯=5/1-1/1)纯化得到中间体1.3(153.4g,收率45%),为米色固体。1HNMR(400MHz,CDCl3)δ(ppm)3.02-3.06(m,1H),3.19-3.21(m,1H),3.83(s,6H),3.88(s,3H),4.04-4.07(m,1H),4.25-4.29(m,1H),4.73-4.75(d,J=4.8Hz,1H),5.14-5.22(m,2H),5.55(brs,1H),5.80-5.84(m,1H),6.51-6.59(m,2H),6.82-6.96(m,1H),7.29(s,1H)。Intermediate 1.2 (106.7g, 0.41mol, 1.0eq), 5-phenolhydroxy-2,2-difluoro-1,3-benzobisoxazole (107.6g, 0.61mol, 1.5eq) and dichloromethane (2000ml) was mixed and stirred, FeCl3 (34.06g, 0.20mol, 0.5eq) was added, the reaction mixture was heated to 40° C. for 2-3 hours, quenched with saturated NaHCO3 , and the aqueous phase was further extracted with dichloromethane. The combined organic phases were washed with saturated brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (200-300 silica gel, petroleum ether: ethyl acetate=5/1-1/1) to obtain Intermediate 1.3 (153.4 g) , yield 45%) as a beige solid.1 HNMR(400MHz, CDCl3 )δ(ppm) 3.02-3.06(m,1H), 3.19-3.21(m,1H), 3.83(s,6H), 3.88(s,3H), 4.04-4.07(m, 1H), 4.25-4.29(m, 1H), 4.73-4.75(d, J=4.8Hz, 1H), 5.14-5.22(m, 2H), 5.55(brs, 1H), 5.80-5.84(m, 1H) , 6.51-6.59(m, 2H), 6.82-6.96(m, 1H), 7.29(s, 1H).
步骤4)、相对-6-((R)-((3R,4R)-2-氧代-4-乙烯基四氢呋喃-3-基)(3,4,5-三甲氧基苯基)甲基)-2,2-二氟苯并[d][1,3]二氧杂-5-基三氟甲磺酸酯(中间体1.4)的合成:Step 4), relative-6-((R)-((3R,4R)-2-oxo-4-vinyltetrahydrofuran-3-yl)(3,4,5-trimethoxyphenyl)methyl )-2,2-difluorobenzo[d][1,3]dioxan-5-yl trifluoromethanesulfonate (intermediate 1.4):
在持续搅拌的中间体1.3(120.0g,0.26mol,1.0eq)和二氯甲烷(1500ml)中加入三乙胺(52.62g,0.52mol,2.0eq),然后在10℃温度下滴加三氟甲磺酸酐(110.0g,0.39mol,1.5eq),混合物在相同温度下继续搅拌混匀30分钟。反应用饱和NaHCO3淬灭,水相进一步以二氯甲烷萃取。合并后的有机相依次用2N HCl和饱和食盐水洗涤,无水硫酸钠干燥,硅胶柱层析(200-300硅胶,石油醚:乙酸乙酯=6/1-3/1)纯化得到中间体1.4(120.0g,收率61%),为白色固体。1HNMR(400MHz,CDCl3)δ(ppm)2.97-3.06(m,1H),3.13-3.17(m,1H),3.84(s,6H),3.85(s,3H),4.03-4.08(m,1H),4.37-4.41(m,1H),4.64-4.66(d,J=8.3Hz,1H),5.08-5.20(dt,2H),5.71-5.80(m,1H),6.58(s,2H),7.04(s,1H),7.23(s,1H)。Triethylamine (52.62g, 0.52mol, 2.0eq) was added to intermediate 1.3 (120.0g, 0.26mol, 1.0eq) and dichloromethane (1500ml) under constant stirring, followed by dropwise addition of trifluoro at 10°C Methanesulfonic anhydride (110.0 g, 0.39 mol, 1.5 eq), and the mixture was continuously stirred for 30 minutes at the same temperature. The reaction was quenched with saturatedNaHCO3 and the aqueous phase was further extracted with dichloromethane. The combined organic phases were washed successively with 2N HCl and saturated brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (200-300 silica gel, petroleum ether:ethyl acetate=6/1-3/1) to obtain the intermediate 1.4 (120.0 g, 61% yield) as a white solid.1 HNMR (400MHz, CDCl3 )δ(ppm) 2.97-3.06(m, 1H), 3.13-3.17(m, 1H), 3.84(s, 6H), 3.85(s, 3H), 4.03-4.08(m, 1H), 4.37-4.41(m, 1H), 4.64-4.66(d, J=8.3Hz, 1H), 5.08-5.20(dt, 2H), 5.71-5.80(m, 1H), 6.58(s, 2H) , 7.04(s, 1H), 7.23(s, 1H).
步骤5)、相对-(5R,5aR,8aS)-2,2-二氟-9-亚甲基-5-(3,4,5-三甲氧基苯基)-5,8,8a,9-四氢呋喃[3',4':6,7]萘并[2,3-d][1,3]二氧杂-6(5aH)-酮(中间体1.5)的合成:Step 5), relative-(5R,5aR,8aS)-2,2-difluoro-9-methylene-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9 - Synthesis of tetrahydrofuran[3',4':6,7]naphtho[2,3-d][1,3]dioxan-6(5aH)-one (intermediate 1.5):
在持续搅拌的中间体1.4(120.0g,0.20mol,1.0eq)和乙腈(1500ml)混合物中加入三苯基膦(15.83g,60.0mmol,0.3eq),K2CO3(82.93g,0.60mol,3.0eq)和Pd(OAc)2(4.49g,20.0mmol,10mol%),加热至80℃20小时。过滤反应物,沉淀用二氯甲烷洗脱。合并后的有机相浓缩干燥,硅胶柱层析(200-300硅胶,石油醚:乙酸乙酯=5/1-1/1)纯化得到中间体1.5(76.13g,收率61%),为白色固体。1HNMR(400MHz,CDCl3)δ(ppm)3.31-3.34(dd,1H),3.65-3.68(t,1H),3.76(s,6H),3.84(s,3H),4.21-4.24(dd,1H),4.57-4.59(m,2H),5.21-5.22(d,1),5.57(s,1H),6.31(s,2H),6.95(s,1H),7.27(s,1H)。To a continuously stirring mixture of intermediate 1.4 (120.0 g, 0.20 mol, 1.0 eq) and acetonitrile (1500 ml) was added triphenylphosphine (15.83 g, 60.0 mmol, 0.3 eq), K2 CO3 (82.93 g, 0.60 mol) , 3.0 eq) and Pd(OAc)2 (4.49 g, 20.0 mmol, 10 mol%), heated to 80° C. for 20 hours. The reaction was filtered and the precipitate was eluted with dichloromethane. The combined organic phases were concentrated and dried, and purified by silica gel column chromatography (200-300 silica gel, petroleum ether: ethyl acetate=5/1-1/1) to obtain Intermediate 1.5 (76.13 g, yield 61%), which was white solid.1 HNMR(400MHz, CDCl3 )δ(ppm) 3.31-3.34(dd,1H), 3.65-3.68(t,1H), 3.76(s,6H), 3.84(s,3H), 4.21-4.24(dd, 1H), 4.57-4.59(m, 2H), 5.21-5.22(d, 1), 5.57(s, 1H), 6.31(s, 2H), 6.95(s, 1H), 7.27(s, 1H).
步骤6)相对-(5aR,8aR,9R)-2,2-二氟-9-(3,4,5-三甲氧基苯基)-5a,6,8a,9-四氢呋喃[3',4':6,7]萘并[2,3-d][1,3]二氧代-5,8-二酮(中间体1.6)的合成:Step 6) Relative-(5aR,8aR,9R)-2,2-difluoro-9-(3,4,5-trimethoxyphenyl)-5a,6,8a,9-tetrahydrofuran[3',4 Synthesis of ':6,7]naphtho[2,3-d][1,3]dioxo-5,8-dione (Intermediate 1.6):
在持续搅拌的中间体1.5(79.04g,0.18mol,1.0eq),4-甲基吗啉-N-氧化物(168.7g,0.72mol,3.0eq)和二氯甲烷(1200ml)中加入OsO4(4.00g,15.7mmol,8mol%),混合物继续在环境温度下搅拌12小时。在反应物中分批加入固态NaIO4(77.00g,0.36mol,2.0eq),继续搅拌1小时。反应在冰浴中用饱和Na2S2O3(300ml)淬灭,水相进一步以二氯甲烷萃取。合并后的有机相用饱和食盐水洗涤,无水硫酸钠干燥,硅胶柱层析(200-300硅胶,石油醚:乙酸乙酯=5/1-1/1)纯化得到中间体1.6(48.80g,收率60%),为白色固体。1HNMR(400MHz,CDCl3)δ(ppm)3.37-3.42(m,2H),3.78(s,6H),3.82(s,3H),4.38-4.42(m,1H),4.79-4.80(d,J=2.8Hz,2H),6.22(s,2H),7.02(s,1H),7.80(s,1H)。To intermediate 1.5 (79.04g, 0.18mol, 1.0eq), 4-methylmorpholine-N-oxide (168.7g, 0.72mol, 3.0eq) and dichloromethane (1200ml) was addedOsO4 with continuous stirring (4.00 g, 15.7 mmol, 8 mol%) and the mixture continued to stir at ambient temperature for 12 hours. Solid NaIO4 (77.00 g, 0.36 mol, 2.0 eq) was added in portions to the reaction, and stirring was continued for 1 hour.The reaction was quenched with saturated Na2S2O3(300 ml) in an ice bath and the aqueous phase was further extracted with dichloromethane. The combined organic phases were washed with saturated brine, dried over anhydrous sodium sulfate, and purified by silica gel column chromatography (200-300 silica gel, petroleum ether: ethyl acetate=5/1-1/1) to obtain Intermediate 1.6 (48.80 g) , yield 60%) as a white solid.1 HNMR (400MHz, CDCl3 )δ(ppm) 3.37-3.42(m, 2H), 3.78(s, 6H), 3.82(s, 3H), 4.38-4.42(m, 1H), 4.79-4.80(d, J=2.8Hz, 2H), 6.22 (s, 2H), 7.02 (s, 1H), 7.80 (s, 1H).
步骤7)相对-(5R,5aS,8aR,9R)-2,2-二氟-9-羟基-5-(3,4,5-三甲氧基苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮(化合物1)的合成:Step 7) Relative-(5R,5aS,8aR,9R)-2,2-difluoro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9- Synthesis of tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one (compound 1):
将中间体1.6(43.30g,98.9mmol,1.0eq)和乙醚(1500ml)混合,氮气保护,在-78℃下搅拌,滴加LiAl(OtBu)3(200ml,197mmol,2.0eq),然后逐渐加热到环境温度过夜。反应在冰浴中用2N HCl淬灭,水相进一步以二氯甲烷萃取。合并后的有机相用饱和食盐水洗涤,无水硫酸钠干燥。残余物用乙酸乙酯重结晶,得到化合物1(33.10g,收率74%),为白色固体。1HNMR(400MHz,CDCl3)δ(ppm)7.38(s,1H),6.53(s,1H),6.47(s,2H),4.67(d,J=0.9Hz,1H),4.55-4.43(m,2H),4.03(d,J=6.4Hz,1H),3.89(s,3H),3.85(s,6H),3.26(q,1H),2.96(d,J=6.9Hz,1H),2.66(m,1H)。Intermediate 1.6 (43.30g, 98.9mmol, 1.0eq) was mixed with ether (1500ml), under nitrogen protection, stirred at -78°C, LiAl(OtBu)3 (200ml, 197mmol, 2.0eq) was added dropwise, and then gradually heated to ambient temperature overnight. The reaction was quenched with 2N HCl in an ice bath and the aqueous phase was further extracted with dichloromethane. The combined organic phases were washed with saturated brine and dried over anhydrous sodium sulfate. The residue was recrystallized from ethyl acetate to obtain compound 1 (33.10 g, yield 74%) as a white solid.1 HNMR (400MHz, CDCl3 )δ(ppm) 7.38(s, 1H), 6.53(s, 1H), 6.47(s, 2H), 4.67(d, J=0.9Hz, 1H), 4.55-4.43(m ,2H),4.03(d,J=6.4Hz,1H),3.89(s,3H),3.85(s,6H),3.26(q,1H),2.96(d,J=6.9Hz,1H),2.66 (m, 1H).
实施例2、相对-(5R,5aS,8aR,9R)-2,2-二氟-8-氧杂-9-(3,4,5-三甲氧基苯基)-5,5a,6,8,8a,9-六氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-5-基乙酸酯(化合物2)Example 2. Relative-(5R,5aS,8aR,9R)-2,2-difluoro-8-oxa-9-(3,4,5-trimethoxyphenyl)-5,5a,6, 8,8a,9-Hexahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxan-5-yl acetate (Compound 2)

将化合物1(1.15g,2.55mmol,1.0eq)和二氯甲烷(30ml)持续搅拌混合,加入Et3N(770mg,7.6mmol,3.0eq)和DMAP(310mg,2.55mmol,1.0eq),然后在冰浴下加入AcCl(400mg,5.1mmol,2.0eq),混合物在环境温度下搅拌12小时。滴加LiAl(OtBu)3(200ml,197mmol,2.0eq),然后逐渐加热到环境温度过夜。反应用饱和NH4Cl淬灭,水相进一步以二氯甲烷萃取。合并后的有机相用饱和食盐水洗涤,无水硫酸钠干燥。残余物用石油醚/乙酸乙酯重结晶,得到化合物2(810mg,收率65%),为白色固体。1HNMR(400MHz,CDCl3)δ(ppm)7.02(s,1H),6.76(s,1H),6.41(s,2H),5.81(d,J=6.3Hz,1H),4.48-4.34(m,3H),3.87(s,3H),3.84(s,6H),3.32(d,1H),3.01(m,1H),2.12(s,3H)。Compound 1 (1.15g, 2.55mmol, 1.0eq) and dichloromethane (30ml) were continuously mixed with stirring,Et3N (770mg, 7.6mmol, 3.0eq) and DMAP (310mg, 2.55mmol, 1.0eq) were added, then AcCl (400 mg, 5.1 mmol, 2.0 eq) was added under ice bath and the mixture was stirred at ambient temperature for 12 hours. LiAl(OtBu)3 (200 ml, 197 mmol, 2.0 eq) was added dropwise, then gradually heated to ambient temperature overnight. The reaction was quenched with saturatedNH4Cl and the aqueous phase was further extracted with dichloromethane. The combined organic phases were washed with saturated brine and dried over anhydrous sodium sulfate. The residue was recrystallized from petroleum ether/ethyl acetate to give compound 2 (810 mg, yield 65%) as a white solid.1 HNMR (400MHz, CDCl3 )δ(ppm) 7.02(s,1H), 6.76(s,1H), 6.41(s,2H), 5.81(d, J=6.3Hz,1H), 4.48-4.34(m , 3H), 3.87(s, 3H), 3.84(s, 6H), 3.32(d, 1H), 3.01(m, 1H), 2.12(s, 3H).
实施例3、相对-(5R,5aS,8aR,9R)-2,2,9-三氟-5-(3,4,5-三甲氧基苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮(化合物3)Example 3. Relative-(5R,5aS,8aR,9R)-2,2,9-trifluoro-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9-tetrahydrofuran Ipo[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one (Compound 3)

将化合物1(700mg,1.60mmol,1.0eq)和二氯甲烷(20ml)持续搅拌混合,滴加入二乙胺基三氟化硫(520mg,3.20mmol,2.0eq),混合物在环境温度下搅拌12小时。反应用饱和NaHCO3淬灭,继续搅拌30分钟。分离出有机相后,水相进一步以二氯甲烷萃取。合并后的有机相用饱和食盐水洗涤,无水硫酸钠干燥。残余物用硅胶柱层析(200-300硅胶,二氯甲烷:甲醇=100/1-50/1)纯化,得到化合物3(130mg,收率18%),为白色固体。1HNMR(400MHz,CDCl3)δ(ppm)7.27(s,1H),6.66(s,1H),6.44(s,2H),5.49-5.34(d,J=50.8Hz,1H),4.60-4.47(m,2H),4.20(d,J=5.0Hz,1H),3.88(s,3H),3.84(s,6H),3.28(m,1H),3.01(m,1H)。Compound 1 (700mg, 1.60mmol, 1.0eq) and dichloromethane (20ml) were continuously mixed with stirring, diethylaminosulfur trifluoride (520mg, 3.20mmol, 2.0eq) was added dropwise, and the mixture was stirred at ambient temperature for 12 Hour. The reaction was quenched with saturatedNaHCO3 and stirring was continued for 30 minutes. After separation of the organic phase, the aqueous phase was further extracted with dichloromethane. The combined organic phases were washed with saturated brine and dried over anhydrous sodium sulfate. The residue was purified by silica gel column chromatography (200-300 silica gel, dichloromethane:methanol=100/1-50/1) to obtain compound 3 (130 mg, yield 18%) as a white solid.1 H NMR (400MHz, CDCl3 )δ(ppm) 7.27(s, 1H), 6.66(s, 1H), 6.44(s, 2H), 5.49-5.34(d, J=50.8Hz, 1H), 4.60-4.47 (m, 2H), 4.20 (d, J=5.0 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 6H), 3.28 (m, 1H), 3.01 (m, 1H).
实施例4、相对-(5R,5aS,8aR,9R)-2,2-二氟-9-甲氧基-5-(3,4,5-三甲氧基苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮(化合物4)Example 4. Relative-(5R,5aS,8aR,9R)-2,2-difluoro-9-methoxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a ,9-Tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one (Compound 4)

将化合物1(600mg,1.33mmol,1.0eq)和二氯甲烷(15ml)持续搅拌混合,加入三甲基氧鎓四氟硼酸盐(246mg,1.66mmol,1.2eq),混合物在环境温度下搅拌20小时。反应用饱和NaHCO3淬灭,继续搅拌30分钟。分离出有机相后,水相进一步以二氯甲烷萃取。合并后的有机相用饱和食盐水洗涤,无水硫酸钠干燥。残余物用硅胶柱层析(200-300硅胶,二氯甲烷:甲醇=100/1-50/1)纯化,得到化合物4(65mg,收率11%),为白色固体。1HNMR(400MHz,CDCl3)δ(ppm)7.16(s,1H),6.74(s,1H),6.41(s,2H),4.45-4.36(m,3H),4.34(d,J=4.3Hz,1H),3.88(s,3H),3.83(s,6H),3.42(d,1H),3.31(s,3H),3.14(m,1H)。Compound 1 (600mg, 1.33mmol, 1.0eq) and dichloromethane (15ml) were continuously mixed with stirring, trimethyloxonium tetrafluoroborate (246mg, 1.66mmol, 1.2eq) was added, and the mixture was stirred at
实施例5、相对-(5R,5aS,8aR,9R)-2,2-二氟-9-羟基-5-(3,4,5-三甲氧基苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮-9-氘代(化合物5)Example 5. Relative-(5R,5aS,8aR,9R)-2,2-difluoro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9 -Tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one-9-deuterated (Compound 5)

将中间体1.6(400mg,0.89mmol,1.0eq)和甲醇(10ml)持续搅拌混合,分批加入NaBD4(37.5mg,0.89mmol,1.0eq),混合物搅拌4小时。反应用水淬灭,浓缩干燥。残余物用水稀释,用乙酸乙酯萃取。合并后的有机相用饱和食盐水洗涤,无水硫酸钠干燥。残余物用石油醚/乙酸乙酯重结晶,得到化合物5(140mg,收率35%),为白色固体。1HNMR(400MHz,CDCl3)δ(ppm)7.38(s,1H),6.57(s,1H),6.47(s,2H),4.66(d,J=9.8Hz,1H),4.47(m,1H),4.06(d,J=6.2Hz,1H),3.89(s,3H),3.87(s,6H),3.27(d,1H),2.68(m,1H),2.44(s,1H)。Intermediate 1.6 (400 mg, 0.89 mmol, 1.0 eq) and methanol (10 ml) were continuously mixed with stirring, NaBD4 (37.5 mg, 0.89 mmol, 1.0 eq) was added in portions, and the mixture was stirred for 4 hours. The reaction was quenched with water and concentrated to dryness. The residue was diluted with water and extracted with ethyl acetate. The combined organic phases were washed with saturated brine and dried over anhydrous sodium sulfate. The residue was recrystallized from petroleum ether/ethyl acetate to give compound 5 (140 mg, yield 35%) as a white solid. 1HNMR(400MHz, CDCl3)δ(ppm)7.38(s,1H),6.57(s,1H),6.47(s,2H),4.66(d,J=9.8Hz,1H),4.47(m,1H), 4.06(d,J=6.2Hz,1H), 3.89(s,3H), 3.87(s,6H), 3.27(d,1H), 2.68(m,1H), 2.44(s,1H).
实施例6、(5R,5aS,8aR,9R)-2,2-二氟-9-羟基-5-(3,4,5-三甲氧基苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮(化合物6)Example 6. (5R,5aS,8aR,9R)-2,2-difluoro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9-tetrahydrofuran Ipo[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one (Compound 6)

化合物6是运用手性柱层析法从化合物1中手性分离得到的。手性柱层析的主要参数如下:仪器:waters SFC200;层析柱:Daicel Chiralcel AD,250×50mm I.D.,10μm;流动相:A为CO2,B为异丙醇;梯度:B 20%;流速:150mL/min;反压:100bar;柱温:38℃;波长:220nm;循环时间:6.5min;样品分离:化合物溶解于~600ml甲醇中;进样体积:每次3ml。1HNMR(400MHz,CDCl3)δ(ppm)7.38(s,1H),6.53(s,1H),6.47(s,2H),4.67(d,J=0.9Hz,1H),4.55-4.43(m,2H),4.03(d,J=6.4Hz,1H),3.89(s,3H),3.85(s,6H),3.26(q,1H),2.96(d,J=6.9Hz,1H),2.66(m,1H);e.e.=98.56%;[α]=11.94°(MeOH,c=0.88g/100ml)。
实施例7、(5R,5aS,8aR,9R)-2,2-二氟-9-羟基-5-(3,4,5-三甲氧基苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮-9-氘代(化合物7)Example 7. (5R,5aS,8aR,9R)-2,2-difluoro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9-tetrahydrofuran Do[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one-9-deuterated (Compound 7)

化合物7是运用手性柱层析法从化合物5中手性分离得到的。手性柱层析的主要参数如下:仪器:waters SFC200;层析柱:Daicel Chiralcel AD,250×50mm I.D.,10μm;流动相:A为CO2,B为异丙醇;梯度:B 20%;流速:150mL/min;反压:100bar;柱温:38℃;波长:220nm;循环时间:6.5min;样品分离:化合物溶解于~600ml甲醇中;进样体积:每次3ml。1HNMR(400MHz,CDCl3)δ(ppm)7.38(s,1H),6.57(s,1H),6.47(s,2H),4.66(d,J=9.8Hz,1H),4.47(m,1H),4.06(d,J=6.2Hz,1H),3.89(s,3H),3.87(s,6H),3.27(d,1H),2.68(m,1H),2.44(s,1H)。
实施例8、相对-(5R,5aS,8aR,9R)-2,2-甲基-9-羟基-5-(3,4,5-三甲氧基-苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮(化合物8)Example 8. Relative-(5R,5aS,8aR,9R)-2,2-methyl-9-hydroxy-5-(3,4,5-trimethoxy-phenyl)-5,8,8a, 9-Tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxan-6(5aH)-one (Compound 8)

化合物8以5-酚羟基-2,2-甲基-1,3-苯并二噁唑作为起始材料,以实施例1中化合物1合成相类似的途径合成得到。1HNMR(400MHz,CDCl3)δ(ppm)6.95(s,1H),6.48(s,2H),6.30(s,1H),4.43-4.53(m,3H),4.12(d,J=5.2Hz,1H),3.87(s,3H),3.84(s,6H),3.25(dd,J=5.2Hz,9.3Hz,1H),2.74-2.80(m,1H),2.18(brs,1H),1.68(s,3H),1.65(s,3H)。
实施例9、相对-(5R,5aS,8aR,9R)-2,2-二氟-9-羟基-5-(3,4,5-三甲氧基-苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮-5,9-二氘代(化合物9)Example 9. Relative-(5R,5aS,8aR,9R)-2,2-difluoro-9-hydroxy-5-(3,4,5-trimethoxy-phenyl)-5,8,8a, 9-Tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one-5,9-dideuterium (compound 9)

化合物9以3,4,5-三甲氧基苯甲醛-d-甲酰作为起始材料,以实施例5中化合物5合成相类似的途径合成得到。1HNMR(400MHz,CDCl3)δ(ppm)7.38(s,1H),6.56(s,1H),6.47(s,2H),4.65(d,J=9.8Hz,1H),4.47(dd,J=6.1Hz,9.7Hz,1H),3.90(s,3H),3.86(s,6H),3.26(d,J=9.4Hz,1H),2.66-2.70(m,1H),2.52(brs,1H)。Compound 9 was synthesized using 3,4,5-trimethoxybenzaldehyde-d-formyl as the starting material, and was synthesized in a similar way to the synthesis of
实施例10、相对-(5R,5aS,8aR,9R)-2,2-二氟-9-羟基-5-(3,4,5-三(氘代甲氧基-d3)苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮-9-氘代(化合物10)Example 10. Relative-(5R,5aS,8aR,9R)-2,2-difluoro-9-hydroxy-5-(3,4,5-tris(deuterated methoxy-d3)phenyl)- 5,8,8a,9-tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one-9-deuterium Generation (Compound 10)

化合物10以3,4,5-三甲氧基苯甲醛-d9作为起始材料,以实施例5中化合物5合成相类似的途径合成得到。1HNMR(400MHz,CDCl3)δ(ppm)7.38(s,1H),6.55(s,1H),6.46(s,2H),4.64(dd,J=1.2Hz,9.8Hz,1H),4.45(dd,J=6.0Hz,9.8Hz,1H),4.04(d,J=6.3Hz,1H),3.26(dd,J=6.4Hz,9.3Hz,1H),2.78(s,1H),2.66(dd,J=6.0Hz,8.5Hz,1H)。
实施例11、(5R,5aS,8aR,9R)-2,2-二氟-9-羟基-5-(3,4,5-三(氘代甲氧基-d3)苯基)-5,8,8a,9-四氢呋喃并[3',4':6,7]萘并[2,3-d][1,3]-二氧杂-6(5aH)-酮-9-氘代(化合物11)Example 11, (5R, 5aS, 8aR, 9R)-2,2-difluoro-9-hydroxy-5-(3,4,5-tris(deuterated methoxy-d3)phenyl)-5, 8,8a,9-tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]-dioxa-6(5aH)-one-9-deuterated ( Compound 11)

化合物11是运用手性柱层析法从化合物10中手性分离得到的。手性柱层析的主要参数如下:仪器:waters SFC200;层析柱:Daicel Chiralcel AD,250×50mm I.D.,10μm;流动相:A为CO2,B为异丙醇;梯度:B 20%;流速:150mL/min;反压:100bar;柱温:38℃;波长:220nm;循环时间:6.5min;样品分离:化合物溶解于~600ml甲醇中;进样体积:每次3ml。1HNMR(400MHz,CDCl3)δ(ppm)7.38(s,1H),6.55(s,1H),6.46(s,2H),4.64(dd,J=1.2Hz,9.8Hz,1H),4.45(dd,J=6.0Hz,9.8Hz,1H),4.04(d,J=6.3Hz,1H),3.26(dd,J=6.4Hz,9.3Hz,1H),2.78(s,1H),2.66(dd,J=6.0Hz,8.5Hz,1H)。Compound 11 was obtained by chiral separation from
本发明的总体设计思路是以苦鬼臼毒素(PPP)分子作为基本骨架,以氟原子替代左侧2处氢原子,提高分子透过血脑屏障的能力;以氘原子替代4处氢原子,延长分子在机体内的半衰期。鉴此,化合物11(命名为PB-020)为本发明的最终目标产物,仅完成氟原子替代的化合物6(命名为PB-004)和完成氟原子替代与一处氘原子替代的化合物7(命名为PB-016)为最重要的结构类似物。这3个化合物均为手性分子,其相应的手性对映体无生物学活性。The overall design idea of the present invention is to take the podophyllotoxin (PPP) molecule as the basic skeleton, and replace the two hydrogen atoms on the left with fluorine atoms to improve the ability of the molecule to pass through the blood-brain barrier; replace the four hydrogen atoms with deuterium atoms, Extend the half-life of molecules in the body. In view of this, compound 11 (named PB-020) is the final target product of the present invention, only compound 6 (named PB-004) with fluorine atom substitution and compound 7 (named PB-004) with fluorine atom substitution and one deuterium atom substitution are completed. Named PB-016) is the most important structural analog. These three compounds are all chiral molecules, and their corresponding chiral enantiomers have no biological activity.
新型取代苦鬼臼毒素衍生物的生物活性检测Biological Activity Detection of Novel Substituted Picropodophyllotoxin Derivatives
以下生物活性检测主要对比化合物11(命名为PB-020)和苦鬼臼毒素(PPP)对体外培养的人源胶质瘤细胞的药效,及其在小鼠体内的代谢动力学、血脑屏障通透性、抗肿瘤药效和安全性等指标,部分试验中同时以化合物6(命名为PB-004)和化合物7(命名为PB-016)作为对照。The following biological activity tests mainly compare the pharmacodynamics of compound 11 (named PB-020) and podophyllotoxin (PPP) on human glioma cells cultured in vitro, and their metabolic kinetics, blood-brain in vivo in mice For indicators such as barrier permeability, antitumor efficacy and safety, compound 6 (named PB-004) and compound 7 (named PB-016) were used as controls in some experiments.
一、化合物体外药效学试验1. In vitro pharmacodynamic test of the compound
1.试验步骤:1. Test steps:
1)细胞来源:人脑星形胶质母细胞瘤/胶质瘤细胞U-87MG(简称U87)购自中国科学院细胞库(上海),货号TCHu138;人胶质瘤细胞DBTRG-05MG(简称DBTRG)来自美国组织培养库ATCC,货号CRL-2020;人胶质瘤细胞KNS-81来自AcceGen Biotech,货号ABC-TC0535。1) Cell source: human brain astrocytoma/glioma cell U-87MG (referred to as U87) was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai), product number TCHu138; human glioma cell DBTRG-05MG (referred to as DBTRG) ) from the American Tissue Culture Bank ATCC, Cat. No. CRL-2020; human glioma cells KNS-81 from AcceGen Biotech, Cat. No. ABC-TC0535.
2)细胞培养和传代:上述细胞均贴壁培养于含完全培养基[含DMEM高糖基础培养液(#SH30243.01B,HyClone)加入终浓度为10%FBS(#1101-500,上海普飞)和Penicillin-Streptomycin双抗(#SV30010,HyClone)]的6cm培养皿(#430166,Corning)或T75培养瓶(#3276,Corning)中,培养皿(瓶)置于37℃,5%CO2,饱和湿度的细胞培养箱(#3111,ThermoFisher Scientific)中培养。传代时,先吸走培养基,用PBS磷酸缓冲盐溶液(#GNM-10944,杭州吉诺)洗涤2次,然后加入适量0.25%胰酶-0.02%EDTA(#25200-072,Gibco),摇晃培养皿(瓶)使之均匀覆盖细胞,置于相差显微镜下观察。待大多数细胞回缩变圆,轻摇即脱落时,迅速加入两倍胰酶体积的完全培养基终止,并轻轻将细胞吹打成单细胞。将细胞悬液移到合适大小的离心管内,800rpm离心5min。弃上清,用新鲜的完全培养重悬细胞团块,重新吹打成单细胞,以1:3-1:6的比率传代接种至新的培养皿(瓶)并补足完全培养基。置于37℃,5%CO2的细胞培养箱中培养。2) Cell culture and passage: The above cells were adhered and cultured in complete medium [containing DMEM high glucose basal medium (#SH30243.01B, HyClone) with a final concentration of 10% FBS (#1101-500, Shanghai Pufei). ) and Penicillin-Streptomycin Dual Antibody (#SV30010, HyClone)] in a 6cm dish (#430166, Corning) or a T75 flask (#3276, Corning) at 37°C, 5%CO2 , cultured in a humidified cell incubator (#3111, ThermoFisher Scientific). When passage, first aspirate the medium, wash twice with PBS phosphate buffered saline (#GNM-10944, Hangzhou Jinuo), then add an appropriate amount of 0.25% trypsin-0.02% EDTA (#25200-072, Gibco), shake The culture dish (flask) was evenly covered with cells and observed under a phase contrast microscope. When most of the cells retracted and became round, and fell off with gentle shaking, quickly add twice the volume of trypsin to complete the medium to stop, and gently pipet the cells into single cells. The cell suspension was transferred to a suitable size centrifuge tube and centrifuged at 800 rpm for 5 min. Discard the supernatant, resuspend the cell pellet with fresh complete culture, re-pipetate into single cells, subculture to a new culture dish (flask) at a ratio of 1:3-1:6, and supplement the complete culture medium. Place in a cell incubator at 37 °C, 5% CO2 .
3)细胞加药处理:将上述每种细胞消化、计数,均按5000细胞/200μl培养液的密度接种至96孔细胞培养板(#3988,Corning)的每个孔中,置于37℃,5%CO2的细胞培养箱中培养24h使细胞充分贴壁。然后分别将含梯度稀释后的各种待测化合物(每种化合物每个浓度设置3个重复孔)以及DMSO(#D5879,Sigma-Aldrich)溶剂对照的完全培养液替换掉原来的培养液,继续培养72h。3) Cell dosing treatment: Digest and count each of the above cells, and inoculate them into each well of a 96-well cell culture plate (#3988, Corning) at a density of 5000 cells/200 μl culture medium, and place them at 37°C. Incubate the cells in a 5% CO2 incubator for 24h to make the cells fully adherent. Then, the original culture medium was replaced with the complete culture medium containing various test compounds after serial dilution (3 replicate wells were set for each concentration of each compound) and DMSO (#D5879, Sigma-Aldrich) solvent control, and continued. Cultivated for 72h.
4)药效学测定和统计:先用倒置相差显微镜(X71,Olympus)对上述化合物处理后的细胞进行形态观察和拍照记录。然后分别将含CCK-8检测试剂(#E606335,上海生工)的无酚红培养液替换掉原来的培养液,培养箱中继续培养2h。然后在多功能酶标仪(168-1130,Bio-Rad)上测量OD450nm吸光值(OD值)。受试化合物处理细胞后,细胞生存率(cellsurvival rate)或细胞增殖率(cell growth rate)的计算公式为:生存率=加药组OD值/对照组OD值×100%;化合物对细胞增殖的抑制率(growth inhibition rate)计算公式为:抑制率=(对照组OD值-加药组OD值)/对照组OD值×100%。进一步根据抑制率数值在SPSS中计算出各化合物的IC50。4) Pharmacodynamics determination and statistics: First, use an inverted phase contrast microscope (X71, Olympus) to observe the morphology of the cells treated with the above compounds and record them by photographing. Then, the phenol red-free culture medium containing CCK-8 detection reagent (#E606335, Shanghai Shenggong) was respectively replaced with the original culture medium, and the culture was continued in the incubator for 2 h. Then, the OD450nm absorbance value (OD value) was measured on a multifunctional microplate reader (168-1130, Bio-Rad). After the cells were treated with the test compound, the calculation formula of cell survival rate or cell growth rate was: survival rate = OD value of drug addition group/OD value of control group × 100%; The calculation formula of growth inhibition rate is: inhibition rate=(OD value of control group-OD value of drug-added group)/OD value of control group×100%. The IC50 of each compound was further calculated in SPSS according to the inhibition rate value.
2.试验结果:2. Test results:
图1是化合物10及其手性分离化合物体外药效学检测结果,可以确定化合物11为化合物10中发挥抗胶质瘤药效的对映体。Figure 1 shows the in vitro pharmacodynamic test results of
PB-020和PPP,PB-004,PB-016体外药效学比较统计如表1所示The in vitro pharmacodynamic comparison statistics of PB-020 and PPP, PB-004 and PB-016 are shown in Table 1.
表1化合物11及其类似物体外药效学检测结果Table 1 In vitro pharmacodynamic test results of compound 11 and its analogs

注:数据为3次独立实验IC50数值的平均值±标准差,Note: The data are the mean ± standard deviation of the IC50 values of 3 independent experiments,
3种人源胶质瘤细胞系均为贴壁培养。All three human glioma cell lines were adherent cultured.
PPP:苦鬼臼毒素;PB-004:化合物6;PB-016:化合PPP: Picropodophyllotoxin; PB-004:
物7;PB-020:化合物11
结果显示,化合物11(PB-020)及其类似物对于体外贴壁培养的人源胶质瘤细胞系具有和PPP接近的抑制增殖作用,IC50均为1μM左右。The results showed that compound 11 (PB-020) and its analogs had an inhibitory effect on the proliferation of adherent human glioma cell lines in vitro, which was close to that of PPP, with IC50 of about 1 μM.
二、化合物作用机制验证2. Validation of the mechanism of action of the compound
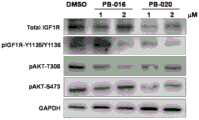
1.试验步骤:1. Test steps:
1)细胞来源:人脑星形胶质母细胞瘤/胶质瘤细胞U-87MG(简称U87)购自中国科学院细胞库(上海),货号TCHu138。1) Cell source: Human brain astrocytoma/glioma cells U-87MG (referred to as U87) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai), with the product number TCHu138.
2)肿瘤微球培养:U87细胞贴壁培养于含完全培养基[含DMEM高糖基础培养液(#SH30243.01B,HyClone)加入终浓度为10%FBS(#1101-500,上海普飞)和Penicillin-Streptomycin双抗(#SV30010,HyClone)]的6cm培养皿(#430166,Corning)或T75培养瓶(#3276,Corning)中,培养皿(瓶)置于37℃,5%CO2,饱和湿度的细胞培养箱(#3111,ThermoFisher Scientific)中培养。将呈对数生长状态的U-87MG细胞用胰蛋白酶消化、收集,用无血清DMEM重悬。用细胞计数仪台盼蓝染色法计数(活细胞比例在90%以上),将105个细胞接种于6cm培养皿中,5ml无血清干细胞培养液(EGF 20ng/ml;bFGF 20ng/ml;LIF 10ng/ml;B27 1:50;PS 1:100)置于37℃,5%CO2,饱和湿度的细胞培养箱培养。每3天补加1ml新鲜的无血清干细胞培养液,一般培养3-5天可观察到典型的肿瘤微球的形成。2) Tumor microsphere culture: U87 cells were adherently cultured in complete medium [containing DMEM high-glucose basal medium (#SH30243.01B, HyClone) with a final concentration of 10% FBS (#1101-500, Shanghai Pufei) and Penicillin-Streptomycin Dual Antibody (#SV30010, HyClone)] in 6cm Petri dishes (#430166, Corning) or T75 flasks (#3276, Corning) at 37°C, 5% CO2, saturated The cells were grown in a humidified cell incubator (#3111, ThermoFisher Scientific). U-87MG cells in logarithmic growth state were trypsinized, harvested, and resuspended in serum-free DMEM. Count with trypan blue staining with a cell counter (the proportion of viable cells is above 90%), inoculate 105 cells in a 6cm dish, and 5ml of serum-free stem cell culture medium (EGF 20ng/ml; bFGF 20ng/ml; LIF 10ng/ml; B27 1:50; PS 1:100) in a cell incubator at 37°C, 5% CO2, and saturated humidity. 1 ml of fresh serum-free stem cell culture medium was added every 3 days, and the formation of typical tumor microspheres could be observed in 3-5 days of culture.
3)细胞加药处理:待上述U-87MG肿瘤微球形成后,分别将含梯度稀释后的各种待测化合物以及DMSO溶剂对照的新鲜培养液替换掉原来的培养液,继续培养1-3天。3) Cell dosing treatment: After the above-mentioned U-87MG tumor microspheres are formed, replace the original culture medium with fresh culture medium containing various compounds to be tested and DMSO solvent control after gradient dilution, and continue to culture for 1-3. sky.
4)IGF1R信号通路生物标志物检测:收集上述化合物处理1-3天后的U-87MG肿瘤微球悬浮液,离心后吸去上清培养液,用冰浴预冷的PBS洗2次,吸干液体。加入200μl细胞裂解液(#P0013,碧云天),剧烈震荡30s,冰上静置5min,重复3次。细胞裂解样品13000rpm,4℃离心6min,取上清与4×Laemmli上样缓冲液(#161-0747,Bio-Rad)3:1体积混合成为细胞裂解液蛋白样品,100℃金属浴变性6min,样品用于下列Western blot检测。将4-15%预制梯度胶(456-8084,Bio-Rad)安装到电泳槽中,加入足量1×SDS-PAGE电泳缓冲液。使用20μl的移液枪加入20μl上述细胞裂解液蛋白样品。将电泳槽盖子盖上,并接通电源,先以80V电压电泳约30min,待样品中溴酚蓝在浓缩胶与分离胶分界线压成一条细线时将电压调至120V,根据目标蛋白以及内参蛋白条带的大小调整电泳持续时间。将预冷1×转膜缓冲液倒入合适大小的容器中,按照说明书组装好泡沫垫-滤纸-胶-PVDF膜-滤纸-泡沫垫的“三明治”结构,装入电泳槽中。加入冰块且整个电泳槽冰浴,连通电源进行转膜,250mA,2h。将PVDF膜(IPVH00010,Millipore)放入由1×TBST配制的5%脱脂奶粉中室温封闭1h。然后依次进行不同一抗(anti-pIGF1R-Y1135/Y1136,#3024S,CST;anti-pAKT-T308,#13038,CST;anti-pAKT-S473,#4060,CST;anti-pIRS1-S302,#2384,CST;anti-GAPDH[HRP],#A00191-40,GenScript)和二抗(anti-Mouse IgG HRP-linked antibody,#7076,CST;anti-Rabbit IgGHRP-linked antibody,#7074,CST)的孵育(分别室温孵育1h)与洗膜(1×TBST洗膜3次,每次5min)。最后,将PVDF膜放至塑料膜中间,加入ECL膜上反应3min,盖上另一层塑料膜,在全自动化学发光/荧光图像分析系统(5200-Multi,天能)中曝光。4) Detection of biomarkers of IGF1R signaling pathway: collect the U-87MG tumor microsphere suspensions treated with the above compounds for 1-3 days, remove the supernatant culture after centrifugation, wash twice with PBS pre-cooled in an ice bath, and blot dry liquid. Add 200 μl of cell lysis solution (#P0013, Biyuntian), shake vigorously for 30 s, let stand on ice for 5 min, and
2.试验结果:2. Test results:
结果如图2所示,化合物11(PB-020)和化合物7(PB-016)对U87肿瘤微球中一系列IGF1R信号通路生物标志物均有剂量依赖型的抑制作用,提示其药靶为IGFIR,并且它们均通过抑制IGF1R下游效应分子而高效抑制U87细胞增殖。尤其值得注意的是:PB-020还能显著降低其靶蛋白IGF1R的蛋白量(total IGF1R,第一行),可能由其促进IGF1R内吞与降解而导致。The results are shown in Figure 2. Compound 11 (PB-020) and compound 7 (PB-016) have dose-dependent inhibitory effects on a series of IGF1R signaling pathway biomarkers in U87 tumor microspheres, suggesting that their drug targets are IGFIR, and they both efficiently inhibit U87 cell proliferation by inhibiting IGF1R downstream effector molecules. It is particularly noteworthy that PB-020 can also significantly reduce the protein amount of its target protein IGF1R (total IGF1R, first row), which may be caused by its promotion of IGF1R endocytosis and degradation.
三、化合物透过血脑屏障试验(生物标志物检测)3. Compounds through the blood-brain barrier test (biomarker detection)
1.试验步骤:1. Test steps:
1)实验动物来源:SPF级雄性BALB/c裸鼠订购自上海斯莱科公司,小鼠周龄为6至8周。1) Source of experimental animals: SPF male BALB/c nude mice were ordered from Shanghai Slack Company, and the mice were 6 to 8 weeks old.
2)实验动物给药处理:将受试化合物溶解在DMSO(#D5879,Sigma-Aldrich)中,配制成50mg/ml母液。用PBS稀释受试化合物母液,以100mg/kg剂量对小鼠进行灌胃,每种受试化合物设置3只小鼠的实验重复。给药前禁食过夜。2) Experimental animal administration treatment: The test compound was dissolved in DMSO (#D5879, Sigma-Aldrich) to prepare a 50 mg/ml stock solution. The test compound stock solution was diluted with PBS, and the mice were gavaged at a dose of 100 mg/kg, and the experiment was repeated with 3 mice for each test compound. Fasting overnight before dosing.
3)检测样品制备:给药3小时后,颈椎脱臼法处死小鼠,取其右脑,加入800μl预冷的细胞裂解液(#P0013,碧云天,新鲜添加入蛋白酶和磷酸酶抑制剂#78444,ThermoFisherScientific),冰浴上充分匀浆,12000rpm离心10分钟后,取上清。用BCA法测定样品的蛋白浓度,加入相应量的4×Laemmli上样缓冲液(#161-0747,Bio-Rad),以3:1体积混合成为脑组织裂解液蛋白样品,100℃金属浴变性8min,12000rpm离心2分钟,取上清进行下列Western blot检测。3) Test sample preparation: 3 hours after administration, the mice were killed by cervical dislocation, the right brain was removed, and 800 μl of pre-cooled cell lysate (#P0013, Biyuntian, freshly added protease and phosphatase inhibitor #78444 was added) , ThermoFisher Scientific), fully homogenized on an ice bath, centrifuged at 12,000 rpm for 10 minutes, and the supernatant was taken. The protein concentration of the sample was determined by the BCA method, the corresponding amount of 4×Laemmli loading buffer (#161-0747, Bio-Rad) was added, and the brain tissue lysate protein sample was mixed at a volume of 3:1, and denatured in a metal bath at 100 °C Centrifuge at 12,000 rpm for 8 min for 2 min, and take the supernatant for the following Western blot detection.
4)IGF1R信号通路生物标志物检测:将4-15%预制梯度胶(456-8084,Bio-Rad)安装到电泳槽中,加入足量1×SDS-PAGE电泳缓冲液。使用20μl的移液枪加入20μl上述细胞裂解液蛋白样品。将电泳槽盖子盖上,并接通电源,先以80V电压电泳约30min,待样品中溴酚蓝在浓缩胶与分离胶分界线压成一条细线时将电压调至120V,根据目标蛋白以及内参蛋白条带的大小调整电泳持续时间。将预冷1×转膜缓冲液倒入合适大小的容器中,按照说明书组装好泡沫垫-滤纸-胶-PVDF膜-滤纸-泡沫垫的“三明治”结构,装入电泳槽中。加入冰块且整个电泳槽冰浴,连通电源进行转膜,250mA,2h。将PVDF膜(IPVH00010,Millipore)放入由1×TBST配制的5%脱脂奶粉中室温封闭1h。然后依次进行不同一抗(anti-pIGF1R-Y1135/Y1136,#3024S,CST;anti-pAKT-T308,#13038,CST;anti-pAKT-S473,#4060,CST;anti-GAPDH[HRP],#A00191-40,GenScript)和二抗(anti-Mouse IgG HRP-linked antibody,#7076,CST;anti-Rabbit IgG HRP-linked antibody,#7074,CST)的孵育(分别室温孵育1h)与洗膜(1×TBST洗膜3次,每次5min)。最后,将PVDF膜放至塑料膜中间,加入ECL膜上反应3min,盖上另一层塑料膜,在全自动化学发光/荧光图像分析系统(5200-Multi,天能)中曝光。4) Detection of IGF1R signaling pathway biomarkers: install a 4-15% prefabricated gradient gel (456-8084, Bio-Rad) into the electrophoresis tank, and add a sufficient amount of 1×SDS-PAGE running buffer. Add 20 μl of the above cell lysate protein sample using a 20 μl pipette. Close the lid of the electrophoresis tank and turn on the power. First, electrophoresis at 80V for about 30min. When the bromophenol blue in the sample is pressed into a thin line between the stacking gel and the separating gel, adjust the voltage to 120V. According to the target protein and The size of the internal reference protein bands adjusts the electrophoresis duration. Pour the pre-cooled 1× transfer buffer into a suitable size container, assemble the “sandwich” structure of foam pad-filter paper-glue-PVDF membrane-filter paper-foam pad according to the instructions, and put it into the electrophoresis tank. Add ice cubes and ice bath the entire electrophoresis tank, connect the power supply to transfer membrane, 250mA, 2h. A PVDF membrane (IPVH00010, Millipore) was placed in 5% nonfat dry milk prepared with 1×TBST and blocked for 1 h at room temperature. Then different primary antibodies (anti-pIGF1R-Y1135/Y1136, #3024S, CST; anti-pAKT-T308, #13038, CST; anti-pAKT-S473, #4060, CST; anti-GAPDH[HRP], # A00191-40, GenScript) and secondary antibody (anti-Mouse IgG HRP-linked antibody, #7076, CST; anti-Rabbit IgG HRP-linked antibody, #7074, CST) incubation (1h at room temperature) and membrane washing ( Membrane was washed 3 times with 1×TBST, 5 min each time). Finally, put the PVDF film in the middle of the plastic film, add the ECL film to react for 3 min, cover with another layer of plastic film, and expose in an automatic chemiluminescence/fluorescence image analysis system (5200-Multi, Tianneng).
2.试验结果:2. Test results:
结果如图3所示,小鼠口服化合物11和化合物7,3小时内就下调了脑组织中的磷酸化IGF1R/AKT等IGF1R信号通路中关键的生物标志物,提示两者均能透过小鼠血脑屏障。而PPP未能有效下调IGF1R信号通路中关键的生物标志物,提示其不能有效透过小鼠血脑屏障。The results are shown in Figure 3. Oral administration of compound 11 and
四、化合物药代动力学4. Pharmacokinetics of Compounds
1.试验步骤:1. Test steps:
1)实验动物来源:雄性或雌性BALB/c品系裸鼠订购自上海斯莱克实验动物有限责任公司,小鼠周龄为7周。1) Source of experimental animals: male or female BALB/c strain nude mice were ordered from Shanghai Slack Laboratory Animal Co., Ltd., and the mice were 7 weeks old.
2)实验动物给药处理:将受试化合物溶解在DMSO(#D5879,Sigma-Aldrich)中,配制成50mg/ml母液。受试化合物最终溶于10%DMSO+90%玉米油(#C8267-500ML,Sigma-Aldrich)以50mg/kg剂量对小鼠进行灌胃。2) Experimental animal administration treatment: The test compound was dissolved in DMSO (#D5879, Sigma-Aldrich) to prepare a 50 mg/ml stock solution. Test compounds were finally dissolved in 10% DMSO + 90% corn oil (#C8267-500ML, Sigma-Aldrich) and administered to mice at a dose of 50 mg/kg.
3)检测样品制备:小鼠分别给药1,3,6,12或24小时后,按250μl/20g体重的剂量腹腔注射4%水合氯醛水溶液,麻醉小鼠,颈动脉取血300μl至肝素管,混匀后,4000rpm5分钟离心,取上清200μl,加入600μl乙腈,混匀超声20分钟,12000rpm离心10分钟,上清4℃静置过夜,然后12000rpm离心10分钟,取上清作为“血浆检测样品”;取0.3-0.5g左右全脑,用预冷的PBS迅速漂洗3次,加1ml乙腈匀浆(PRIMA手持式匀浆机PB100,35000rpm,1分钟),超声20分钟,12000rpm离心10分钟,上清4℃静置过夜,12000rpm离心10分钟,取上清作为“脑组织检测样品”。3) Preparation of test samples: 1, 3, 6, 12 or 24 hours after administration to mice, 4% chloral hydrate aqueous solution was injected intraperitoneally at a dose of 250 μl/20 g body weight, the mice were anesthetized, and 300 μl of blood was collected from the carotid artery to heparin Tube, after mixing, centrifuge at 4000rpm for 5 minutes, take 200μl of supernatant, add 600μl of acetonitrile, mix for 20 minutes, centrifuge at 12,000rpm for 10 minutes, let the supernatant stand at 4°C overnight, then centrifuge at 12,000rpm for 10 minutes, take the supernatant as "plasma". "Test sample"; take about 0.3-0.5 g of the whole brain, rinse it three times with pre-cooled PBS, add 1 ml of acetonitrile to homogenize (PRIMA hand-held homogenizer PB100, 35,000 rpm, 1 minute), ultrasonicate for 20 minutes, and centrifuge at 12,000 rpm for 10 minutes. minutes, the supernatant was allowed to stand at 4°C overnight, centrifuged at 12,000 rpm for 10 minutes, and the supernatant was taken as a "brain tissue test sample".
4)HPLC检测:取上述“血浆检测样品”和“脑组织检测样品”,设置各受试化合物的标准品对照、HPLC流动相空白对照等,用高效液相色谱仪(Hitachi Chromaster-5430检测器,Hitachi Chromaster-5310柱温箱,Hitachi Chromaster-5210自动进样器,HitachiChromaster-5110泵)分析样品中受试化合物的含量。4) HPLC detection: Take the above-mentioned "plasma test sample" and "brain tissue test sample", set the standard control of each test compound, HPLC mobile phase blank control, etc., and use high performance liquid chromatograph (Hitachi Chromaster-5430 detector). , Hitachi Chromaster-5310 column oven, Hitachi Chromaster-5210 autosampler, Hitachi Chromaster-5110 pump) to analyze the content of the test compounds in the samples.
2.试验结果:2. Test results:
结果如图4所示,与PPP相比,化合物11(PB-020)和化合物7(PB-016)在血浆和脑组织中的峰值浓度都高得多,提示PPP在小鼠体内代谢较快,并且几乎无法透过血脑屏障,有可能因为PPP能够透过血脑屏障前几乎已被代谢掉了。化合物11在血浆和脑组织中的峰值浓度又都显著高于化合物7。在血浆和脑组织中,化合物7在给药后1小时达到峰值,而化合物11在给药后3小时达到峰值。因此,药代动力学结果提示化合物11在血浆和脑组织中的代谢都比化合物7更慢,因而也更稳定。The results are shown in Figure 4. Compared with PPP, the peak concentrations of compound 11 (PB-020) and compound 7 (PB-016) in both plasma and brain tissue were much higher, suggesting that PPP is metabolized faster in mice , and almost cannot pass through the blood-brain barrier, probably because PPP has almost been metabolized before it can pass through the blood-brain barrier. The peak concentrations of compound 11 in both plasma and brain tissue were significantly higher than those of
五、化合物短期安全性评价V. Short-term safety evaluation of compounds
BALB/c雄性裸鼠,以DMSO作为对照,以50,100,200,400mg/kg四个剂量的化合物11(PB-020)进行灌胃给药,每天给药1次,分别于第2,4,6,8天称量体重,并观察形态和行为。BALB/c male nude mice, with DMSO as a control, were given four doses of compound 11 (PB-020) at 50, 100, 200, 400 mg/kg by gavage, once a day, on the 2nd, 4th, 6th, and 8th, respectively. Body weights were weighed every day, and morphology and behavior were observed.
结果如图5所示,灌胃给药后8天内,所有剂量组小鼠均未见体重减轻和明显的形态与行为异常,提示化合物11具有较好的安全性。下述小鼠体内药效学试验均采用50mg/kg和100mg/kg两个较低剂量的化合物11进行。The results are shown in FIG. 5 , within 8 days after oral administration, there was no weight loss and obvious morphological and behavioral abnormalities in mice in all dose groups, suggesting that compound 11 has good safety. The following in vivo pharmacodynamic experiments in mice were all performed with two lower doses of
六、化合物体内药效学试验6. In vivo pharmacodynamic test of the compound
(一)化合物11单药以及化合物11+替莫唑胺对原位接种于裸鼠脑部的人源胶质瘤细胞增殖的抑制作用(1) Inhibitory effect of compound 11 single drug and compound 11+temozolomide on the proliferation of human glioma cells orthotopically inoculated into the brain of nude mice
1.试验步骤:1. Test steps:
1)建模及分组:选取10周龄雌性BALB/c品系裸鼠40只,每只小鼠颅腔内注射5μl含3x105 U87-Luc细胞(Cat.#BW124577,Perkin Elmer)的悬液,待小鼠恢复几天后,通过活体成像术检测所接种U87-Luc细胞的荧光强度来判断其增殖情况。根据U87-Luc细胞的荧光强度,将接种成功的小鼠随机分成4组,每组8只小鼠用于给药实验。1) Modeling and grouping: 40 nude mice of 10-week-old female BALB/c strain were selected, and 5 μl of suspension containing 3×105 U87-Luc cells (Cat.#BW124577, Perkin Elmer) was injected into the cranial cavity of each mouse. Several days after the mice recovered, the fluorescence intensity of the inoculated U87-Luc cells was detected by in vivo imaging to judge their proliferation. According to the fluorescence intensity of U87-Luc cells, the successfully inoculated mice were randomly divided into 4 groups, with 8 mice in each group for the drug administration experiment.
2)给药:Vehicle组:以200μl 10%DMSO+90%玉米油(#C8267-500ML,Sigma-Aldrich)对每只小鼠灌胃,每天1次,连续给药14-19天(直至小鼠死亡);PB-020组:PB-020(化合物11)溶解于10%DMSO+90%玉米油,以50mg/kg剂量、总体积200μl对小鼠进行灌胃,每天1次,连续给药17-21天(直至小鼠死亡);TMZ组:TMZ(替莫唑胺)溶解于PBS,以20mg/kg剂量、总体积200μl对小鼠进行腹腔注射,每天1次,连续给药5天;PB-020+TMZ组:按上述单药的给药方式和剂量分别给药。2) Administration: Vehicle group: gavage each mouse with 200 μl of 10% DMSO+90% corn oil (#C8267-500ML, Sigma-Aldrich), once a day, for 14-19 days (until small Mice died); PB-020 group: PB-020 (compound 11) was dissolved in 10% DMSO + 90% corn oil, and the mice were given 50 mg/kg dose by gavage in a total volume of 200 μl, once a day, continuously administered 17-21 days (until the mice died); TMZ group: TMZ (temozolomide) was dissolved in PBS, and the mice were intraperitoneally injected at a dose of 20 mg/kg and a total volume of 200 μl, once a day, for 5 consecutive days; PB- 020+TMZ group: The patients were administered according to the above-mentioned single drug administration mode and dosage.
3)药效评价:小鼠开始给药后,每3天用活体成像术测定所接种U87-Luc细胞的荧光强度,以此来判断U87-Luc细胞在小鼠脑部的增殖情况和待测药物对其增殖的抑制程度;每天称量记录小鼠体重,一天内小鼠体重下降超过10%时暂停给药;每天观察记录小鼠的形态与行为特征;记录小鼠死亡时间,作出生存曲线。3) Efficacy evaluation: After the mice began to be administered, the fluorescence intensity of the inoculated U87-Luc cells was measured by in vivo imaging every 3 days, so as to judge the proliferation of U87-Luc cells in the mouse brain and the conditions to be tested. The inhibition degree of the drug on its proliferation; the weight of the mice is recorded every day, and the drug is suspended when the body weight of the mice drops more than 10% within one day; the morphology and behavioral characteristics of the mice are observed and recorded every day; the time of death of the mice is recorded, and the survival curve is drawn. .
2.试验结果:2. Test results:
结果如图6所示,给药1周内,化合物11(PB-020)和TMZ均有效抑制了U87细胞在小鼠脑部的快速增殖。超过1周后,化合物11的药效显著下降,而TMZ持续有效,并能小幅缩减移植瘤的体积。给药10天内,化合物11和TMZ联用比单药效果更明显,超过10天后,与TMZ单药作用趋近。相比对照组,化合物11能够有效延长移植瘤小鼠的生存期,p=0.02。在该实验模型中,由于TMZ单药已有出色药效,化合物11与之联用的药效很难有效展现。The results are shown in Figure 6. Within 1 week of administration, both Compound 11 (PB-020) and TMZ effectively inhibited the rapid proliferation of U87 cells in the mouse brain. After more than 1 week, the efficacy of compound 11 decreased significantly, while TMZ continued to be effective and could slightly reduce the size of the transplanted tumor. Within 10 days of administration, the combined use of compound 11 and TMZ had more obvious effect than the single drug, and after more than 10 days, the effect approached the effect of TMZ single drug. Compared with the control group, compound 11 can effectively prolong the survival period of the tumor-transplanted mice, p=0.02. In this experimental model, due to the excellent efficacy of TMZ as a single drug, the efficacy of compound 11 in combination with it is difficult to effectively demonstrate.
(二)化合物11对接种于裸鼠的人源乳腺癌细胞向脑部转移的抑制作用(2) Inhibitory effect of compound 11 on brain metastasis of human breast cancer cells inoculated in nude mice
1.试验步骤:1. Test steps:
1)建模及分组:5周龄雌性Balb/c裸鼠24只,适应性饲养5-7天,开始实验。MDA-MB-231-Luc细胞复苏,扩大培养。待细胞数量满足实验要求后,消化收集细胞,将细胞混入PBS中,制成单细胞悬液,调整细胞密度为2×106/ml,用于实验。阿佛丁麻醉小鼠,左心室注射细胞。细胞接种量为100μl/只。接种后腹腔注射D-Luciferin,IVIS检测接种效果,选择20只用于实验。待小鼠苏醒后,返回IVC系统继续饲养。接种后第14天进行活体成像,根据成像效果,将小鼠随机分为2组,每组8只。1) Modeling and grouping: 24 5-week-old female Balb/c nude mice were reared adaptively for 5-7 days before the experiment was started. MDA-MB-231-Luc cells were recovered and expanded. After the number of cells met the experimental requirements, the cells were digested and collected, and the cells were mixed into PBS to prepare a single-cell suspension, and the cell density was adjusted to 2×106 /ml for the experiment. Mice were anesthetized with Avertin, and cells were injected into the left ventricle. The cell inoculation volume was 100 μl/cell. After inoculation, D-Luciferin was intraperitoneally injected, and the inoculation effect was detected by IVIS, and 20 animals were selected for the experiment. After the mice woke up, they returned to the IVC system to continue feeding. Live imaging was performed on the 14th day after inoculation, and the mice were randomly divided into 2 groups with 8 mice in each group according to the imaging effect.
2)给药:Vehicle组:以100μl 10%DMSO+90%玉米油对每只小鼠灌胃,每天1次,连续给药30天;PB-020组:PB-020(化合物11)溶解于10%DMSO+90%玉米油,以50mg/kg剂量、总体积100μl对小鼠进行灌胃,每天1次,连续给药30天。2) Administration: Vehicle group: gavage each mouse with 100 μl of 10% DMSO+90% corn oil, once a day, for 30 consecutive days; PB-020 group: PB-020 (compound 11) was dissolved in 10% DMSO + 90% corn oil, 50 mg/kg dose,
3)药效评价:小鼠开始给药后,第0,5,16,24,30天进行活体成像,测定小鼠脑部所接种MDA-MB-231-Luc细胞的荧光强度,以此来判断MDA-MB-231-Luc细胞向小鼠脑部的转移情况和化合物11对其转移/增殖的抑制程度;每天称量记录小鼠体重,一天内小鼠体重下降超过10%时暂停给药;每天观察记录小鼠的形态与行为特征。3) Efficacy evaluation: After the mice started administration, in vivo imaging was performed on the 0, 5, 16, 24, and 30 days, and the fluorescence intensity of the MDA-MB-231-Luc cells inoculated in the mouse brain was measured. To judge the transfer of MDA-MB-231-Luc cells to the mouse brain and the degree of inhibition of the transfer/proliferation by compound 11; the weight of the mice was recorded every day, and the administration was suspended when the weight of the mice decreased more than 10% within one day. ; Observe and record the morphological and behavioral characteristics of mice every day.
2.试验结果:2. Test results:
结果如图7所示,转移至小鼠脑部的MDA-MB-231-Luc细胞增殖相对缓慢,给药后24天才进入快速增殖期。在给药后24天到30天的快速增殖期,化合物11(PB-020)较为显著地抑制了MDA-MB-231-Luc细胞增殖。由于该转移模型致死率很低,无法制作生存曲线。The results are shown in FIG. 7 , the MDA-MB-231-Luc cells transferred to the mouse brains proliferated relatively slowly, and entered a
(三)化合物11与PD-1抗体联用对免疫正常小鼠中肺癌细胞增殖的抑制作用(3) Inhibitory effect of compound 11 combined with PD-1 antibody on the proliferation of lung cancer cells in immunized normal mice
1.试验步骤:1. Test steps:
1)建模及分组:32只5周龄C57BL/6雌性免疫正常小鼠,适应性饲养1周,用于实验。小鼠LLC肺癌细胞常规培养,满足实验要求后,制成密度为1x107/ml的单细胞悬液,待用。将LLC细胞接种于C57BL/6小鼠右侧腋下,返回继续饲养。待肿瘤长至50-150mm3时,将小鼠随机分为4组,每组8只。1) Modeling and grouping: 32 5-week-old C57BL/6 female immunized normal mice, adaptively reared for 1 week, were used for the experiment. Mouse LLC lung cancer cells were routinely cultured, and after meeting the experimental requirements, a single-cell suspension with a density of 1×107 /ml was prepared for use. LLC cells were inoculated into the right armpit of C57BL/6 mice and returned to continue feeding. When the tumors grew to 50-150 mm3 , the mice were randomly divided into 4 groups with 8 mice in each group.
2)给药:Vehicle组:以200μl 10%DMSO+90%玉米油(#C8267-500ML,Sigma-Aldrich)对每只小鼠灌胃,每天1次,连续给药直至小鼠死亡;PB-020组:PB-020(化合物11)溶解于10%DMSO+90%玉米油,以50mg/kg剂量、总体积200μl对小鼠进行灌胃,每天1次,连续给药直至小鼠死亡;PD1组:将200μg anti-mouse PD-1(RMP1-14,Bio X Cell)溶解于100μl PBS,腹腔注射每只小鼠,每天1次,每3天给药1次;PB-020+PD1组:按上述单药的给药方式和剂量分别给药。2) Administration: Vehicle group: gavage each mouse with 200 μl of 10% DMSO+90% corn oil (#C8267-500ML, Sigma-Aldrich), once a day, continuously administered until the mice died; PB- Group 020: PB-020 (compound 11) was dissolved in 10% DMSO + 90% corn oil, and the mice were intragastrically administered at a dose of 50 mg/kg and a total volume of 200 μl, once a day, continuously administered until the mice died; PD1 Group: Dissolve 200 μg anti-mouse PD-1 (RMP1-14, Bio X Cell) in 100 μl PBS, intraperitoneally inject each mouse, once a day, once every 3 days; PB-020+PD1 group: According to the above-mentioned single drug administration mode and dosage, it is administered separately.
3)药效评价:小鼠开始给药后,跟踪测量肿瘤体积大小,每周2次;每天称量记录小鼠体重,一天内小鼠体重下降超过10%时暂停给药;每天观察记录小鼠的形态与行为特征;记录小鼠死亡时间,肿瘤体积超过2000mm3时,根据动物伦理要求,认定小鼠死亡,至Vehicle组小鼠全部死亡,绘制小鼠生存曲线。3) Efficacy evaluation: After the mice started administration, the tumor volume was tracked and measured twice a week; the weight of the mice was weighed and recorded every day, and the administration was suspended when the weight of the mice decreased by more than 10% within one day; Morphological and behavioral characteristics of the mice; the time of death of the mice was recorded. When the tumor volume exceeded 2000 mm3 , the mice were considered dead according to the requirements of animal ethics. When all the mice in the Vehicle group died, the survival curves of the mice were drawn.
2.试验结果:2. Test results:
结果如图8所示,化合物11(PB-020)单药能有效抑制鼠源LLC肺癌细胞在免疫正常小鼠体内的增殖,其效果略优于PD-1抗体单药;化合物11与PD-1抗体联用具有一定的协同作用,能高效抑制LLC细胞增殖,显著延长小鼠的生存期。The results are shown in Figure 8, compound 11 (PB-020) single drug can effectively inhibit the proliferation of mouse-derived LLC lung cancer cells in immune normal mice, and its effect is slightly better than that of PD-1 antibody single drug; 1 The combination of antibodies has a certain synergistic effect, which can effectively inhibit the proliferation of LLC cells and significantly prolong the survival period of mice.
综上所述,本发明设计合成了靶向胰岛素样生长因子-1受体的新型小分子抑制剂化合物11(PB-020)及其系列类似物。相对于目前处于临床试验阶段的苦鬼臼毒素,化合物11在维持其作用机理和生化特性的同时,具有更高的血脑屏障通透性和更长的体内半衰期。化合物11在单独使用时,能有效抑制多种裸鼠移植瘤(尤其是胶质瘤)的增殖和脑转移;化合物11与替莫唑胺联用能高效抑制原位移植胶质瘤细胞的增殖,显著延长小鼠的生存期;化合物11和PD-1抗体联用具有一定的协同作用,能高效抑制LLC细胞在免疫正常小鼠体内的增殖,显著延长小鼠的生存期。In conclusion, the present invention designed and synthesized a novel small molecule inhibitor compound 11 (PB-020) targeting insulin-like growth factor-1 receptor and its series of analogs. Compared with podophyllotoxin currently in clinical trials, compound 11 has higher blood-brain barrier permeability and longer in vivo half-life while maintaining its mechanism of action and biochemical properties. When used alone, compound 11 can effectively inhibit the proliferation and brain metastasis of a variety of nude mice xenografts (especially gliomas); the combination of compound 11 and temozolomide can effectively inhibit the proliferation of orthotopic transplanted glioma cells, significantly prolonging the Survival period of mice; the combination of compound 11 and PD-1 antibody has a certain synergistic effect, which can effectively inhibit the proliferation of LLC cells in immunized normal mice, and significantly prolong the survival period of mice.
最后,需要指出的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。Finally, it should be pointed out that the above list is only a few specific embodiments of the present invention. Obviously, the present invention is not limited to the above embodiments, and many modifications are possible. All modifications that those of ordinary skill in the art can directly derive or associate from the disclosure of the present invention shall be considered as the protection scope of the present invention.
Claims (8)




Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011040572.4ACN112125916A (en) | 2020-09-28 | 2020-09-28 | Novel small molecule inhibitors of insulin-like growth factor-1 receptor and their uses |
| CN202111128797.XACN114315848B (en) | 2020-09-28 | 2021-09-26 | Small molecule inhibitors of insulin-like growth factor-1 receptor and uses thereof |
| CN202111130655.7ACN114276362B (en) | 2020-09-28 | 2021-09-26 | Use of IGF-1R small molecule inhibitors in the preparation of combination drugs for the treatment and/or prevention of cancer |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202011040572.4ACN112125916A (en) | 2020-09-28 | 2020-09-28 | Novel small molecule inhibitors of insulin-like growth factor-1 receptor and their uses |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112125916Atrue CN112125916A (en) | 2020-12-25 |
Family
ID=73843316
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202011040572.4APendingCN112125916A (en) | 2020-09-28 | 2020-09-28 | Novel small molecule inhibitors of insulin-like growth factor-1 receptor and their uses |
| CN202111130655.7AActiveCN114276362B (en) | 2020-09-28 | 2021-09-26 | Use of IGF-1R small molecule inhibitors in the preparation of combination drugs for the treatment and/or prevention of cancer |
| CN202111128797.XAActiveCN114315848B (en) | 2020-09-28 | 2021-09-26 | Small molecule inhibitors of insulin-like growth factor-1 receptor and uses thereof |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111130655.7AActiveCN114276362B (en) | 2020-09-28 | 2021-09-26 | Use of IGF-1R small molecule inhibitors in the preparation of combination drugs for the treatment and/or prevention of cancer |
| CN202111128797.XAActiveCN114315848B (en) | 2020-09-28 | 2021-09-26 | Small molecule inhibitors of insulin-like growth factor-1 receptor and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (3) | CN112125916A (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114315848A (en)* | 2020-09-28 | 2022-04-12 | 浙江大学 | Small molecule inhibitor of insulin-like growth factor-1 receptor and its use |
| WO2023088257A1 (en)* | 2021-11-19 | 2023-05-25 | 杭州拿因生物科技有限责任公司 | Picropodophyllotoxin derivative, pharmaceutical composition comprising same and use thereof |
| CN118178447A (en)* | 2024-03-15 | 2024-06-14 | 北京悦康科创医药科技股份有限公司 | Application of antisense oligonucleotide in preparing medicament for treating thyroid abnormality-induced diseases |
| WO2024206858A1 (en) | 2023-03-30 | 2024-10-03 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| WO2024229406A1 (en) | 2023-05-04 | 2024-11-07 | Revolution Medicines, Inc. | Combination therapy for a ras related disease or disorder |
| WO2025034702A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Rmc-6291 for use in the treatment of ras protein-related disease or disorder |
| WO2025080946A2 (en) | 2023-10-12 | 2025-04-17 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104672249A (en)* | 2013-11-26 | 2015-06-03 | 上海医药工业研究院 | Podophyllotoxin derivative, preparation method thereof, medicinal composition and application thereof |
| CN110669054A (en)* | 2018-07-03 | 2020-01-10 | 南京药石科技股份有限公司 | Insulin-like growth factor-1 receptor tyrosine kinase inhibitors and uses thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2009006466A (en)* | 2006-12-13 | 2009-06-26 | Schering Corp | Methods of treatment. |
| CN112125916A (en)* | 2020-09-28 | 2020-12-25 | 杭州仁者生物科技有限公司 | Novel small molecule inhibitors of insulin-like growth factor-1 receptor and their uses |
- 2020
- 2020-09-28CNCN202011040572.4Apatent/CN112125916A/enactivePending
- 2021
- 2021-09-26CNCN202111130655.7Apatent/CN114276362B/enactiveActive
- 2021-09-26CNCN202111128797.XApatent/CN114315848B/enactiveActive
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104672249A (en)* | 2013-11-26 | 2015-06-03 | 上海医药工业研究院 | Podophyllotoxin derivative, preparation method thereof, medicinal composition and application thereof |
| CN110669054A (en)* | 2018-07-03 | 2020-01-10 | 南京药石科技股份有限公司 | Insulin-like growth factor-1 receptor tyrosine kinase inhibitors and uses thereof |
Non-Patent Citations (1)
| Title |
|---|
| 宋振玉: "《药物代谢研究》", 31 July 1990* |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114315848A (en)* | 2020-09-28 | 2022-04-12 | 浙江大学 | Small molecule inhibitor of insulin-like growth factor-1 receptor and its use |
| CN114315848B (en)* | 2020-09-28 | 2023-09-01 | 杭州拿因生物科技有限责任公司 | Small molecule inhibitors of insulin-like growth factor-1 receptor and uses thereof |
| WO2023088257A1 (en)* | 2021-11-19 | 2023-05-25 | 杭州拿因生物科技有限责任公司 | Picropodophyllotoxin derivative, pharmaceutical composition comprising same and use thereof |
| WO2024206858A1 (en) | 2023-03-30 | 2024-10-03 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| WO2024229406A1 (en) | 2023-05-04 | 2024-11-07 | Revolution Medicines, Inc. | Combination therapy for a ras related disease or disorder |
| WO2025034702A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Rmc-6291 for use in the treatment of ras protein-related disease or disorder |
| WO2025080946A2 (en) | 2023-10-12 | 2025-04-17 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| CN118178447A (en)* | 2024-03-15 | 2024-06-14 | 北京悦康科创医药科技股份有限公司 | Application of antisense oligonucleotide in preparing medicament for treating thyroid abnormality-induced diseases |
Also Published As
| Publication number | Publication date |
|---|---|
| CN114315848B (en) | 2023-09-01 |
| CN114276362B (en) | 2023-09-01 |
| CN114315848A (en) | 2022-04-12 |
| CN114276362A (en) | 2022-04-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112125916A (en) | Novel small molecule inhibitors of insulin-like growth factor-1 receptor and their uses | |
| CN111484477B (en) | Benzopyridone heterocyclic compound and application thereof | |
| CN113105454B (en) | FGFR4 inhibitor, its preparation method and application | |
| JP7128345B2 (en) | Diaryl macrocyclic compound, pharmaceutical composition and use thereof | |
| JP6951406B2 (en) | Flabagulin derivative | |
| EA029768B1 (en) | Heterocyclyl compounds | |
| JP2021525264A (en) | 3-Indazolinone compounds, their preparation methods, and their use in pharmaceuticals | |
| CN113698415A (en) | Novel oridonin analogue and derivative, preparation method and medical application thereof | |
| CN110669054B (en) | Insulin-like growth factor-1 receptor tyrosine kinase inhibitor and use thereof | |
| US20060160773A1 (en) | Combretastatin derivatives with cytotoxic action | |
| JP2022513101A (en) | Spiro ring compounds and their pharmaceutical uses | |
| CN107216283B (en) | A kind of beta-elemene derivatives and its preparation method and application containing dihydropyridine structure | |
| AU2015349306B2 (en) | New type of cytidine derivative dimer and application thereof | |
| CN112384508B (en) | Tricyclic ASK1 inhibitor and application thereof | |
| WO2019184919A1 (en) | Adamantane-containing compound and use thereof in treating cancer | |
| CN115960106B (en) | Mitochondrial RNA polymerase inhibitors and derivatives thereof, pharmaceutical compositions and medicinal uses | |
| WO2019228170A1 (en) | Octahydropentene compound, preparation method therefor, and pharmaceutical application thereof | |
| CN114380780B (en) | A new type of citrin A derivative, its preparation method and medicinal use | |
| WO2019091046A1 (en) | Preparation method for lenalidomide derivative and application thereof | |
| CN110526907B (en) | Benzoxazinone derivatives and their applications | |
| KR20220104734A (en) | Oleanane cinnamamide derivatives, preparation method and use thereof | |
| JP2017537108A (en) | 4H-pyrido [1,2-a] pyrimidin-4-one compounds | |
| WO2023088257A1 (en) | Picropodophyllotoxin derivative, pharmaceutical composition comprising same and use thereof | |
| WO2021215522A1 (en) | Linderapyrone, and analogue thereof | |
| WO2021164749A1 (en) | Insulin-like growth factor-1 receptor tyrosine kinase inhibitor and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| TA01 | Transfer of patent application right | Effective date of registration:20210729 Address after:310014 No. 866, yuhangtang Road, Xihu District, Hangzhou City, Zhejiang Province Applicant after:ZHEJIANG University Address before:310026 room a0101-31, building 2, 452, Baiyang street, Hangzhou Economic and Technological Development Zone, Zhejiang Province Applicant before:Hangzhou Renzhe Biotechnology Co.,Ltd. | |
| TA01 | Transfer of patent application right | ||
| WD01 | Invention patent application deemed withdrawn after publication | Application publication date:20201225 | |
| WD01 | Invention patent application deemed withdrawn after publication |