CN109416927B - System and method for secondary analysis of nucleotide sequencing data - Google Patents
System and method for secondary analysis of nucleotide sequencing dataDownload PDFInfo
- Publication number
- CN109416927B CN109416927BCN201780040788.0ACN201780040788ACN109416927BCN 109416927 BCN109416927 BCN 109416927BCN 201780040788 ACN201780040788 ACN 201780040788ACN 109416927 BCN109416927 BCN 109416927B
- Authority
- CN
- China
- Prior art keywords
- nucleotide subsequence
- sequencing
- processing
- reference sequence
- alignment
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images







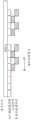






Classifications
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G16B30/10—Sequence alignment; Homology search
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G16B30/20—Sequence assembly
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
- G16B40/30—Unsupervised data analysis
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B50/00—ICT programming tools or database systems specially adapted for bioinformatics
- G16B50/30—Data warehousing; Computing architectures
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Physics & Mathematics (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Medical Informatics (AREA)
- Analytical Chemistry (AREA)
- Bioinformatics & Computational Biology (AREA)
- Evolutionary Biology (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Theoretical Computer Science (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Data Mining & Analysis (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Software Systems (AREA)
- Databases & Information Systems (AREA)
- Epidemiology (AREA)
- Computer Vision & Pattern Recognition (AREA)
- Bioethics (AREA)
- Artificial Intelligence (AREA)
- Public Health (AREA)
- Evolutionary Computation (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
Abstract
Description
Translated fromChinese相关申请related application
本申请要求2016年10月7日提交的美国临时申请No.62/405824的优先权;其内容通过引用整体并入本文。This application claims priority to US Provisional Application No. 62/405824, filed October 7, 2016; the contents of which are incorporated herein by reference in their entirety.
背景技术Background technique
技术领域technical field
本公开一般涉及DNA测序领域,并且更具体地涉及用于进行下一代测序应用的实时二级分析的系统和方法。The present disclosure relates generally to the field of DNA sequencing, and more particularly to systems and methods for performing real-time secondary analysis for next generation sequencing applications.
相关技术的描述Description of related technologies
可以通过鉴定序列读段中-相对于参考序列-的变异来鉴定遗传突变。为了鉴定变异,可以使用测序仪器对来自受试者的样品进行完全测序以获得序列读段。在获得序列读段之后,可以在变异识别之前组装或比对序列读段。因此,鉴定变异包括依序进行的不同步骤,并且在测序过程结束后执行可能是耗时的。Genetic mutations can be identified by identifying variations in sequence reads - relative to a reference sequence. To identify variants, a sample from a subject can be fully sequenced using a sequencing instrument to obtain sequence reads. After sequence reads are obtained, the sequence reads can be assembled or aligned prior to variant calling. Therefore, identifying variants involves different steps that are performed sequentially and can be time-consuming to perform after the sequencing process is complete.
发明概述Summary of the invention
本文公开了用于对多核苷酸进行测序的系统和方法。在一个实施方案中,该系统包括:存储器,其包括参考核苷酸序列;处理器,其被配置为执行指令,该指令进行包括以下的方法:从测序系统接收读段的第一核苷酸子序列;使用第一比对路径处理第一核苷酸子序列,以确定参考序列上读段的第一多个候选位置;根据确定的候选位置确定第一核苷酸子序列是否比对至参考序列;从测序系统接收第二核苷酸子序列;使用以下路径处理第二核苷酸子序列以确定比对至参考序列的读段的第二多个候选位置:如果读段比对至参考序列,则使用第二比对路径,并且如果不是这样,则使用第一比对路径,其中第二比对路径比第一比对路径计算效率更高,以确定读段的第二多个候选位置。Disclosed herein are systems and methods for sequencing polynucleotides. In one embodiment, the system comprises: a memory comprising a reference nucleotide sequence; a processor configured to execute instructions performing a method comprising: receiving a first nucleotide of a read from a sequencing system subsequence; processing the first nucleotide subsequence using a first alignment path to determine a first plurality of candidate positions for the read on the reference sequence; determining whether the first nucleotide subsequence aligns to a reference sequence; receiving a second nucleotide subsequence from the sequencing system; processing the second nucleotide subsequence to determine a second plurality of candidate positions for reads that align to the reference sequence using the following pathway: if the read aligns to reference sequence, the second alignment path is used, and if not, the first alignment path is used, where the second alignment path is more computationally efficient than the first alignment path to determine a second more Candidate position.
在一个实施方案中,该方法包括:在测序运行期间从测序系统接收第一核苷酸子序列;并且使用第一分析路径或第二分析路径基于参考序列进行读段的第一核苷酸子序列的二级分析,其中第二分析路径在进行二级分析方面比第一处理路径计算效率更高。In one embodiment, the method comprises: receiving a first nucleotide subsequence from a sequencing system during a sequencing run; and performing the first nucleotide subsequence of the read based on a reference sequence using a first analysis path or a second analysis path. Secondary analysis of a sequence, wherein the second analysis path is more computationally efficient at performing the secondary analysis than the first processing path.
附图简述Brief description of the drawings
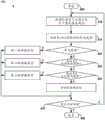
图1是示出用于进行实时分析的示例测序系统的示意图。Figure 1 is a schematic diagram illustrating an example sequencing system for performing real-time analysis.
图2示出了用于进行实时分析的示例计算机系统的功能框图。Figure 2 shows a functional block diagram of an example computer system for performing real-time analysis.
图3是通过边合成边测序的示例方法的流程图。3 is a flowchart of an example method by sequencing by synthesis.
图4是用于进行碱基识别的示例方法的流程图。4 is a flowchart of an example method for performing base calling.
图5A和图5B示出了示例迭代比对和变异识别。Figures 5A and 5B illustrate example iterative alignments and variant calling.
图6是用于进行实时二级序列分析的示例方法的流程图。6 is a flowchart of an example method for performing real-time secondary sequence analysis.
图7A和图7B是将二级分析的传统方法(图7A)与二级分析的迭代方法(图7B)进行比较的示意图。7A and 7B are schematic diagrams comparing a traditional method of secondary analysis (FIG. 7A) with an iterative method of secondary analysis (FIG. 7B).
图8是以16个碱基间隔生成读段的示意图。Figure 8 is a schematic diagram of generating reads at 16 base intervals.
图9A是用于进行实时二级分析的示例方法的流程图。图9B是示出按K-Mer处理的数据的预测线图。图9C是示出运行时间的条形图。9A is a flowchart of an example method for performing real-time secondary analysis. Figure 9B is a line graph showing predictions for K-Mer processed data. Figure 9C is a bar graph showing elapsed time.
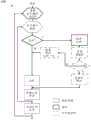
图10是用于进行实时二级分析的示例方法的另一流程图。10 is another flowchart of an example method for performing real-time secondary analysis.
图11A和图11B将现有变异识别器(variant caller)(图11A)与使用如本文所述的高置信度低处理路径的变异识别器(图11B)进行比较。Figures 11A and 11B compare an existing variant caller (Figure 11A) with a variant caller (Figure 11B) that uses a high confidence low processing path as described herein.
发明详述Detailed description of the invention
在以下详述中,参考了附图,附图形成了其的一部分。在附图中,除非上下文另有指示,否则类似的符号通常标识类似的组件。在详述、附图和权利要求中描述的说明性实施方案不旨在限制。在不脱离本文提出的主题的精神或范围的情况下,可以利用其他实施方案,并且可以进行其他改变。容易理解的是,如本文一般描述的和附图中所示的本公开的方面可以以各种不同的配置来布置、替换、组合、分离和设计,所有这些都是本文明确考虑的。In the following detailed description, reference is made to the accompanying drawings, which form a part hereof. In the drawings, similar symbols typically identify similar components, unless context dictates otherwise. The illustrative embodiments described in the detailed description, drawings, and claims are not intended to be limiting. Other embodiments may be utilized, and other changes may be made, without departing from the spirit or scope of the subject matter presented here. It is readily understood that aspects of the disclosure as generally described herein and illustrated in the drawings may be arranged, substituted, combined, separated and designed in various different configurations, all of which are expressly contemplated herein.
本文公开了用有时效性的方式进行核苷酸测序数据的二级分析的系统和方法。在一些实施方案中,该方法包括迭代地进行二级分析,而序列读段由测序系统生成。二级分析可以包括序列读段与参考序列(如,人参考基因组序列)的比对以及利用该比对来检测样品和参考之间的差异。二级分析可以能够检测遗传差异、变异检测和基因分型,鉴定单核苷酸多态性(SNP)、小插入和缺失(indel)以及DNA中的结构变化,诸如拷贝数变异(CNV)和染色体重排。Disclosed herein are systems and methods for secondary analysis of nucleotide sequencing data in a time-sensitive manner. In some embodiments, the method includes iteratively performing secondary analysis while the sequence reads are generated by the sequencing system. Secondary analysis can include the alignment of sequence reads to a reference sequence (eg, a human reference genome sequence) and using the alignment to detect differences between the sample and the reference. Secondary analysis can enable detection of genetic differences, variant detection and genotyping, identification of single nucleotide polymorphisms (SNPs), small insertions and deletions (indels), and structural changes in DNA such as copy number variations (CNVs) and Chromosomal rearrangement.
通过在生成序列读段的同时进行二级分析,该系统和方法可以实时(或以零或低延时)迭代地确定初步变异识别。变异确定的最终结果可在测序运行结束后不久(或紧接其后)获得。或者,如果在运行期间以足够的置信度获得变异识别,则可以提前终止测序运行。在一些实施方案中,仅将与变异确定(如,变异识别)有关的信息从测序系统传送出去。与在外部系统中进行的变异确定相比,这可以减少或最小化所需的数据带宽。另外,可以仅将变异信息发送到计算系统(如,云计算系统)以进行进一步处理。在该实施方案中,可以在完成整个测序过程之前终止测序运行。例如,如果在测序运行的多个测序循环后确定目标病原体的身份,则可以终止测序运行。因此,可以减少特定应答(如,病原体鉴定)的时间。在一个实施方案中,系统的输出和中间结果可以包括以下的直方图:重复、精确匹配、单和双SNP以及单和双插入缺失。By performing secondary analysis while sequence reads are being generated, the systems and methods can iteratively determine preliminary variant calls in real-time (or with zero or low latency). The final results of variant calling are available shortly after (or immediately after) the completion of the sequencing run. Alternatively, a sequencing run can be terminated early if variant calls are obtained with sufficient confidence during the run. In some embodiments, only information relevant to variant determination (eg, variant identification) is communicated from the sequencing system. This can reduce or minimize the required data bandwidth compared to variant determination performed in an external system. Alternatively, the mutation information may only be sent to a computing system (eg, a cloud computing system) for further processing. In this embodiment, the sequencing run can be terminated before the entire sequencing process is complete. For example, a sequencing run can be terminated if the identity of the pathogen of interest is determined after multiple sequencing cycles of the sequencing run. Thus, the time to a specific response (eg, pathogen identification) can be reduced. In one embodiment, the output and intermediate results of the system may include histograms of: duplicates, exact matches, single and double SNPs, and single and double indels.
定义definition
除非另外定义,否则本文使用的技术和科学术语具有与本公开所属领域的普通技术人员通常理解的含义相同的含义。参见,如Singleton等人,Dictionary ofMicrobiology and Molecular Biology第2版,J.Wiley&Sons(New York,NY 1994);Sambrook等人,Molecular Cloning,A Laboratory Manual,Cold Springs Harbor Press(Cold Springs Harbor,NY 1989)。出于本公开的目的,以下术语定义如下。Unless defined otherwise, technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure belongs. See, eg, Singleton et al., Dictionary of Microbiology and Molecular Biology 2nd Edition, J. Wiley & Sons (New York, NY 1994); Sambrook et al., Molecular Cloning, A Laboratory Manual, Cold Springs Harbor Press (Cold Springs Harbor, NY 1989) . For the purposes of this disclosure, the following terms are defined below.
用于进行实时二级分析的测序仪Sequencers for real-time secondary analysis
本文公开了用于以时间和/或计算资源有效方式迭代地进行二级分析的系统和方法。二级分析可以包括序列读段与参考序列(比,人参考基因组序列)的比对和利用该比对来检测样品和参考之间的差异。二级分析可以能够检测遗传差异、变异检测和基因分型,鉴定单核苷酸多态性(SNP)、小插入和缺失(indel)以及DNA中的结构变化,诸如拷贝数变异(CNV)和染色体重排。可以对一个测序循环进行二级分析,同时为下一个测序循环生成测序数据。Disclosed herein are systems and methods for iteratively performing secondary analysis in a time and/or computational resource efficient manner. Secondary analysis can include alignment of sequence reads to a reference sequence (alignment, human reference genome sequence) and use of this alignment to detect differences between the sample and the reference. Secondary analysis can enable detection of genetic differences, variant detection and genotyping, identification of single nucleotide polymorphisms (SNPs), small insertions and deletions (indels), and structural changes in DNA such as copy number variations (CNVs) and Chromosomal rearrangement. Secondary analysis can be performed on one sequencing cycle while sequencing data is generated for the next sequencing cycle.
图1是示出用于进行实时二级分析的示例测序系统100的示意图。由测序系统100利用的测序方法的非限制性实例可包括边合成边测序和Heliscope单分子测序。测序系统100可以包括光学系统102,其被配置为使用由作为测序系统100的一部分的射流系统104供应的测序试剂来生成原始测序数据。原始测序数据可以包括由光学系统102捕获的荧光图像。作为测序系统100的一部分的计算机系统106可以被配置为经由通信通道108a和108b控制光学系统102和射流系统104。例如,光学系统102的计算机接口110可以被配置为通过通信通道108a与计算机系统106通信。FIG. 1 is a schematic diagram illustrating an
在测序反应期间,射流系统104可以将试剂流通过一个或多个试剂管112引导入位于安装台116上的流动池114并从位于安装台116上的流动池114引导出。试剂可以是,例如荧光标记的核苷酸、缓冲液、酶和裂解试剂。流动池114可包括至少一个流体通道。流动池114可以是图案化阵列流动池或随机阵列流动池。流动池114可包括多个单链多核苷酸簇,以在至少一个流体通道中测序。多核苷酸的长度可以是变化的,例如为200个碱基至1000个碱基。多核苷酸可以附接至流动池114的一个或多个流体通道。在一些实施方案中,流动池114可以包括多个珠粒,其中每个珠粒可以包括待测序的多核苷酸的多个拷贝。安装台116可以被配置为允许流动池114相对于光学系统102的其他组件适当地比对和移动。在一个实施方案中,安装台116可用于将流动池114与透镜118比对。During a sequencing reaction, fluidics system 104 may direct reagent flow through one or
光学系统102可包括多个激光器120,其被配置为生成预定波长的光。由激光器120生成的光可以穿过纤维光缆122以激发流动池114中的荧光标签。安装在聚焦器124上的透镜118可以沿z轴移动。聚焦的荧光发射可以由检测器126检测,例如电荷耦合装置(CCD)传感器或互补金属氧化物半导体(CMOS)传感器。Optical system 102 may include a plurality of
光学系统102的过滤器总成128可以被配置为过滤流动池114中的荧光标签的荧光发射。过滤器总成128可包括第一过滤器和第二过滤器。每个过滤器可以是长通过滤器、短通过滤器或带通过滤器,这取决于系统中使用的荧光分子的类型。第一过滤器可以被配置为通过检测器126检测第一荧光标签的荧光发射。第二过滤器可以被配置为通过检测器126检测第二荧光标签的荧光发射。利用过滤器总成128中的两种过滤器,检测器126可以检测到两种不同波长的荧光发射。
在一些实施方案中,光学系统102可以包括分色器,其经配置以分离荧光发射。光学系统102可包括两个检测器,与用于检测第一波长处的荧光发射的第一过滤器耦合的第一检测器,以及与用于检测第二波长处的荧光发射的第二过滤器耦合的第二检测器。In some embodiments, optical system 102 can include a color splitter configured to separate fluorescent emissions. Optical system 102 may include two detectors, a first detector coupled to a first filter for detecting fluorescent emissions at a first wavelength, and a second filter coupled to a second filter for detecting fluorescent emissions at a second wavelength. coupled second detector.
在使用中,将具有待测序的多核苷酸的样品加载到流动池114中并放置在安装台116中。然后计算机系统106激活射流系统104以开始测序循环。在测序反应期间,计算机系统106通过通信通道108b指示射流系统104向流动池114供应试剂,如核苷酸类似物。通过通信通道108a和计算机接口110,计算机系统106被配置为控制光学系统102的激光器120以生成预定波长的光并照射到与荧光标签耦接的核苷酸类似物上,所述荧光标签掺入与被测序的多核苷酸杂交的生长引物中。计算机系统106控制光学系统102的检测器126以捕获荧光图像中的核苷酸类似物的发射光谱。计算机系统106从检测器126接收荧光图像并处理接收的荧光图像以确定被测序的多核苷酸的核苷酸序列。In use, a sample with polynucleotides to be sequenced is loaded into the
计算机系统computer system
如上所讨论,测序系统100的计算机系统106可以被配置为控制光学系统102和射流系统104。计算机系统106可以有许多配置,图2示出了一个实施方案。如图2所示,计算机系统106可包括处理器202,其与存储器204、存储装置206和通信接口208电通信。在一个实施方案中,计算机系统106包括现场可编程门阵列(FPGA)、图形处理单元(GPU)和/或矢量中央处理单元(CPU),以进行序列比对并生成变异识别。As discussed above, the
处理器202可以被配置为执行指令,该指令使射流系统104在测序反应期间向流动池114供应试剂。处理器202可以执行控制光学系统102的激光器120以产生预定波长的光的指令。处理器202可以执行控制光学系统102的检测器126并从检测器126接收数据的指令。处理器202可以执行指令以处理从检测器126接收的数据例如荧光图像,并基于从检测器126接收的数据确定多核苷酸的核苷酸序列。Processor 202 may be configured to execute instructions that cause fluidics system 104 to supply reagents to flow
存储器204可以被配置为存储指令,所述指令用于配置处理器202以在测序系统100通电时执行计算机系统106的功能。当测序系统100断电时,存储装置206可以存储指令,所述指令用于配置处理器202以执行计算机系统106的功能。通信接口208可以被配置为促进计算机系统106、光学系统102和射流系统104之间的通信。Memory 204 may be configured to store instructions for configuring processor 202 to perform functions of
计算机系统106可以包括用户接口210,其被配置为与用于显示测序系统100的测序结果(包括二级分析(诸如变异识别)的结果)的显示设备(未示出)通信。用户接口210可以被配置为接收来自测序系统100的用户的输入。计算机系统106的光学系统接口212和射流系统接口214可被配置为通过图1所示的通信通道108a和108b控制光学系统102和射流系统104。例如,光学系统接口212可以通过通信通道108a与光学系统102的计算机接口110通信。
计算机系统106可包括核酸碱基确定器216,其被配置为使用从检测器126接收的数据确定多核苷酸的核苷酸序列。核酸碱基确定器216可以使用由检测器126捕获的荧光图像在流动池114中生成多核苷酸簇的位置的模板。核酸碱基确定器216可以基于所生成的位置模板在由检测器126捕获的荧光图像中记录流动池114中的多核苷酸簇的位置。核酸碱基确定器216可以从荧光图像中提取荧光发射的强度以生成提取的强度。核酸碱基确定器216可以从提取的强度确定多核苷酸的碱基。核酸碱基确定器216可以确定所确定的多核苷酸碱基的质量评分。
计算机系统106可以包括迭代比对器218和变异识别器220,诸如Strelka变异识别器(sites.google.com/site/strelkasomaticvariantcaller/home/faq)。在测序循环期间,迭代比对器218可以将由核酸碱基确定器216所确定的序列读段与参考序列比对。比对的序列读段可以具有相关的评分。评分可以是序列读段已正确地比对至参考序列的概率(如,错配百分比)。在一些实施方式中,计算机系统106可以包括硬件,诸如现场可编程门阵列(FPGA)或图形处理单元(GPU),用于将序列读段与参考序列比对并用于确定变异识别。在一些实施方案中,迭代比对器218和变异识别器220可以由与计算机系统106不同的计算机系统来实现。在一些实施方案中,计算机系统106可以是测序系统100的集成组件。在一些实施方案中,光学系统102、射流系统104和/或计算机系统106可以集成到一个机器中。The
边合成边测序Sequencing by Synthesis
图3是用于利用测序系统100的边合成边测序的示例方法300的流程图。在框305处开始方法300之后,在框310处接收包括片段化双链多核苷酸片段的流动池114。片段化的双链多核苷酸片段可以由脱氧核糖核酸(DNA)样品生成。DNA样品可以来自各种来源,例如生物样品、细胞样品、环境样品或其任意组合。DNA样品可包括来自患者的生物流体、组织和细胞中的一种或多种。例如,DNA样品可以取自或包括血液、尿液、脑脊髓液、胸膜液、羊水、精液、唾液、骨髓、活检样品或其任意组合。FIG. 3 is a flowchart of an
DNA样品可包括来自目标细胞的DNA。目标细胞可以变化,并且在一些实施方案中表达恶性表型。在一些实施方案中,目标细胞可包括肿瘤细胞骨髓细胞、癌细胞、干细胞内皮细胞、病毒感染的病原体细胞、寄生生物体细胞或其任意组合。A DNA sample can include DNA from cells of interest. Target cells can vary, and in some embodiments express a malignant phenotype. In some embodiments, target cells may include tumor cells, myeloid cells, cancer cells, stem cells, endothelial cells, virus-infected pathogen cells, parasitic organism cells, or any combination thereof.
片段化的双链多核苷酸片段的长度可以为200个碱基至1000个碱基。当在框310处接收包括片段化的双链多核苷酸片段的流动池114,方法300进行至框315,其中将双链多核苷酸片段桥扩增至附接于流动池(例如流动池114)的一个或多个通道的内表面的双链多核苷酸片段簇。流动池的一个或多个通道的内表面可以包括两种类型的引物,例如第一引物类型(P1)和第二引物类型(P2),并且DNA片段可以通过熟知的方法扩增。Fragmented double-stranded polynucleotide fragments can be 200 bases to 1000 bases in length. When the
在流动池114内生成簇之后,方法300可以开始边合成边测序过程。边合成边测序过程可以包括确定单链多核苷酸片段的簇的核苷酸序列。为了确定具有序列5’-P1-F-A2R-3’的单链多核苷酸片段的簇的序列,可以添加具有序列A2F(其是序列A2R的互补序列)的引物,并在框320处用具有0个、1个或2个标签的核苷酸类似物通过DNA聚合酶延伸以形成生长引物-多核苷酸。After the clusters are generated within the
在每个测序循环期间,可以添加四种类型的核苷酸类似物并将其掺入生长的引物-多核苷酸中。四种类型的核苷酸类似物可以具有不同的修饰。例如,第一类型的核苷酸可以是不与任何荧光标记缀合的脱氧鸟苷三磷酸(dGTP)的类似物。第二类型的核苷酸可以是经由接头与第一类型的荧光标签缀合的脱氧胸苷三磷酸(dTTP)的类似物。第三类型的核苷酸可以是经由接头与第二类型的荧光标签缀合的脱氧胞苷三磷酸(dCTP)的类似物。第四类型的核苷酸可以是经由一个或多个接头与第一类型的荧光标签和第二类型的荧光标签缀合的脱氧腺苷三磷酸(dATP)的类似物。接头可包含一种或多种裂解基团。在随后的测序循环之前,可以从核苷酸类似物中去除荧光标签。例如,将荧光标签附接至核苷酸类似物的接头可以包含叠氮化物和/或烷氧基基团,例如在相同的碳上,使得接头可以在每次掺入循环后被膦试剂裂解,从而从随后的测序循环中释放荧光标签。During each sequencing cycle, four types of nucleotide analogs can be added and incorporated into the growing primer-polynucleotide. The four types of nucleotide analogs can have different modifications. For example, the first type of nucleotide may be an analog of deoxyguanosine triphosphate (dGTP) that is not conjugated to any fluorescent label. The second type of nucleotide may be an analog of deoxythymidine triphosphate (dTTP) conjugated to the first type of fluorescent tag via a linker. The third type of nucleotide may be an analog of deoxycytidine triphosphate (dCTP) conjugated to the second type of fluorescent tag via a linker. The fourth type of nucleotide may be an analog of deoxyadenosine triphosphate (dATP) conjugated to the first type of fluorescent tag and the second type of fluorescent tag via one or more linkers. A linker may contain one or more cleavage groups. Fluorescent tags can be removed from the nucleotide analogs prior to subsequent sequencing cycles. For example, a linker that attaches a fluorescent tag to a nucleotide analog may contain azide and/or alkoxy groups, e.g., on the same carbon, such that the linker can be cleaved by a phosphine reagent after each cycle of incorporation , thereby releasing the fluorescent tags from subsequent sequencing cycles.
核苷酸三磷酸可以在3’位置处被可逆地阻断,使得控制测序,并且每个循环中每个延伸的引物-多核苷酸上可以添加不超过一个核苷酸类似物。例如,核苷酸类似物的3’核糖位置可以包含烷氧基和叠氮基官能团,其可以通过用膦试剂裂解去除,从而产生可以进一步延伸的核苷酸。在掺入核苷酸类似物之后,射流系统104可以洗涤流动池114的一个或多个通道,以便去除任何未掺入的核苷类似物和酶。在随后的测序循环之前,可以去除可逆的3’嵌段,从而可以将另一种核苷酸类似物添加到每个延伸的引物-多核苷酸上。Nucleotide triphosphates can be reversibly blocked at the 3' position so that sequencing is controlled and no more than one nucleotide analog can be added per extended primer-polynucleotide per cycle. For example, the 3' ribose sugar position of a nucleotide analog can contain alkoxy and azido functional groups, which can be removed by cleavage with a phosphine reagent, thereby yielding a nucleotide that can be further extended. After incorporation of the nucleotide analogs, the fluidics system 104 can wash one or more channels of the
在框325处,激光器诸如激光器120可以在预定的波长处激发两个荧光标签。在框330处,可以检测来自荧光标签的信号。检测荧光标签可以包括例如通过使用两个过滤器的检测器126在两个荧光图像中捕获第一波长和第二波长处的荧光发射。第一荧光标签的荧光发射可以在第一波长处或周围,且第二荧光标签的荧光发射可以在第二波长处或周围。可以存储荧光图像以供以后离线处理。在一些实施方案中,可以处理荧光图像以实时确定每个簇中生长的引物-多核苷酸的序列。At
在在线实时荧光成像处理中,可以在框335处理包含检测到的荧光信号的荧光图像,并且可以确定掺入的核苷酸的碱基。对于确定的每个核苷酸碱基,可以在框340处确定质量评分。可以在决策框345处确定是否基于例如信号的质量或在预定数量的碱基之后检测更多的核苷酸。如果要检测更多的核苷酸,则可以在框320处进行下一个测序循环的核苷酸确定。在一些实施方案中,标记的核苷酸可以添加到对应于簇的DNA链的一端。标记的核苷酸也可以添加到对应于簇的DNA链的另一端。DNA链的一端上的读段通常称为读段1组,而DNA链的另一端上的读段通常称为读段2组。允许确定来自单个多核苷酸双链体上的两个位置的序列的两个或更多个读段的测序技术被称为配对末端(PE)测序。来自单个多核苷酸双链体上的两个位置的序列的两个或更多个读段被称为读段1组、读段2组等。配对末端测序已在美国专利申请No.14/683,580中描述;其内容通过引用整体并入本文。配对末端方法的优点在于,从单个模板对两个链段进行测序,相比以随机方式对两个独立模板中的每一个进行测序,可获得更多的信息。In the online real-time fluorescence imaging process, the fluorescence image containing the detected fluorescence signal can be processed at
在下一个测序循环之前,可以从核苷酸类似物中去除荧光标签,并且可以去除可逆的3’嵌段,使得可以将另一种核苷酸类似物添加至每个延伸的引物-多核苷酸上。在处理完所有荧光图像之后,方法300可以在框350处终止。Before the next sequencing cycle, the fluorescent tag can be removed from the nucleotide analog and the reversible 3' block can be removed so that another nucleotide analog can be added to each extended primer-polynucleotide superior. After all fluorescence images have been processed,
碱基识别base calling
碱基识别可以指确定掺入正在测序的生长引物-多核苷酸簇中的核苷酸的碱基为鸟嘌呤(G)、胸腺嘧啶(T)、胞嘧啶(C)或腺嘌呤(A)的过程。图4是用于利用测序系统100进行碱基识别的示例方法400的流程图。在图3所示的框335处处理检测到的信号可以包括进行方法400的碱基识别。在框405处开始之后,可以使用激光器生成预定波长的光。所生成的光可以在框410处照射到核苷酸类似物上。例如,计算机系统106通过其光学系统接口212和通信通道108a可以使激光器120生成预定波长的光。Base calling can refer to determining whether the base of the nucleotide incorporated into the growing primer-polynucleotide cluster being sequenced is guanine (G), thymine (T), cytosine (C) or adenine (A) the process of. FIG. 4 is a flowchart of an
激光器生成的光可以照射到核苷酸类似物上,所述核苷酸类似物合并入附接至流动池(例如流动池114)的一个或多个通道的内表面上的生长引物-多核苷酸中的核苷酸类似物上。引物-多核苷酸可以包括与测序引物杂交的单链多核苷酸片段的簇。核苷酸类似物各自可包含0个、1个或2个荧光标签。两个荧光标签可以是第一荧光标签和第二荧光标签。荧光标签在被激光器生成的光激发后,可以发出荧光发射。例如,第一荧光标签可以在第一波长处产生荧光发射,其可以在例如第一荧光图像中被捕获。第二荧光标签可以在第二波长处产生荧光发射,其可以在例如第二荧光图像中被捕获。The light generated by the laser can be shone onto the nucleotide analogs incorporated into the growth primer-polynucleoside on the inner surface of one or more channels attached to the flow cell (e.g., flow cell 114) on nucleotide analogs in acid. A primer-polynucleotide may comprise a cluster of single-stranded polynucleotide fragments to which a sequencing primer hybridizes. The nucleotide analogs may each
核苷酸类似物可包括第一类型的核苷酸、第二类型的核苷酸、第三类型的核苷酸和第四类型的核苷酸。第一类型的核苷酸,例如脱氧鸟苷三磷酸(dGTP)的类似物,不缀合至第一荧光标签或第二荧光标签。第二类型的核苷酸,例如脱氧胸苷三磷酸(dTTP)的类似物,可以与第一类型的荧光标签缀合,而不是与第二类型的荧光标签缀合。第三类型的核苷酸,例如脱氧胞苷三磷酸(dCTP)的类似物,可以与第二类型的荧光标签缀合,而不与第一类型的荧光标签缀合。第四类型的核苷酸,例如脱氧腺苷三磷酸(dATP)的类似物,可以与第一类型的荧光标签和第二类型的荧光标签两者缀合。Nucleotide analogs may include nucleotides of the first type, nucleotides of the second type, nucleotides of the third type and nucleotides of the fourth type. Nucleotides of the first type, such as analogs of deoxyguanosine triphosphate (dGTP), are not conjugated to either the first fluorescent tag or the second fluorescent tag. A second type of nucleotide, such as an analog of deoxythymidine triphosphate (dTTP), can be conjugated to the first type of fluorescent tag, but not to the second type of fluorescent tag. A third type of nucleotide, such as an analog of deoxycytidine triphosphate (dCTP), can be conjugated to the second type of fluorescent tag, but not to the first type of fluorescent tag. A fourth type of nucleotide, such as an analog of deoxyadenosine triphosphate (dATP), can be conjugated to both the first type of fluorescent tag and the second type of fluorescent tag.
在框415处,可以使用至少一种检测器检测核苷酸类似物在第一波长和第二波长处的荧光发射。例如,检测器126可以捕获两个荧光图像,第一波长处的第一荧光图像和第二波长处的第二荧光图像。在从光学系统102接收到两个荧光图像之后,核酸碱基确定器216可以确定两个荧光图像中荧光发射的存在或不存在。At
由于第一类型的核苷酸不与第一荧光标签或第二荧光标签缀合,第一类型的核苷酸在第一波长处或在第二波长处不能产生或产生最小的荧光发射。在决策框420处,如果未检测到荧光发射,则可以确定核苷酸是第一类型的核苷酸,例如dGTP。如果检测到任何或多于最小的荧光发射,则方法400可以进行至决策框425。Since the nucleotides of the first type are not conjugated to the first fluorescent label or the second fluorescent label, the nucleotides of the first type produce no or minimal fluorescent emission at the first wavelength or at the second wavelength. At
由于第二类型的核苷酸与第一类型的荧光标签缀合而不与第二类型的荧光标签缀合,第二类型的核苷酸可以在第一波长处产生荧光发射而在第二波长处不产生或产生最小荧光发射。在决策框425处,如果在第二荧光图像中没有检测到第二波长处的荧光发射,并且从决策框420,在第一波光图像中检测到第一波长处的荧光发射,则核苷酸可以被确定为第二类型的核苷酸,例如dTTP。如果在第二波长处检测到荧光发射,则方法400可以进行至决策框430。Since the nucleotides of the second type are conjugated to the fluorescent tags of the first type but not to the fluorescent tags of the second type, the nucleotides of the second type can produce fluorescent emission at the first wavelength but not at the second wavelength. produce no or minimal fluorescence emission. At
由于第三类型的核苷酸与第二类型的荧光标签缀合而不与第一类型的荧光标签缀合,所以第三类型的核苷酸可以在第二波长处产生荧光发射而在第一波长处没有产生或产生最小的荧光发射。在决策框430处,如果在第一荧光图像中没有检测到第一波长处的荧光发射,并且从决策框425处,在第二荧光图像中检测到第二波长处的荧光发射,则核苷酸可以被确定为第三类型的核苷酸,例如dCTP。Since the third type of nucleotides are conjugated to the second type of fluorescent tags but not the first type of fluorescent tags, the third type of nucleotides can produce fluorescent emission at the second wavelength while at the first No or minimal fluorescence emission occurs at wavelengths. At
由于第四类型的核苷酸与第一类型的荧光标签和第二类型的荧光标签两者缀合,所以第四类型的核苷酸可以在第一波长处或第二波长处产生荧光发射。在决策框430处,如果在第一荧光图像中在第一波长处检测到荧光发射,并且从决策框425处,可以在第二荧光图像中在第二波长处检测到荧光发射,则核苷酸可以被确定为第四类型的核苷酸,例如dATP。Since the fourth type of nucleotide is conjugated to both the first type of fluorescent label and the second type of fluorescent label, the fourth type of nucleotide can produce fluorescent emission at the first wavelength or at the second wavelength. At
流动池114可以包括待测序的生长引物-多核苷酸的簇。在决策框435处,对于给定的测序循环,如果还存在至少一个具有荧光发射的簇待处理,则方法400可以在框410处继续。如果没有更多单链多核苷酸簇待处理,则方法400可以在框440处结束。The
测序方法Sequencing method
本文描述的方法可以与多种核酸测序技术结合使用。特别适用的技术是其中核酸附接在阵列中的固定位置使得它们的相对位置不改变并且其中阵列被重复成像的那些。其中在不同颜色通道中获得图像(例如,其与用于将一种核苷酸碱基类型与另一种核苷酸碱基类型区分开的不同标签相一致)的实施方案是特别适用的。在一些实施方案中,确定靶核酸的核苷酸序列的过程可以是自动化过程。优选的实施方案包括边合成边测序(“SBS”)技术。The methods described herein can be used in conjunction with a variety of nucleic acid sequencing technologies. Particularly suitable techniques are those in which the nucleic acids are attached to fixed positions in the array such that their relative positions do not change and in which the array is imaged repeatedly. Embodiments in which images are obtained in different color channels (eg, to correspond to different labels used to distinguish one nucleotide base type from another) are particularly useful. In some embodiments, the process of determining the nucleotide sequence of a target nucleic acid can be an automated process. A preferred embodiment involves sequencing by synthesis ("SBS") technology.
“边合成边测序(“SBS”)技术”通常涉及通过针对模板链迭代添加核苷酸来酶促延伸新生核酸链。在SBS的传统方法中,可以在每次递送中在聚合酶存在下将单个核苷酸单体提供给靶核苷酸。然而,在本文所述的方法中,可以在递送中在聚合酶存在下向靶核酸提供一种以上类型的核苷酸单体。"Sequencing by synthesis ("SBS") techniques" generally involve enzymatic extension of a nascent nucleic acid strand by iterative addition of nucleotides to a template strand. In the traditional method of SBS, a single nucleomonomer can be provided to a target nucleotide in each delivery in the presence of a polymerase. However, in the methods described herein, more than one type of nucleomonomer may be provided to the target nucleic acid during delivery in the presence of a polymerase.
迭代比对和变异识别Iterative Alignment and Variant Calling
图5A和图5B示出了根据一个实施方案的示例迭代比对和变异识别过程。在对一定数量的最小测序循环进行成像后,可以进行实时初步分析,以确定每个未比对读段的碱基识别和质量评分。在图5A中,显示的最小测序循环数是3。在一些实施方案中,最小测序循环可以是16、32或更多个循环。以上参照图3说明了碱基识别和质量评分确定。每个读段可以选择最可能的比对以比对至参考序列,然后读段可以堆叠在堆积(pile-up)中并且可以进行变异识别。5A and 5B illustrate an example iterative alignment and variant calling process, according to one embodiment. After imaging a certain number of minimal sequencing cycles, real-time primary analysis can be performed to determine base calls and quality scores for each unaligned read. In Figure 5A, the minimum number of sequencing cycles shown is 3. In some embodiments, the minimum sequencing cycle can be 16, 32 or more cycles. Base calling and quality score determination are explained above with reference to FIG. 3 . The most probable alignment can be selected for each read to align to a reference sequence, the reads can then be stacked in a pile-up and variation calling can be performed.
在图5A中,主要分析包括从流动池上显示的16个簇确定未比对的序列读段,诸如CCA 504a、TTA 504d和TAG 504k。在一级分析标题下,每个簇表示为一行字母,每个字母代表经测序的多核苷酸。当对最小循环数(如3个循环)进行测序,二级分析可以包括将16个序列读段与图5A中的二级分析标题下所示的参考序列(GATTACATAAGATTCTTTCATCG 508)进行比对。在二级分析图中,在参考序列下比对的序列构成了多核苷酸的堆积。作为一个实例,序列读段CCA 504a(“一级分析”标题下的第1行)、TTA 504d(第4行)和TAG 504k(第11行)可以分别比对至参考序列508的TTACAT 512子序列内的序列ACA、TTA和TAC,其分别具有1个、0个和1个错配。因此,TTACAT 512子序列的第三位置可以具有一定的正确概率地被确定为C516a而不是参考序列508中的A,并且TTACAT 512子序列的第四位置可以具有一定的正确概率地被确定为G 516b而不是参考序列中的C。可以类似地确定参考序列的其他变异。In Figure 5A, the main analysis included determining unaligned sequence reads from 16 clusters displayed on the flow cell, such as
当进行新的测序循环并确定碱基识别时,可以改进比对概率,并且读段比对可以转变到新的最可能的比对。这种转变将触发在受影响的区域中进行新的变异识别。在图5B中,在第四测序循环之后,来自第三测序循环的测序读段CCA 504a、TTA 504d和TAG 504k分别变为CCAT 504a’(“一级分析”标题下的第1行)、TTAC 504d’(第4行)和TAGG 504k’(第11行)。序列读段CCAT 504a’和TTAC 504d’仍然可以与参考序列508的TTACAT 512子序列比对,分别具有1个和0个错配。对于序列读段CCAT 504a’和TTAC 504d’,比对位置在图5A所示的迭代和图5B所示的迭代之间不改变;TTACAT 512子序列的第三位置可以确定为C 516a而不是参考序列中的A。为了使读段TAGG 504k’比对至TTACAT 512子序列,需要两个错配。然而,序列读段TAGG 504k’可以以更高的概率比对至参考序列508的TAAG 520,是因为该比对仅具有一个错配。图5A和图5B的实例示出比对位置可以随着测序运行进行转变,并且可以改善变异识别。When a new sequencing cycle is performed and base calls are determined, the alignment probability can be improved and the read alignment can be shifted to a new most probable alignment. This shift will trigger the identification of new variants in the affected regions. In Figure 5B, after the fourth sequencing cycle, the sequencing reads
在一个实施方案中,将序列读段与参考序列比对包括对于每个序列读段将最可能比对的列表保持为节点上的叶。每个叶可以具有相关的概率。可以修剪具有低于某个阈值的概率的叶。In one embodiment, aligning the sequence reads to the reference sequence includes maintaining, for each sequence read, a list of most likely alignments as a leaf on the node. Each leaf can have an associated probability. Leaves with probabilities below a certain threshold can be pruned.
实时二级分析Real-time secondary analysis
图6是用于进行实时二级序列分析的示例方法600的流程图。在方法600在框605开始之后,可以在框610处接收测序循环的成像数据。例如,计算机系统106可以从检测器126接收成像数据。在框615处,可以确定碱基并且可以确定碱基的质量评分。参考图3-图4,说明了生成成像数据并确定所确定的碱基和质量。在每个测序循环后,测序读段的长度变为长一个核苷酸。例如,在第31个测序循环后,测序读段的长度为31个核苷酸,并且在第32个测序循环后,测序读段变为长一个核苷酸至长度为32个核苷酸。6 is a flowchart of an
在决策框620处,可以确定是否已经进行了一定数量的最小测序循环。最小测序循环可以是16、32或更多个循环。如果进行的测序循环的数量低于所需的最小测序循环,则方法600进行至框610。如果进行的测序循环的数量至少是所需的最小测序循环,则方法600进行至框625。At
在框625处,确定的序列读段可以比对至参考序列。方法600可以在不同的实施方式中使用不同的比对方法。比对方法的非限制性实例包括全局比对(诸如Needleman–Wunsch算法)、局部比对、动态编程(诸如Smith–Waterman算法)、启发式算法或概率方法、渐进方法、迭代方法、基序发现或谱分析、遗传算法、模拟退火、成对比对、多序列比对。At
在框630处,可以确定变异。只有在达到预定的变异阈值之后,才可以识别初始变异。由于可能的PCR或测序错误,变异阈值可能是重要的。变异阈值可以基于与参考序列位置的碱基比对(不同于参考序列的对应位置处的碱基)。At
在图5A中,变异阈值是一个观察值。因此,TTACAT的第三位置可以被确定为参考序列中的C而不是A。如果变异阈值是两个或更多,则在特定测序循环的框630处不会识别C变异。在图5B中,如果变异阈值最多是两个观察值,则TTACAT的第三位置可以被确定为参考序列中的C而不是A。在一些实施方案中,变异阈值可以是对比至参考序列的特定位置的所有碱基的百分比,诸如1%、5%、10%、25%、50%或更大。如下面进一步详细描述的,对于每个序列读段,最可能的比对可以作为叶存储在节点上。每个叶可以具有相关的概率。可以修剪具有低于某个阈值的概率的叶。因此,在参考序列上识别核苷酸位置的变异可以被改进或者可以在随后的循环期间减少。In Figure 5A, the variation threshold is an observation. Therefore, the third position of TTACAT could be determined as C instead of A in the reference sequence. If the variation threshold is two or more, no C variation will be identified at
可以在决策框635处确定是否有更多的核苷酸待读取或是否所有测序循环完整。该确定可以基于例如信号的质量或者在预定数量的碱基之后。如果有更多的核苷酸待读取并且并非所有测序循环都完整,则方法600进行至框610,其中可以为下一个测序循环生成测序数据。如果没有更多的核苷酸待读取并且所有测序循环都完整,则方法600在框650处结束。A determination can be made at
在一些实施方案中,可以在已经进行最小数量的测序循环之后并行地进行框625和框630以及框610和框615。例如,在进行32个测序循环之后,该方法可以进行至框625以进行长度为32个核苷酸的序列读段的比对。虽然方法600在框625进行比对并且在框630处进行变异识别,但是可以进行下一个测序循环(即,第33个测序循环)。因此,可以在完成第33个测序循环之前在框630处确定变异。并且方法600可以在进行测序循环时,实时地(或以零或低延时地)实现比对和变异识别。此外,可以在后续循环期间改进在早期测序循环期间识别的变异。因此,图6中所示的变异识别可以是迭代过程。例如,在第32个测序循环后或在第33个测序循环期间识别的变异可以是识别的初始变异。在随后的测序循环期间,可以改进所识别的变异(包括在特定核苷酸位置上先前识别的变异不再被识别和减少)。作为另一个实例,如图5A和图5B所示,TTACAT的第四个位置的变异在第三个循环之后被识别为G,而在第四个位置之后没有识别该位置的变异。In some embodiments, blocks 625 and 630 and blocks 610 and 615 may be performed in parallel after a minimum number of sequencing cycles have been performed. For example, after performing 32 sequencing cycles, the method can proceed to block 625 to perform an alignment of sequence reads that are 32 nucleotides in length. While the
在另一个实施方案中,可以在所有测序循环完整之前终止测序过程。例如,如果在完成所有测序循环之前鉴定出特定的靶变异,则测序过程可以终止。这使得该系统比在进行靶变异识别之前需要完成所有循环的系统节省试剂成本并且更早地提供期望的结果。In another embodiment, the sequencing process can be terminated before all sequencing cycles are complete. For example, the sequencing process can be terminated if a specific target variant is identified before all sequencing cycles are completed. This allows the system to save on reagent costs and provide the desired results earlier than systems that require all cycles to be completed before proceeding to target variant calling.
在一些实施方案中,比对可以不在框625处执行,并且在每个测序循环在框630处识别变异。可以进行比对,并且每个第n个测序循环识别变异,其中n是1、2、3、4、5、10、20或更多个测序循环。在一些实施方案中,在框625处进行的比对和在框630处识别的变异的频率可以基于在先前的测序循环中识别的变异的数量。例如,如果在一个测序循环中识别大量变异,则可以更频繁地(例如,下一个循环)或更不频繁地进行比对和变异识别。作为另一个实例,如果在一个测序循环中没有识别变异或没有识别新变异,则可以更频繁地或更不频繁地进行比对和变异识别(如,不是下一个循环)。In some embodiments, an alignment may not be performed at
在一些实施方案中,可以对于参考序列的区域选择性地进行框630处的变异识别。参考序列的比对部分在不同的实施方式中可以是不同的。例如,对于参考序列的区域,可以选择性地进行变异识别,其中序列读段与参考序列的比对在先前的测序循环(如,紧邻的先前测序循环)期间已经改变。作为另一个实例,可以基于已知的单核苷酸多态性(SNP)位置确定参考序列的比对区域。In some embodiments, the variant calling at
在一些实施方案中,用于进行实时二级序列分析的方法600可以基于每个读段的树结构。树根可以用“$”标记,表示序列的开始。根的子节点对应于四个可能的碱基识别:‘A’、‘C’、‘G’和‘T’。树中的每个节点都可以有三个与之关联的变量:从根通至此节点的当前分支的序列的差异总数(称为序列S)、来自当前读段的碱基(称为序列W)、以及用于与序列S匹配的参考中的所有位置的参考序列的Burrows-Wheeler变换(BWT)中的开始和停止指数。BWT的一个重要特性是保证具有共同起始序列的所有行在变换中是连续的,而不是将各个指数的列表保持在与序列S匹配的参考中,这足以追踪起始和停止指数。因为存在非常多的重复区域,这在将读段作图至人类参考基因组的情况下是有价值的。In some embodiments, the
然后,根的每个子节点也将具有其自己的4个子节点,也对应于四个可能的碱基‘A’、‘C’、‘G’和‘T’。同样,可以追踪与当前读段的序列的差异的数量W。例如,如果前两个循环的读段是‘C’然后是‘T’,则读段可以具有通过由根->C->T定义的树的路径。因此,对于最后的T节点,总累积差将为零。相反,对于由根->A->G定义的路径,G节点处的总累积差将为2,这是因为A和G都不匹配当前读段中的对应循环。Each child node of the root will then also have its own 4 child nodes, also corresponding to the four possible bases 'A', 'C', 'G' and 'T'. Likewise, the number W of differences from the sequence of the current read can be tracked. For example, if the reads of the first two cycles are 'C' and then 'T', the reads may have a path through the tree defined by root->C->T. Therefore, for the last T-node, the total cumulative difference will be zero. Conversely, for a path defined by root->A->G, the total cumulative difference at the G node will be 2 because neither A nor G match the corresponding loop in the current read.
在一些实施方案中,可以定义与可接受的参考的差异数量的限值。一旦达到该限值,该分支就会死亡,并且在后续循环中将不再对其进行分析。具有适当指数的BWT变换可用于在常量O(1)时间内进行每个节点所必需的计算。计算所需的存储器的数量以及树中的节点数受允许的错误阈值总数的影响。在一些实施方案中,可以实现对小插入和缺失的支持。In some embodiments, a limit on the number of differences from an acceptable reference may be defined. Once that limit is reached, the branch dies, and it will no longer be analyzed in subsequent loops. A BWT transformation with appropriate exponents can be used to perform the computations necessary for each node in constant O(1) time. The amount of memory required for the computation and the number of nodes in the tree is affected by the total number of allowed error thresholds. In some embodiments, support for small insertions and deletions can be achieved.
在一些实施方案中,将通过多个种子处理更复杂的重排。也就是说,如果发现特定读段在任何地方都不匹配,则该过程可以在稍后的某些循环再次开始,期望读段的另一部分将作图至某处。可以追踪所有这些读段,并且当存在可用的计算能力时,可以进行更复杂的分析(如,像Smith-Waterman算法的动态编程方法)。In some embodiments, more complex rearrangements will be addressed by multiple seeds. That is, if a particular read is found to not match anywhere, the process can start again some later cycle, with the expectation that another portion of the read will map somewhere. All these reads can be tracked, and when there is computing power available, more complex analyzes can be performed (eg, dynamic programming methods like the Smith-Waterman algorithm).
可替代的实施方案Alternative implementation
另外的实施方案是用于二级分析的系统和方法,其包括对测序读段的迭代处理。二级分析可以包括序列读段与参考序列(如,人类参考基因组序列)的比较,并利用该比对来检测样品和参考之间的差异,诸如变异检测和识别。在一个实施方式中,可以在测序仪完成运行之前获得比对和变异识别结果。例如,可以根据可用的计算资源按时间间隔提供这些结果。这可以通过使用来自当前迭代的比对结果来扩展来自先前迭代的中间比对结果来实现。通过将当前迭代的新测序的碱基与先前比对的位置处的参考序列的碱基进行比较,产生来自当前迭代的比对结果。比较结果与来自先前迭代的比对结果相结合,并且结合的输出被存储用于下一次迭代。Additional embodiments are systems and methods for secondary analysis that include iterative processing of sequencing reads. Secondary analysis can include comparison of sequence reads to a reference sequence (eg, human reference genome sequence) and use of this alignment to detect differences between the sample and the reference, such as variant detection and identification. In one embodiment, the alignment and variant calling results can be obtained before the sequencer completes the run. For example, these results may be provided at time intervals according to available computing resources. This can be achieved by extending intermediate alignment results from previous iterations with alignment results from the current iteration. The alignment from the current iteration is generated by comparing the newly sequenced bases of the current iteration to the bases of the reference sequence at previously aligned positions. The comparison results are combined with alignment results from previous iterations, and the combined output is stored for the next iteration.
图7A和图7B是将传统的二级分析方法(图7A)与本公开的实施方案的二级分析(图7B)进行比较的示意图。图7A说明对于传统的二级分析方法,比对直到读段中的全套碱基被测序才进行。比对过程可以包括多个比对处理步骤。第一比对处理步骤等待读段中的全套测序碱基可用。在比对过程完成之后,可以开始变异识别器过程,其包括多个变异识别器处理步骤。第一变异识别器处理步骤等待全套比对数据可用。7A and 7B are schematic diagrams comparing a conventional secondary analysis method (FIG. 7A) with the secondary analysis of an embodiment of the present disclosure (FIG. 7B). Figure 7A illustrates that for traditional secondary analysis methods, alignment is not performed until the full set of bases in a read has been sequenced. The alignment process may include multiple alignment processing steps. The first alignment processing step waits for the full set of sequenced bases in the read to be available. After the alignment process is complete, the variant caller process can begin, which includes a number of variant caller processing steps. The first variant caller processing step waits for the full set of alignment data to become available.
图7B说明了根据本公开的一个实施方案的二级分析的迭代方法。如所示,比对和变异识别实时运行并生成临时结果。可以按固定间隔安排处理。固定间隔可以包括N个碱基的子序列的到达,其中N是正整数,诸如16。例如,处理可以以16个碱基的间隔发生。作为另一个实例,处理可以以1、2、4、8、16、32、64、128、151或更多个碱基的间隔发生。在一个实施方式中,处理可以以1至152之间的任意数量的间隔发生,最优选地以16+/-8的间隔发生。在一个实施方案中,间隔可以从一次迭代改变到另一次迭代。如图8所示,测序系统,诸如图1中的测序系统100,可以以16个碱基的间隔生成序列读段。可替代地,每个处理间隔中的碱基数可以不同。例如,在对16个碱基进行测序后可以处理第一间隔,并且可以在对18个碱基进行测序后处理第二次迭代。迭代中的碱基数可以低至1或高至读段中碱基的数量。Figure 7B illustrates an iterative approach to secondary analysis according to one embodiment of the present disclosure. Alignment and variant calling run in real time and generate interim results as shown. Processing can be scheduled at regular intervals. The fixed interval may include the arrival of a subsequence of N bases, where N is a positive integer, such as 16. For example, processing can occur at intervals of 16 bases. As another example, the treatments can occur at intervals of 1, 2, 4, 8, 16, 32, 64, 128, 151 or more bases. In one embodiment, treatment may occur at any number of intervals between 1 and 152, most preferably at 16+/-8 intervals. In one embodiment, the interval can vary from one iteration to another. As shown in FIG. 8, a sequencing system, such as
当使用配对的末端测序技术时,图7B中描述的过程可以应用于读段1组或读段2组。另外,在处理读段1组时捕获的信息可以应用于读段2组。例如,可能的是在对读段1组进行测序期间或之后使用常规方法执行比对步骤,并且该信息可以用于在对读段2多核苷酸进行测序时处理读段2组。When paired-end sequencing technology is used, the process described in Figure 7B can be applied to the
现在参考图8,可以从测序仪器产生单链多核苷酸的多个读段804a-804d。这些单链多核苷酸的长度可以是151个碱基,称为碱基0至碱基150。这些单链多核苷酸的序列可以通过上述边合成边测序来确定。在16个测序循环的迭代0(第一次迭代)之后,通过测序系统确定16个碱基的序列读段。例如,对于读段0(804a,)生成碱基0至碱基15的序列读段,并且对于读段1(804b)确定碱基0至碱基15的序列读段等。在另一16个测序循环的迭代1(第二次迭代)之后,为每个读段确定16个额外的序列碱基。例如,为读段0(804a)生成碱基16至碱基31。测序系统可以继续以16个间隔间隔生成读段,直到在迭代8处生成每个簇的碱基128至碱基143的序列读段。测序系统可以在迭代9(最后一次迭代)处生成每个簇的碱基144至碱基151的读段。在一个可替代的实施方案中,在每次迭代时生成的碱基数可以不同,每次迭代的碱基数由可用计算资源确定。例如,第一处理间隔可以由16个碱基组成,而第二处理间隔可以由18个碱基组成。处理间隔中最小的碱基数是1,并且处理间隔中最大的碱基数等于读段的长度。Referring now to FIG. 8, a plurality of
参考图7B,如所示,比对可以以16个碱基的间隔发生。变异识别可以在比对完成后以16的间隔发生。例如,用于实时二级分析的测序系统每1.3小时可输出16个碱基的序列读段。对于读段-时间的二级分析,进行比对和变异识别所需的总时间应在1.3小时内,以便用户可以访问在序列读段的下一个16个碱基可用之前进行的变异识别。Referring to Figure 7B, as shown, alignments can occur at intervals of 16 bases. Variant calling can occur in intervals of 16 after the alignment is complete. For example, a sequencing system for real-time secondary analysis can output sequence reads of 16 bases every 1.3 hours. For read-time secondary analysis, the total time required to perform alignment and variant calling should be within 1.3 hours so that users can access variant calling performed before the next 16 bases of sequence reads are available.
在一个实施方案中,处理可以在可用的计算机资源上尽可能快地连续发生,没有固定的迭代步骤。分析可以自我调整,并尽可能接近测序进度。比对和变异识别结果可以随时按需生成。In one embodiment, processing may occur in succession as rapidly as possible on available computer resources, with no fixed iterative steps. The analysis can self-adjust and come as close as possible to the sequencing schedule. Alignment and variant calling results can be generated on-demand at any time.
可替代的实施方案-比对Alternative Embodiment - Alignment
图9A是用于进行实时二级分析的示例方法900的流程图。方法900包括两个路径:传统二级分析方法的低置信度、高计算处理路径和根据本公开的一个实施方案的高置信度、低计算处理路径。低置信度、高处理路径以及高置信度、低处理路径在本文中分别称为蓝色路径和黄色路径。FIG. 9A is a flowchart of an
低置信度、高计算处理路径可包括每个读段比对至参考序列的序列。对于该路径,来自读段的可用迭代的所有碱基用于将读段比对至参考序列。例如,如果迭代0和迭代1各自由16个碱基组成,则比对器将处理32个碱基。许多常见的比对技术之一可以用于低置信度、高计算路径。一旦序列比对完成,就可以存储作图和比对位置并进行评分。在所有读段比对后,可以识别变异。The low-confidence, high-computational processing path may include a sequence that aligns each read to a reference sequence. For this path, all bases from the available iterations of the read are used to align the read to the reference sequence. For example, if
方法900通过添加高置信度、低计算处理路径来改进传统的二级分析方法。在迭代0处,方法900等待多个测序循环完成以生成每个读段的多个碱基。例如,方法900可以等待16个测序循环完成以生成每个读段的16个碱基。在迭代0期间,按照低置信度、高计算处理路径分析和处理每个读段的16个碱基。传统方法在本文中称为蓝色路径。在迭代1和任何后续迭代期间,按照低置信度、高计算处理路径或高置信度、低计算处理路径分析每个读段的接下来的16个碱基。如果读段在紧邻的先前迭代中以足够的置信度比对,则按照高置信度、低计算处理路径分析当前迭代的16个碱基。否则,按照高置信度、低计算处理路径分析当前迭代的16个碱基。
如果读段在紧邻的先前迭代中以足够的置信度比对,则当前迭代的16个碱基与参考序列的接下来的16个碱基比对。这种比对在本文中被称为简单比对,与常见的序列比对相比,它需要更少的处理。代替与整个参考序列的序列比对,可以确定当前迭代的16个碱基与参考序列的接下来的16个碱基之间的错配数。如果错配数高于阈值,则16个碱基的处理可以返回到低置信度、高计算处理路径。在返回低置信度、高处理路径时,比对(isAligned)变量可以设置为0或错误。可以相对于当前迭代的16个碱基或当前迭代和先前一次或多次迭代的所有碱基确定错配数。If the reads aligned with sufficient confidence in the immediately preceding iteration, the 16 bases of the current iteration are aligned to the next 16 bases of the reference sequence. Such alignments, referred to herein as simple alignments, require less processing than common sequence alignments. Instead of a sequence alignment to the entire reference sequence, the number of mismatches between the 16 bases of the current iteration and the next 16 bases of the reference sequence can be determined. If the number of mismatches is above a threshold, the processing of 16 bases can be returned to the low confidence, high computational processing path. The alignment (isAligned) variable can be set to 0 or false when returning low confidence, high processing paths. The number of mismatches can be determined relative to the 16 bases of the current iteration or to all bases of the current iteration and one or more previous iterations.
如果错配数低于阈值,则16个碱基的处理可以保持在高置信度、低计算处理路径中,并且可以存储特定读段的比对结果。可以制定替代度量以确定比对(isAligned)变量是否设置为0或错误。例如,如果错配数低于阈值,则可以计算(作图质量)MapQ评分。MapQ评分可以等于-10log10 Pr{作图位置错误},四舍五入到最接近的整数。因此,如果正确作图一些随机读段的概率为0.99,则MapQ评分应为20(即log10为0.01*-10)。如果正确匹配的概率增加到0.999,则MapQ评分将增加到30。相反,由于正确匹配的概率趋于零,MapQ评分也是如此。If the number of mismatches is below a threshold, the processing of 16 bases can be kept in the high-confidence, low-computational processing path, and the read-specific alignment can be stored. A surrogate metric can be formulated to determine if the alignment (isAligned) variable is set to 0 or false. For example, a (mapping quality) MapQ score can be calculated if the number of mismatches is below a threshold. A MapQ score may be equal to -10log10 Pr{mapping position error}, rounded to the nearest whole number. Therefore, if the probability of correctly mapping some random reads is 0.99, the MapQ score should be 20 (ie log10 is 0.01*-10). If the probability of a correct match increases to 0.999, the MapQ score increases to 30. Conversely, as the probability of a correct match tends to zero, so does the MapQ score.
当16个碱基的处理保持在高置信度、低计算处理路径中时,读段可以有助于堆积(当多个读段比对至参考序列的相似位置时,使得这些读段彼此“堆积”在参考序列顶部上。当16个碱基的处理返回到低置信度、高计算处理路径时,读段可以从堆积中移除。在一个实施方案中,只有当候选物的数量、序列比对位置的总数低于阈值诸如1000时,才在低置信度、高计算处理路径中处理读段。当处理读段时,存储比对的结果。When the 16-base processing is kept in a high-confidence, low-computational processing path, reads can facilitate stacking (when multiple reads align to similar positions in a reference sequence, making the reads "stack" with each other ” on top of the reference sequence. Reads can be removed from the stack when processing of 16 bases returns to the low-confidence, high-computational processing path. In one embodiment, only when the number of candidates, sequence ratio Reads are only processed in the low confidence, high computational processing path when the total number of pairs of positions is below a threshold, such as 1000. As reads are processed, the results of the alignment are stored.
图9B是使用图9A中所示的方法900由两个处理路径处理的数据量的概念图。在16个测序循环之后,通过测序系统生成每个读段的16个碱基。读段全部在迭代0期间在低置信度、高计算处理路径中处理。在32个测序循环之后,在迭代1之后约75%的候选物被认为是比对的(aligned)。这些候选物在迭代2期间在高置信度、低计算处理路径中处理。在迭代2之后,大约90%的候选物被认为是比对的(aligned)并且在迭代3期间在高置信度、低计算处理路径中处理。当在高置信度、低计算处理路径中处理读段时,需要较少的计算和处理,这是因为只需要简单的比对。由于在高置信度、低计算处理路径中处理大量数据并且在该路径中需要更少的处理,所需的总时间低于仅在低置信度、高计算处理路径中处理读段的情况。因此,可以在测序仪完成运行之前获得比对和变异识别结果。可以根据可用计算资源按时间间隔向用户提供这些结果。因此,方法900可以以时间有效的方式进行二级分析以实现实时二级分析。Figure 9B is a conceptual diagram of the amount of data processed by the two processing paths using the
图9C示出了图10中描述的比对器的预测运行时间改进。仅使用图10中的“现有处理”(常规或蓝色路径)生成“碱基”数据。“加载读段1”数据示出了当来自读段1组的数据被比对、预先存储、然后用以加速读段2组中的数据处理时的减少的处理循环。方法900可以实现两种类型的用于高置信度、低计算处理路径的简单比对器中的一种:跳过精确匹配的简单比对器或跳过单个错配的简单比对器。跳过单个匹配的简单比对器允许0个或1个错配。“跳过精确匹配”数据示出了当如果当前迭代的16个碱基与参考序列的16个碱基在先前确定的参考位置处完全匹配,就跳过常规(蓝色)路径时减少的处理循环。“跳过单一错配”数据显示当如果当前迭代的16个碱基在先前确定的参考位置处比对至参考序列的16个碱基且具有至多1个错配就跳过常规(蓝色)路径时减少的处理循环。图9C示出了与基线相比,当方法900利用在高置信度、低计算处理路径中检测到单个错配,就跳过常规处理的简单比对器时,运行时间减少三倍。请注意,这些数字是由原型处理器生成,所述原型处理器不包括所有处理步骤并因此是预期的预测。FIG. 9C shows the predicted runtime improvement of the aligner described in FIG. 10 . Only "base" data was generated using "existing processing" (regular or blue path) in Figure 10. "
图10是用于进行实时二级分析的示例方法1000的另一流程图。图9A中所示的方法1000和方法900可以实现相同的低置信度、高计算处理路径和不同的高置信度、低计算处理路径。方法1000的高置信度、低计算处理路径在简单比对之后生成MapQ评分并且使用MapQ评分来确定是否在高置信度、低计算处理路径中继续处理或返回到低置信度、高处理路径。10 is another flowchart of an
高百分比的运行时间发生在小百分比的读段上。在一个实施方案中,如果使用度量所确定的成功置信度低,则方法900或1000的低置信度、高计算处理路径可以跳过比对和存储步骤。在一个实施方案中,可以生成度量,其指示子序列可以比对至参考序列的候选位置的数量。如果候选物质数量很高,比对成功的置信度将是低的。在第二个实施方案中,如果序列中碱基的多样性低,则比对成功的置信度是低的。碱基的多样性可以例如通过计算子序列中的独特n聚体的数量来确定,其中n聚体是子序列中的碱基序列,其长度小于或等于子序列本身的长度。A high percentage of run time occurs on a small percentage of reads. In one embodiment, the low confidence, high computational processing path of
可替代的实施方案–变异识别器Alternative implementation – variant caller
图11A和图11B示出了现有变异识别方法,Strelka小的变异识别器(图11A)和本公开的变异识别方法(图11B)的简化流程图。图11A示出了小的变异识别器使用从比对器生成的堆积信息作为输入。从堆积中,小的变异识别器鉴定被称为活性区域的序列变异区域。接下来,可以将从头重新组装应用于活性区域。在每个基因组位置,生成概率用以确定基因组位置处的测序的多核苷酸为A、C、T或G的可能性。根据这些概率,可以检测变异。11A and 11B show simplified flowcharts of existing variant calling methods, the Strelka small variant caller ( FIG. 11A ) and the variant calling method of the present disclosure ( FIG. 11B ). Figure 11A shows that the small variant identifier uses as input the stacking information generated from the aligner. From the stack, small variation callers identify regions of sequence variation known as active regions. Next, de novo reassembly can be applied to active regions. At each genomic position, a probability is generated to determine the likelihood that the sequenced polynucleotide at the genomic position is A, C, T, or G. From these probabilities, variants can be detected.
图11B示出了如本发明中公开的变异识别器的实施方案。在该实施方案中,生成度量用以确定是否可以高置信度地确定基因组位置处的多核苷酸。例如,如果给定基因组位置处的所有多核苷酸都相同,则可以生成高置信度决策。或者,如果基因组位置处相同类型的多核苷酸的数量高于阈值,则可以生成高置信度决策。还可以实现用于确定高置信度的替代度量。如果可以以高置信度确定多核苷酸,则可以跳过概率的公式并且可以执行简单的变异识别步骤。例如,简单的变异识别器可以识别以高置信度检测的任何变异。Figure 1 IB shows an embodiment of a variant caller as disclosed in the present invention. In this embodiment, a metric is generated to determine whether a polynucleotide at a genomic location can be identified with high confidence. For example, high confidence decisions can be generated if all polynucleotides at a given genomic location are identical. Alternatively, a high confidence decision can be generated if the number of polynucleotides of the same type at a genomic location is above a threshold. Alternative metrics for determining high confidence can also be implemented. If the polynucleotide can be identified with high confidence, the formulation of probabilities can be skipped and a simple variant calling step can be performed. For example, a simple variant caller can identify any variant detected with high confidence.
现有变异识别方法的概率生成步骤和变异识别步骤可以组合需要高达40%的变异识别器的计算和处理。图11B示出了变异识别方法1100,其实现了现有变异识别方法的低置信度、高计算处理路径和高置信度、低计算处理路径。通过添加高置信度、低计算处理路径,Strelka变异识别器得到优化,并且处理降低了近40%。可以将高置信度、低计算处理路径添加到可替代的变异识别器。The probability generation step and the variant calling step of existing variant calling methods can combine to require up to 40% of the computation and processing of the variant caller. FIG. 11B illustrates a
如图7B所示,可以在迭代处理窗口内执行变异识别器。图11A或图11B的变异识别器可以在迭代处理窗口内迭代地执行。另外,可以在迭代处理窗口内执行多于一种类型的变异识别器。例如,小的变异识别器,如Strelka,以及可替代的变异识别器,诸如结构变异识别器或拷贝数变异识别器,可以在迭代处理窗口内执行。As shown in Figure 7B, the variant caller can be executed within an iterative processing window. The variation identifier of FIG. 11A or FIG. 11B may be executed iteratively within an iterative processing window. Additionally, more than one type of variant caller can be executed within an iterative processing window. For example, small variation callers, such as Strelka, and alternative variation callers, such as structural variation callers or copy number variation callers, can be executed within an iterative processing window.
在至少一些先前描述的实施方案中,实施方案中所使用的一个或多个要素可以互换地用于另一个实施方案中,除非这种替换在技术上不可行。本领域技术人员将理解,在不脱离所要求保护的主题的范围的情况下,可以对上述方法和结构进行各种其他省略、添加和修改。所有此类修改和变化都旨在落入由所附权利要求限定的主题的范围内。In at least some of the previously described embodiments, one or more elements used in one embodiment may be used interchangeably in another embodiment, unless such substitution is not technically feasible. Those skilled in the art will appreciate that various other omissions, additions and modifications may be made to the methods and structures described above without departing from the scope of the claimed subject matter. All such modifications and changes are intended to fall within the scope of the subject matter defined by the appended claims.
关于本文中基本上任何复数和/或单数术语的使用,本领域技术人员可以根据上下文和/或应用适当地从复数转换为单数和/或从单数转换为复数。为清楚起见,本文可以明确地阐述各种单数/复数排列。With respect to the use of substantially any plural and/or singular term herein, one skilled in the art can switch from the plural to the singular and/or from the singular to the plural as appropriate depending on the context and/or application. For the sake of clarity, various singular/plural permutations may be explicitly set forth herein.
本领域技术人员将理解,通常,本文并且尤其是所附的权利要求(如,所附权利要求的主题)使用的术语通常旨在作为“开放式”术语(如,术语“包括”应当被解释为“包括但不限于”,术语“具有”应该被解释为“至少具有”,术语“包括”应该被解释为“包括但不限于”等)。本领域技术人员将进一步理解,如果意图列举特定数量的引入的权利要求,则在权利要求中将明确地叙述这样的意图,并且在没有这样的叙述的情况下,不存在这样的意图。例如,作为对理解的帮助,以下所附权利要求可包含使用引导短语“至少一个”和“一个或多个”以引入权利要求叙述。然而,这些短语的使用不应被解释为暗示由不定冠词“一”或“一个”引入权利要求叙述将包含这种引入的权利要求叙述的任何特定权利要求限制于仅包含一个这样的叙述的实施方案,即使当相同的权利要求包括引入短语“一个或多个”或“至少一个”和不定冠词诸如“一”或“一个”(如,“一”和/或“一个”应解释为意指“至少一个”或“一个或多个”);对于使用用于引入权利要去叙述的定冠词也是如此。另外,即使明确地叙述了特定数量的引入的权利要求叙述,本领域技术人员将认识到,这种叙述应该被解释为意指至少所叙述的数字(如,仅详细叙述“两个叙述”而没有其他修饰语意指至少两个叙述或两个或多个叙述)。此外,在使用类似于“A、B和C等中的至少一个”的惯例的那些情况下,通常这样的构造意图与本领域技术人员将理解该惯例的一样(如,“具有A、B和C中的至少一个的系统”将包括但不限于具有单独的A、单独的B、单独的C、A和B一起、A和C一起、B和C一起和/或A、B和C一起等的系统)。在使用类似于“A、B或C等中的至少一个”的惯例的那些情况下,通常这样的构造意图与本领域技术人员将理解该惯例的一样(如,“具有A、B或C中的至少一个的系统”将包括但不限于具有单独的A、单独的B、单独的C、A和B一起、A和C一起、B和C一起和/或A、B和C一起等的系统)。本领域技术人员将进一步理解,实际上任何呈现两个或更多个替代术语的析取词和/或短语,无论是在说明书、权利要求书还是在附图中,都应该被理解为考虑包括这些术语之一、任何一个术语或两个术语的可能性。例如,短语“A或B”将被理解为包括“A”或“B”或“A和B”的可能性。Those skilled in the art will appreciate that, generally, the terms used herein and especially in the appended claims (e.g., the subject matter of the appended claims) are generally intended to be interpreted as "open-ended" terms (e.g., the term "comprising") as "including but not limited to", the term "having" should be interpreted as "having at least", the term "including" should be interpreted as "including but not limited to", etc.). It will be further understood by those within the art that if a specific number of an introduced claim is intended to recite, such an intent will be explicitly recited in the claim, and in the absence of such recitation no such intent is present. For example, as an aid to understanding, the following appended claims may contain usage of the introductory phrases "at least one" and "one or more" to introduce claim recitations. However, use of these phrases should not be construed as implying that introduction of a claim recitation by the indefinite article "a" or "an" limits any particular claim containing such an introduced claim recitation to those containing only one of such recitation. embodiment, even when the same claim includes the introductory phrase "one or more" or "at least one" and an indefinite article such as "a" or "an" (eg, "a" and/or "an" should be construed as meaning "at least one" or "one or more"); the same is true for the use of definite articles used to introduce a claim to be recited. Additionally, even if a particular number of an introduced claim recitation is expressly recited, those skilled in the art will recognize that such recitation should be construed to mean at least that recited number (eg, only "two recitations" are recited in detail and No other modifier means at least two statements or two or more statements). Furthermore, in those cases where a convention like "at least one of A, B, and C, etc." is used, generally such constructions are intended as one skilled in the art would understand the convention (e.g., "having A, B, and A system of at least one of C" would include, but not be limited to, having A alone, B alone, C alone, A and B together, A and C together, B and C together, and/or A, B and C together, etc. system). In those cases where a convention similar to "at least one of A, B, or C, etc." is used, generally such constructions are intended as one skilled in the art would understand the convention (e.g., "having A system of at least one of "will include, but is not limited to, systems having A alone, B alone, C alone, A and B together, A and C together, B and C together, and/or A, B and C together, etc. ). Those skilled in the art will further appreciate that virtually any disjunction and/or phrase that presents two or more alternative terms, whether in the specification, claims, or drawings, should be understood to include The possibility of one of these terms, either term, or both terms. For example, the phrase "A or B" will be understood to include the possibilities of "A" or "B" or "A and B."
另外,在根据马库什组描述本公开的特征或方面的情况下,本领域技术人员将认识到,本公开也因此以马库什组的任何单个成员或成员子组的形式描述。In addition, where features or aspects of the disclosure are described in terms of Markush groups, those skilled in the art will recognize that the disclosure is also thereby described in terms of any individual member or subgroup of members of the Markush group.
如本领域技术人员将理解的,出于任何和所有目的,就提供书面描述而言,本文所公开的所有范围还涵盖任何和所有可能的子范围及其子范围的组合。任何列出的范围可以容易地被识别为充分描述并且使得相同的范围被分解为至少相等的一半、三分之一、四分之一、五分之一、十分之一等。作为非限制性实例,本文讨论的每个范围可以容易地被分解为下三分之一、中三分之一和上三分之一等。如本领域技术人员还将理解,所有措辞诸如“高达”、“至少”、“大于”、“小于”以及类似措辞包括所述的数字并且指的是可以随后分解成如上所讨论的子范围的范围。最后,如本领域技术人员将理解,范围包括每个单独的成员。因此,例如,具有1-3个物品的组是指具有1、2或3个物品的组。类似地,具有1-5个物品的组是指具有1、2、3、4或5个物品的组,等等。As will be understood by those skilled in the art, for any and all purposes in terms of providing a written description, all ranges disclosed herein also encompass any and all possible subranges and combinations of subranges thereof. Any listed range can be readily identified as sufficiently descriptive and such that the same range is broken down into at least equal halves, thirds, quarters, fifths, tenths, etc. As a non-limiting example, each range discussed herein can be easily broken down into lower thirds, middle thirds, upper thirds, etc. As will also be understood by those skilled in the art, all expressions such as "up to," "at least," "greater than," "less than," and similar expressions include the stated number and refer to a range that can then be broken down into sub-ranges as discussed above. scope. Finally, as will be understood by those skilled in the art, a range includes each individual member. Thus, for example, a set of 1-3 items refers to a set of 1, 2 or 3 items. Similarly, a group having 1-5 items refers to groups having 1, 2, 3, 4, or 5 items, and so on.
虽然本文已经公开了各种方面和实施方案,但是其他方面和实施方案对于本领域技术人员来说将是显而易见的。本文公开的各个方面和实施方案是出于说明的目的而不是限制性的,真正的范围和精神由所附权利要求指示。Although various aspects and embodiments have been disclosed herein, other aspects and embodiments will be apparent to those skilled in the art. The various aspects and embodiments disclosed herein are for purposes of illustration and not limitation, with the true scope and spirit being indicated by the appended claims.
Claims (27)
Translated fromChinesePriority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202211557451.6ACN115810396A (en) | 2016-10-07 | 2017-10-06 | Systems and methods for secondary analysis of nucleotide sequencing data |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662405824P | 2016-10-07 | 2016-10-07 | |
| US62/405,824 | 2016-10-07 | ||
| PCT/US2017/055653WO2018068014A1 (en) | 2016-10-07 | 2017-10-06 | System and method for secondary analysis of nucleotide sequencing data |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202211557451.6ADivisionCN115810396A (en) | 2016-10-07 | 2017-10-06 | Systems and methods for secondary analysis of nucleotide sequencing data |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN109416927A CN109416927A (en) | 2019-03-01 |
| CN109416927Btrue CN109416927B (en) | 2023-05-02 |
Family
ID=60480359
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202211557451.6APendingCN115810396A (en) | 2016-10-07 | 2017-10-06 | Systems and methods for secondary analysis of nucleotide sequencing data |
| CN201780040788.0AExpired - Fee RelatedCN109416927B (en) | 2016-10-07 | 2017-10-06 | System and method for secondary analysis of nucleotide sequencing data |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202211557451.6APendingCN115810396A (en) | 2016-10-07 | 2017-10-06 | Systems and methods for secondary analysis of nucleotide sequencing data |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US11646102B2 (en) |
| EP (2) | EP4586258A1 (en) |
| JP (3) | JP6898441B2 (en) |
| KR (3) | KR102515638B1 (en) |
| CN (2) | CN115810396A (en) |
| AU (3) | AU2017341069A1 (en) |
| BR (1) | BR122023004154A2 (en) |
| CA (1) | CA3027179C (en) |
| IL (2) | IL300135B2 (en) |
| MX (2) | MX2018015412A (en) |
| MY (2) | MY193917A (en) |
| RU (1) | RU2741807C2 (en) |
| SG (2) | SG11201810924WA (en) |
| WO (1) | WO2018068014A1 (en) |
| ZA (2) | ZA201808277B (en) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110016499B (en) | 2011-04-15 | 2023-11-14 | 约翰·霍普金斯大学 | Safe sequencing system |
| CA2889937C (en) | 2012-10-29 | 2020-12-29 | The Johns Hopkins University | Papanicolaou test for ovarian and endometrial cancers |
| WO2017027653A1 (en) | 2015-08-11 | 2017-02-16 | The Johns Hopkins University | Assaying ovarian cyst fluid |
| CN110268072B (en)* | 2016-12-15 | 2023-11-07 | Illumina公司 | Method and system for determining paralogous genes |
| WO2019067092A1 (en) | 2017-08-07 | 2019-04-04 | The Johns Hopkins University | Methods and materials for assessing and treating cancer |
| AU2019369302A1 (en) | 2018-10-31 | 2021-01-21 | Illumina, Inc. | Systems and methods for grouping and collapsing sequencing reads |
| US11210554B2 (en) | 2019-03-21 | 2021-12-28 | Illumina, Inc. | Artificial intelligence-based generation of sequencing metadata |
| IL279558B2 (en) | 2019-05-24 | 2025-02-01 | Illumina Inc | Flexible seed extension for hash table genomic mapping |
| KR102292599B1 (en)* | 2019-11-06 | 2021-08-23 | 주식회사 뷰웍스 | Optical analysis device and optical analysis method |
| BR112022015194A2 (en)* | 2020-03-11 | 2022-10-11 | Illumina Inc | INCREMENTAL SECONDARY ANALYSIS OF NUCLEIC ACID SEQUENCES |
| US12006539B2 (en)* | 2020-03-17 | 2024-06-11 | Western Digital Technologies, Inc. | Reference-guided genome sequencing |
| CN113436683B (en)* | 2020-03-23 | 2024-08-16 | 北京合生基因科技有限公司 | Method and system for screening candidate inserts |
| AU2022202798A1 (en)* | 2021-05-26 | 2022-12-15 | Genieus Genomics Pty Ltd | Processing sequencing data relating to amyotrophic lateral sclerosis |
| CN113299344A (en)* | 2021-06-23 | 2021-08-24 | 深圳华大医学检验实验室 | Gene sequencing analysis method, gene sequencing analysis device, storage medium and computer equipment |
| IL320228A (en)* | 2022-10-13 | 2025-06-01 | Element Biosciences Inc | Separating sequencing data in parallel with a sequencing run in next generation sequencing data analysis |
| CN119360966B (en)* | 2024-12-24 | 2025-04-04 | 苏州大学 | Genome assembly method and system based on iterative k-mer decomposition |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101137991A (en)* | 2005-02-11 | 2008-03-05 | 智明基因有限责任公司 | Computer-implemented method and computer-based system for validating DNA sequencing data |
| CN101278295A (en)* | 2005-08-01 | 2008-10-01 | 454生命科学公司 | Methods of amplifying and sequencing nucleic acids |
| US20110270533A1 (en)* | 2010-04-30 | 2011-11-03 | Life Technologies Corporation | Systems and methods for analyzing nucleic acid sequences |
| CN104462211A (en)* | 2014-11-04 | 2015-03-25 | 北京诺禾致源生物信息科技有限公司 | Re-sequencing data processing method and processing device |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2357263A1 (en) | 2001-09-07 | 2003-03-07 | Bioinformatics Solutions Inc. | New methods for faster and more sensitive homology search in dna sequences |
| US20120203792A1 (en) | 2011-02-01 | 2012-08-09 | Life Technologies Corporation | Systems and methods for mapping sequence reads |
| US10424394B2 (en)* | 2011-10-06 | 2019-09-24 | Sequenom, Inc. | Methods and processes for non-invasive assessment of genetic variations |
| EP2764458B1 (en)* | 2011-10-06 | 2021-04-07 | Sequenom, Inc. | Methods and processes for non-invasive assessment of genetic variations |
| KR101394339B1 (en)* | 2012-03-06 | 2014-05-13 | 삼성에스디에스 주식회사 | System and method for processing genome sequence in consideration of seed length |
| US10504613B2 (en)* | 2012-12-20 | 2019-12-10 | Sequenom, Inc. | Methods and processes for non-invasive assessment of genetic variations |
| KR101481457B1 (en) | 2012-10-29 | 2015-01-12 | 삼성에스디에스 주식회사 | System and method for aligning genome sequence considering entire read |
| US20140238250A1 (en) | 2013-02-28 | 2014-08-28 | Wki Holding Company, Inc. | Microwavable Heating Element and Composition |
| US20160034638A1 (en)* | 2013-03-14 | 2016-02-04 | University Of Rochester | System and Method for Detecting Population Variation from Nucleic Acid Sequencing Data |
| WO2014186604A1 (en)* | 2013-05-15 | 2014-11-20 | Edico Genome Corp. | Bioinformatics systems, apparatuses, and methods executed on an integrated circuit processing platform |
| US10191929B2 (en) | 2013-05-29 | 2019-01-29 | Noblis, Inc. | Systems and methods for SNP analysis and genome sequencing |
| RU2539038C1 (en) | 2013-11-02 | 2015-01-10 | Общество с ограниченной ответственностью "Гамма" | Dna sequencing method and device therefor (versions) |
- 2017
- 2017-10-06MXMX2018015412Apatent/MX2018015412A/enunknown
- 2017-10-06ILIL300135Apatent/IL300135B2/enunknown
- 2017-10-06SGSG11201810924WApatent/SG11201810924WA/enunknown
- 2017-10-06EPEP25150789.3Apatent/EP4586258A1/enactivePending
- 2017-10-06MYMYPI2018002632Apatent/MY193917A/enunknown
- 2017-10-06USUS16/311,141patent/US11646102B2/enactiveActive
- 2017-10-06CNCN202211557451.6Apatent/CN115810396A/enactivePending
- 2017-10-06CACA3027179Apatent/CA3027179C/enactiveActive
- 2017-10-06AUAU2017341069Apatent/AU2017341069A1/ennot_activeAbandoned
- 2017-10-06RURU2018143972Apatent/RU2741807C2/enactive
- 2017-10-06EPEP17804976.3Apatent/EP3458993A1/ennot_activeWithdrawn
- 2017-10-06WOPCT/US2017/055653patent/WO2018068014A1/ennot_activeCeased
- 2017-10-06CNCN201780040788.0Apatent/CN109416927B/ennot_activeExpired - Fee Related
- 2017-10-06ILIL263512Apatent/IL263512B2/enunknown
- 2017-10-06KRKR1020227011278Apatent/KR102515638B1/enactiveActive
- 2017-10-06SGSG10201911912XApatent/SG10201911912XA/enunknown
- 2017-10-06BRBR122023004154-2Apatent/BR122023004154A2/ennot_activeApplication Discontinuation
- 2017-10-06KRKR1020237010257Apatent/KR102694651B1/enactiveActive
- 2017-10-06KRKR1020187038172Apatent/KR102384832B1/enactiveActive
- 2017-10-06JPJP2019519631Apatent/JP6898441B2/enactiveActive
- 2018
- 2018-12-07ZAZA2018/08277Apatent/ZA201808277B/enunknown
- 2018-12-11MXMX2022011757Apatent/MX2022011757A/enunknown
- 2018-12-17MYMYPI2022003491Apatent/MY209503A/enunknown
- 2020
- 2020-05-27JPJP2020091991Apatent/JP7051937B2/enactiveActive
- 2020-07-22AUAU2020207826Apatent/AU2020207826B2/ennot_activeCeased
- 2021
- 2021-03-15ZAZA2021/01720Apatent/ZA202101720B/enunknown
- 2021-12-01AUAU2021277671Apatent/AU2021277671B2/enactiveActive
- 2022
- 2022-02-22JPJP2022025557Apatent/JP7387777B2/enactiveActive
- 2023
- 2023-04-13USUS18/300,343patent/US20230410945A1/enactivePending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101137991A (en)* | 2005-02-11 | 2008-03-05 | 智明基因有限责任公司 | Computer-implemented method and computer-based system for validating DNA sequencing data |
| CN101278295A (en)* | 2005-08-01 | 2008-10-01 | 454生命科学公司 | Methods of amplifying and sequencing nucleic acids |
| US20110270533A1 (en)* | 2010-04-30 | 2011-11-03 | Life Technologies Corporation | Systems and methods for analyzing nucleic acid sequences |
| CN104462211A (en)* | 2014-11-04 | 2015-03-25 | 北京诺禾致源生物信息科技有限公司 | Re-sequencing data processing method and processing device |
Non-Patent Citations (2)
| Title |
|---|
| Assembly algorithms for next-generation sequencing data;Jason R. Miller 等;《Genomics》;20100306;第317页第1栏倒数第2段-323页第2栏倒数第1段* |
| 基因组浏览器底层数据的分析与集成;王振兴;《中国优秀硕士学位论文全文数据库 基础科学辑》;20140315;正文第1-41页* |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN109416927B (en) | System and method for secondary analysis of nucleotide sequencing data | |
| US20250191678A1 (en) | Systems and methods for determining copy number variation | |
| US20210358572A1 (en) | Methods, systems, and computer-readable media for calculating corrected amplicon coverages | |
| US20240274241A1 (en) | Methods for compression of molecular tagged nucleic acid sequence data | |
| JP7171709B2 (en) | Methods for Detection of Fusions Using Compacted Molecularly Tagged Nucleic Acid Sequence Data | |
| NZ793021A (en) | System and method for secondary analysis of nucleotide sequencing data | |
| EP4588049A1 (en) | Methods for detecting allele dosages in polyploid organisms | |
| Baruzzo | Improving the RNA-Seq analysis pipeline: read alignment and expression level quantification |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | Granted publication date:20230502 |