CN105671078B - Nano non-viral gene delivery system and application thereof - Google Patents
Nano non-viral gene delivery system and application thereofDownload PDFInfo
- Publication number
- CN105671078B CN105671078BCN201610023604.7ACN201610023604ACN105671078BCN 105671078 BCN105671078 BCN 105671078BCN 201610023604 ACN201610023604 ACN 201610023604ACN 105671078 BCN105671078 BCN 105671078B
- Authority
- CN
- China
- Prior art keywords
- pei
- cells
- cyd
- gene
- transfection
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images











Classifications
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0663—Bone marrow mesenchymal stem cells (BM-MSC)
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Biomedical Technology (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Developmental Biology & Embryology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Cell Biology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Plant Pathology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Translated fromChineseDescription
The invention is a divisional application of the invention patent application with the application number of CN 201010292778.6.
Technical Field
The invention belongs to the field of biotechnology; more specifically, the invention relates to a nano non-viral gene delivery system and application thereof.
Background
The bone marrow mesenchymal stem cells have very important position in bone tissue engineering and are the main source of seed cells in the bone tissue engineering. Synovial cells and chondrocytes play an important role in the development and progression of osteoarthritis, and they are also frequently used as target cells for gene therapy. However, the bone joint related cells have the problem of low gene transfection efficiency at present, which becomes a bottleneck of technical progress and greatly limits the development of clinical gene therapy.
Gene therapy refers to the introduction of exogenous genes into the cells of a patient by genetic engineering techniques to correct or compensate for diseases caused by pathogenic genes. The introduction of foreign genes into cells requires the use of a certain technical method. The current methods of gene transfection can be mainly divided into three major categories, one is biological, one is physical and the other is chemical. The biological method mainly refers to a technique of introducing a foreign gene into a cell using a virus as a vector. The viral vectors most studied at present are retroviral vectors, lentiviral vectors, adenoviral vectors, adeno-associated viral vectors, herpes simplex viral vectors and the like. The physical method mainly refers to a technique of introducing a foreign gene into a cell by physical means. The current common methods include microinjection, electric breakdown membrane, pressure penetration, ultrasonic spraying, particle bombardment and the like. The chemical method refers to a technique of introducing a foreign gene into a cell using a chemical agent. The transfection reagents that are currently more commonly used are a series of derivatives using cationic liposomes as the parent material. In addition, there are many reports on a series of derivatives using Polyethyleneimine (PEI) and Chitosan (Chitosan) as matrix materials. The chemical transfection reagents are various, and the chemical transfection reagents used by different cells are different in consideration of transfection efficiency, toxicity and the like. Therefore, for a particular cell, it is desirable to find the most suitable chemical transfection reagent.
The above three methods have advantages and disadvantages, and experimenters often use different methods to introduce foreign genes into cells according to different requirements. When using different introduction methods, the following factors are mainly taken into account: safety, efficiency, operability, and cost factors. For example, in the production of transgenic animals, exogenous genes are often introduced into fertilized egg cells, or sperm and oocytes, by microinjection. However, this method requires a great deal of operating experience and skilled operating skill of the laboratory operator, and the microinjection method requires a relatively high instrument, which is not suitable for general cell transfection.
Gene vectors are vector systems for introducing foreign genes into cells, and can be generally classified into viral vectors and non-viral vectors. These two types of vectors have their own advantages and disadvantages. The virus vector has certain immunogenicity and cytotoxicity, carcinogenesis caused by random integration into host cell genome, complex preparation and other defects, and has broad spectrum of host cells and high transfection efficiency on primary cells, which is difficult to reach by non-virus vectors. The non-viral vector has better biocompatibility, lower cytotoxicity and almost no immunogenicity. However, the transfection efficiency of the gene has great difference on different target cells. This difference is mainly reflected by the relatively low transfection efficiency on primary cells, which limits their clinical application to some extent. An ideal gene vector should be low in toxicity, efficient, low in cost, and operable.
The low cytotoxicity, low immunogenicity and better biocompatibility of non-viral gene vectors have been considered as promising alternatives to viral vectors. However, the lower transfection efficiency compared to viral gene vectors has been a great obstacle to their use. There is a need to improve the transfection efficiency of non-viral gene vectors by chemical modification and the like.
Non-viral gene vectors can be divided into two broad classes, one being cationic liposomes and derivatives thereof and the other being cationic polymers and derivatives thereof, depending on the parent material. Cationic liposomes, mainly Lipo2000, Fugene6, etc., tend to have stable and high transfection efficiency in vitro for some cell lines, which is one of the major reasons why some of them can become commercial transfection reagents. However, the lower transfection efficiency of cationic liposomes for some primary cells does limit their use in some areas. In addition, when the cationic liposome is used for gene therapy in vivo, the cationic liposome is often coagulated and precipitated by serum albumin in blood, thereby greatly reducing the transfection efficiency.
There are many factors that affect the transfection efficiency of non-viral gene vectors, and they can be discussed and analyzed from the material and biological perspectives, respectively.
The ideal non-viral gene vector should first be able to have good solubility at physiological pH and physiological ionic concentration. Secondly, the complex has better complexing ability with plasmid, and the formed complex has more proper particle size and surface potential. It has been reported in the literature that particles having a particle size of less than 200 μm generally enter cells via clathrin-mediated endocytosis, whereas particles having a particle size between 200 μm and 500 μm generally enter cells via cytoplasmic vesicle-mediated endocytosis. These two different endocytic modes directly lead to different fates of the complex after entry into the cell. The former, through clathrin-mediated endocytosis, the complex must pass through lysosomes to enter the cytoplasm, while the latter can directly enter the cytoplasm without passing through lysosomes. For the surface potential of the complex, it is presently considered desirable to have a small amount of surface charge (around 10 mV). Because too high surface charge is likely to aggregate with serum albumin during in vivo blood circulation, while too low surface charge is not conducive to adsorption of these complexes by negatively charged membrane proteins on the cell surface. The particle size and surface potential of the complex are largely related to the ratio of non-viral vector material to foreign gene, i.e., the nitrogen-to-phosphorus ratio (N/P). In some cases, the shape of the complex also affects transfection efficiency.
In many studies, it has been found that the same non-viral gene vector often has significantly different transfection efficiencies in different cell types. These differences are manifested firstly in that the cytotoxicity of the non-viral gene vectors varies among cells, and secondly in that the physiological properties of each cell itself vary.
In conclusion, the non-viral vectors of choice are often different for different cells and the art needs to investigate osteoarticular related cells in order to develop suitable transfection reagents for them.
Disclosure of Invention
The invention aims to provide a nano non-viral gene delivery system and application thereof.
In a first aspect of the present invention, there is provided a method of transfecting an exogenous gene into a bone joint-related cell, comprising:
(1) mixing an expression system for expressing the exogenous gene with a gene delivery system to obtain a transfection solution;
(2) adding the transfection solution to the bone joint-related cells, whereby the foreign gene is transfected into the cells;
the gene delivery system is a complex of polyethyleneimine and cyclodextrin with low molecular weight (molecular weight less than 2000 Da; preferably molecular weight between 400-1500 Da; more preferably between 500-1000 Da; more preferably between 500-800; most preferably molecular weight 600 Da).
In a preferred embodiment, the expression system for expressing the exogenous gene forms a complex with the gene delivery system, and the complex is cultured in a serum-free culture medium to obtain a transfection solution.
In another preferred embodiment, the gene delivery system has a complex having the structure of formula I,

wherein m is1A positive integer of 8 to 50 (preferably 5 to 40; more preferably 5 to 25), m2A positive integer of 4 to 25 (preferably 5 to 20; more preferably 5 to 15), n is a positive integer of 10 to 20 (preferably 10 to 18; more preferably 14 to 18); and m is1:m2=2:1。
In another preferred embodiment, the bone joint-related cells are selected from the group consisting of: synoviocytes, chondrocytes, bone marrow mesenchymal stem cells (BMSCs).
In another preferred embodiment, the bone joint related cells are bone marrow mesenchymal stem cells.
In another preferred embodiment, the method for mixing the expression system of the exogenous gene and the gene delivery system comprises the following steps: mixing the water solution containing the gene delivery system with the water solution containing the expression system for expressing the exogenous gene, so that the ratio (N/P) of available nitrogen in the gene delivery system to available phosphorus in the exogenous gene is 20-35.
In another preferred embodiment, the ratio of nitrogen in the gene delivery system to phosphorus in the foreign gene is 20 to 30.
In another preferred embodiment, the ratio (N/P) of available nitrogen in the gene delivery system to available phosphorus in the foreign gene is 25 to 35 (preferably 28 to 32, most preferably 30) relative to bone marrow mesenchymal stem cells or synoviocytes.
In another preferred embodiment, the ratio (N/P) of available nitrogen in the gene delivery system to available phosphorus in the foreign gene is 20 to 30 (preferably 22 to 27, most preferably 25) relative to chondrocytes.
In another aspect of the invention, there is provided the use of a complex of the structure of formula I for the preparation of an agent for transfecting a foreign gene into a cell associated with a bone joint.

Wherein m is1Is a positive integer of 8 to 50, m2Is a positive integer of 4-25, n is a positive integer of 10-20; and m is1:m2=2:1。
In another preferred embodiment, the bone joint-related cells are selected from the group consisting of: synoviocytes, chondrocytes, bone marrow mesenchymal stem cells.
In another preferred embodiment, the bone joint related cells are bone marrow mesenchymal stem cells.
Other aspects of the invention will be apparent to those skilled in the art in view of the disclosure herein.
Drawings
FIG. 1 non-viral vector PEI600-cytotoxicity of CyD on BMSC.
A:PEI600-cytotoxicity of CyD, PEI-25kDa for 4 h;
B:PEI600CyD, PEI-25kDa cytotoxicity for 24 h.
FIG. 2 non-viral vector PEI600-cytotoxicity of CyD against primary synoviocytes.
A:PEI600-cytotoxicity of CyD, PEI-25kDa for 4 h;
B:PEI600CyD, PEI-25kDa cytotoxicity for 24 h.
FIG. 3 non-viral vector PEI600-cytotoxicity of CyD against primary chondrocytes.
A:PEI600-cytotoxicity of CyD, PEI-25kDa for 4 h;
B:PEI600CyD, PEI-25kDa cytotoxicity for 24 h.
FIG. 4 non-viral vector PEI600Efficiency of transfection of CyD for BMSC.
A: BMSC transfection effect graph under a fluorescence microscope;
b: quantitative analysis of BMSC transfection efficiency, PEI600-CyD group N/P30, PEI-25kDa group N/P10; c: correlation of BMSC transfection efficiency with N/P.
FIG. 5 non-viral vector PEI600Efficiency of transfection of CyD for primary synoviocytes.
A: primary synovial cell transfection effect picture under a fluorescence microscope;
b: quantitative analysis of primary synovial cell transfection efficiency, PEI600-CyD group N/P30, PEI-25kDa group N/P10;
c: the transfection efficiency of primary synoviocytes is related to N/P.
FIG. 6 non-viral vector PEI600Efficiency of transfection of CyD for primary chondrocytes.
A: primary chondrocyte transfection effect graph under a fluorescence microscope;
b: quantitative analysis of Primary chondrocyte transfection efficiency, PEI600-CyD group N/P25, PEI-25kDa group N/P10;
c: the transfection efficiency of primary chondrocytes is related to N/P.
FIG. 7 depicts the distribution of pEGFP in BMSC cells 4h after transfection using quantum dot labeling. In the figure, red is quantum dot labeled plasmid, and blue is Hochest stained cell nucleus.
FIG. 8, PEI600Comparison of the transfection efficiencies of CyD and lipofectamine 2000 in different cells (293T, Hela, chondrocytes, bone marrow mesenchymal cells, synoviocytes).
Fig. 9, transfection efficiency of different non-viral vectors for primary bone marrow mesenchymal stem cells.
Fig. 10, transfection efficiency of different non-viral vectors for primary chondrocytes.
FIG. 11, transfection efficiency of different non-viral vectors for primary synoviocytes.
Detailed Description
The inventor is dedicated to the molecular therapy research of the bone joint for a long time, and finds a suitable gene transfection reagent aiming at the characteristics of the cell for the first time. The transfection reagent is a compound of low molecular weight Polyethyleneimine (PEI) and cyclodextrin (CyD), and has high transfection efficiency and low toxicity when being used for transfecting exogenous genes into osteoarticular related cells.
Term(s) for
As used herein, an "exogenous gene (gene of interest)" is any gene of interest that needs to be transfected into a cell in order to be expressed in the cell or to exert a corresponding effect. The transfection is in vivo or in vitro.
As used herein, the "expression system for expressing a foreign gene" is generally an expression vector containing an expression cassette for a foreign gene, which, when transferred into a suitable cell, can express the foreign gene in the cell.
As used herein, the "gene delivery system" is also referred to as a "drug delivery system" which is a system that facilitates delivery of an expression system expressing a foreign gene into a cell after being mixed (homogeneously mixed) with the expression system expressing the foreign gene and being brought into contact with the cell. It belongs to a non-viral vector, which itself is preferably low or no immunogenic, low or no toxic, so as not to have an additional effect on the cells.
As used herein, unless otherwise indicated, by "low molecular weight PEI" is meant PEI having a molecular weight of less than 2000Da (preferably a molecular weight between 400-1500 Da; more preferably between 500-1000 Da; more preferably between 500-800; most preferably a molecular weight of 600 Da).
As used herein, unless otherwise indicated, "high molecular weight PEI" refers to a molecular weight greater than 2000 Da; such as PEI ranging between 5kDa and 25 kDa.
As used herein, the "nitrogen to phosphorus ratio (N/P)" refers to: the ratio of available nitrogen content in the gene delivery system to available phosphorus content in the expressed exogenous gene alone. In PEI600CyD, a carrier material (gene delivery system), 13.5mg/ml 100nmol/L available N, 1ug/ul of the expressed single exogenous gene corresponds to 3nmol/L available phosphorus.
Composite material
PEI was used as a transfection reagent, first in 1998, discovered and applied by Boussif et al [ Sato T, Ishii T, Okahata Y. in vitro gene delivery by chromatography. Effect of pH, serum, and molecular mass of chromatography on the transfection efficiency. biomaterials 2001; 22(15):2075-80].
PEI is a high molecular polymer with better water solubility. It can be classified into high molecular weight PEI and low molecular weight PEI according to the difference of molecular weight. Among them, PEI-25kDa (high molecular weight PEI) is a transfection reagent which has been commercialized at present. Primary amines (-NH) in PEI2-) and a secondary amine (-NH-) tend to bind H in aqueous solution+So that the high molecular polymer has higher positive charge and can be mutually attracted and compounded with the phosphate skeleton with negative charge on the plasmid through the mutual electrostatic attraction between the positive charge and the negative charge. By which means to increase the transfection efficiency of low molecular weight PEI is currently a matter of ongoing research in the art.
Cyclodextrin is a natural cyclic high molecular compound without immunogenicity. The molecular formula is as follows:

in a preferred embodiment of the present invention, the molecular weight of Polyethyleneimine (PEI) in the complex of low molecular weight polyethyleneimine and cyclodextrin is less than 2000 Da.
In a more preferred mode of the invention, the compound of the low molecular weight polyethyleneimine and the cyclodextrin has a structure shown in formula I,

wherein m is1Is a positive integer of 8 to 50, m2Is a positive integer of 4-25, n is a positive integer of 10-20; and m is1:m2=2:1。
Transfection method
Aiming at the technology of transferring exogenous genes into cells, the inventor finds that various transfection reagents have great difference on the transfection efficiency of cells due to the great difference of cell structures or properties (such as the difference of cell membranes, nuclear membrane structures and components; the difference of components and compositions of cytoplasm and organelles contained in the cytoplasm, and the like); and the effects on cells (e.g., toxicity) are also different. Thus, for one type or specific type of cell (e.g., osteoarticular-related cells), the selection of transfection reagent is important. Better transfection efficiency can only be achieved if a suitable transfection reagent is found.
It has been shown (Felgner PL et al Lipofection: a highlyeffective, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci USA 1987; 84:7413-7417.) that the transfection efficiency of liposomes on different cells varies by a factor of 100 depending on the cell type. Bone joint-related cells have been classified as cells that are difficult to transfer genes. In order to transfect a foreign gene in a bone joint-related cell, the present inventors have tried a wide variety of transfection reagents. Finally, the compound of the low molecular weight polyethyleneimine and the cyclodextrin has good transfection efficiency for osteoarticular related cells; and has low toxicity to cells. In the course of the experiments of the present invention, the influence of N/P on the transfection efficiency of different joint-associated primary cells was mainly considered. The following are found: on the one hand, different N/P will produce different toxicity to cells, and on the other hand, different N/P will have different transfection efficiency to cells. There is no relevant study on bone joint-related cells.
Accordingly, the present invention provides a method for transfecting a foreign gene into a cell associated with a bone joint, comprising: (1) mixing an expression system for expressing the exogenous gene with a gene delivery system to obtain a transfection solution; and (2) adding the transfection solution to the bone joint-related cells, whereby the foreign gene is transfected into the cells.
In a preferred embodiment of the present invention, the bone joint-related cells are selected from the group consisting of: synoviocytes, chondrocytes, bone marrow mesenchymal stem cells. The inventor surprisingly found that the low molecular weight polyethyleneimine and cyclodextrin complex has very excellent transfection efficiency for mesenchymal stem cells, which is significantly higher than Lipo 2000. Therefore, more preferably, the bone joint-related cell is a bone marrow mesenchymal stem cell.
In order to obtain a better transfection effect, the inventor also optimizes the nitrogen-phosphorus ratio of the mixture of the expression system of the exogenous gene and the gene delivery system. Therefore, in a preferred embodiment of the present invention, the ratio (N/P) of available nitrogen in the gene delivery system to available phosphorus in the expressed foreign gene alone is 20 to 35.
As a preferred mode of the invention, the nitrogen-phosphorus ratio of different bone joint related cells has a certain difference, and is preferably 25-35 relative to synovial cells and bone marrow mesenchymal stem cells; more preferably 28-32 and most preferably 30. Preferably 20-30, more preferably 22-27, most preferably 25 for chondrocytes.
The main advantages of the invention are:
(1) for the first time, the reagent suitable for carrying out the gene transfection efficiency of bone joint related cells (including bone marrow mesenchymal stem cells, synoviocytes and chondrocytes; particularly the bone marrow mesenchymal stem cells) is found. Provides a solid experimental foundation for the subsequent gene therapy of osteoarthritis in vivo.
(2) Optimizes the condition of carrying out bone joint related cell transfection by using the compound of low molecular weight polyethyleneimine and cyclodextrin as a gene delivery system.
The invention will be further illustrated with reference to the following specific examples. It should be understood that these examples are for illustrative purposes only and are not intended to limit the scope of the present invention. Experimental procedures without specific conditions noted in the following examples, molecular cloning is generally performed according to conventional conditions such as Sambrook et al: the conditions described in the laboratory Manual (New York: Cold Spring Harbor laboratory Press, 1989), or according to the manufacturer's recommendations. Unless otherwise indicated, percentages and parts are by weight.
Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art. In addition, any methods and materials similar or equivalent to those described herein can be used in the practice of the present invention. The preferred embodiments and materials described herein are intended to be exemplary only.
I. Materials and methods
PEI as the material used in this example600-CyD(PEI600- β -CyD) obtained from professor Tougiping, college of materia Medica, Zhejiang university, and having the following structural formula (wherein m is1Is 10, m2Has a value of 5), and m1:m22:1, mainly considering the ratio of amino groups, n ═ 16) since, from the molecular weight of PEI and cyclodextrin, it is again 1: the ratio of 1 is combined, and the present inventors dialyzed with a 15000 dialysis membrane at the time of synthesis, so that the molecular weight is generally higher than 15000, but the molecular weight is too large to easily cause crosslinking, and thus such a limitation is imposed.

Extraction and culture of rat BMSC
Reagent:
DMEM(Hyclone);
fetal bovine serum (Hyclone);
PBS buffer: 8g NaCl, 0.2g KCl, 3.5g Na2HPO4·12H2O,0.24g KH2PO4Dissolving with 1L of ultrapure water, adjusting pH to 7.2-7.4, and autoclaving;
0.25% (w/v) Trypsin-EDTA solution (Gibco)TM,Invitrogen);
Double resistance: cyan-streptomycin solution (100X) containing penicillin sodium salt 5,000 units/ml, streptomycin 5,000. mu.g/ml (Hyclone).
Extraction and culture of rat BMSCs in vitro:
healthy SD rats, 250 + -10 g, 8 + -1 week, were sacrificed by cervical dislocation and then sterilized in 75% (v/v) alcohol for 10 minutes. Dissecting rat, respectively taking out femur and tibia of hind limb at two sides of rat, removing muscle attached to bone with surgical scissors and forceps, cutting one end of femur and tibia, sucking 3ml of fetal calf serum containing 10% (v/v) with disposable 5ml syringeDMEM, then the culture medium is injected into the marrow cavities of the femur and the tibia, and the marrow is flushed out and flows into the culture dish along with the liquid. One mouse corresponds to one dish of cells. The volume of the medium in the dish was made up to 10 ml. Then at 5% (v/v) CO2Culturing in an incubator at 37 deg.C for 4 days under saturated humidity, discarding nonadherent cells with PBS when changing culture medium, and continuously culturing while changing culture medium (10% (v/v) FBS + DMEM) every 3 days. By day 7, when the cells were substantially full of monolayer, they were digested with 0.25% (w/v) pancreatin for 1 minute at 37 ℃, after most of the cells had shrunk and rounded, the digestion was stopped by adding medium, and after centrifugation at 1000rpm for 4 minutes, the cells were harvested. Adding culture medium, gently blowing and beating to make cells fully suspended to obtain primary BMSC suspension, taking 10 μ l for cell counting, and counting by 1 × 104/cm2Subculture at the density of (2). All other experiments were taken from 3-6 passages of cells as primary cells.
Extraction and culture of rat primary synovial membrane
Reagent:
DMEM(Hyclone);
fetal bovine serum (Hyclone);
PBS buffer: 8g NaCl, 0.2g KCl, 3.5g Na2HPO4·12H2O,0.24g KH2PO4Dissolving with 1L of ultrapure water, adjusting pH to 7.2-7.4, and autoclaving;
0.25% Trypsin-EDTA solution (Gibco)TM,Invitrogen);
Collagenase type I (Sigma);
double resistance: cyan-streptomycin solution (100X) containing penicillin sodium salt 5,000 units/ml, streptomycin 5,000. mu.g/ml (Hyclone).
Extracting and culturing primary synovial cells of rats in vitro:
healthy SD rats 250 + -10 g, 8 + -1 week, were sacrificed by cervical dislocation and then sterilized in 75% ethanol for 10 minutes. The rat is dissected, the patellar ligament is cut open, the joint cavity is opened from the front of the joint cavity, and the synovial tissues at the two sides are taken out. The fat and part of the connective tissue were cut off in a 10cm petri dish containing PBS and a smooth synovial membrane group was isolatedAnd (5) weaving. Transferred to another petri dish, washed 3 times with PBS, and then the synovial tissue was cut to 1-2mm using sterile ophthalmic scissors3Size pieces, immersed in PBS throughout the procedure. Transferring the fragments into 50ml centrifuge tube, adding 10ml of type I collagenase 0.4% (w/v), 37 deg.C, 5% CO2The incubator digests for 5 h. After 5 hours, after the debris had been substantially digested, the filter was filtered through a sterile 120 mesh filter, the filtrate was pipetted into a 50ml centrifuge tube, and the cells attached to the bottom of the filter and petri dish were washed with PBS and collected. After collection, the mixture was centrifuged at 1000rpm for 10 minutes, and the supernatant was discarded. And adding a fresh culture medium, slightly blowing and beating to enable the cells to be fully suspended, adding the cells into a 10cm culture dish for culture, and replacing the culture solution once in 3 days later to obtain the primary synoviocytes. Passage method of synovial cells: after the cells grow over the bottom of the culture dish, the culture medium is removed, the cells are washed once by PBS, then 0.25% pancreatin is added to be digested for 2 minutes at 37 ℃, after most of the cells shrink and become round, the culture medium is added to stop the digestion, and the cells are collected after centrifugation for 4 minutes at 1000 rpm. Adding culture medium, gently blowing to make cells fully suspended, taking 10 μ l for cell counting, and counting at 1 × 104/cm2Subculture at the density of (2). All other experiments were taken from 3-6 passages of cells as primary cells.
Extraction and culture of rat primary cartilage
Reagent:
α-MEM(Hyclone);
fetal bovine serum (Hyclone);
PBS buffer: 8g NaCl, 0.2g KCl, 3.5g Na2HPO4·12H2O,0.24g KH2PO4Dissolving with 1L of ultrapure water, adjusting pH to 7.2-7.4, and autoclaving;
0.25% Trypsin-EDTA solution (Gibco)TM,Invitrogen);
Collagenase type II (Sigma);
double resistance: cyan-streptomycin solution (100X) containing penicillin sodium salt 5,000 units/ml, streptomycin 5,000. mu.g/ml (Hyclone).
Extracting and culturing primary chondrocytes of rats in vitro:
a healthy SD rat, 250 + -10 g, 8 + -1 week, is sacrificed by cervical dislocation, and is placed in 75% alcohol for 10 minutes for sterilization, the rat is dissected, patellar ligament is cut, joint cavity is opened from the front of the joint cavity, cartilage tissue is cut from the tibial plateau and the cartilage surface of femoral condyle by a sterilized surgical cutter, the cartilage tissue is placed in α -MEM without serum, all cartilage pieces are washed three times by PBS after being taken, non-cartilage tissue such as synovial tissue or fibrous connective tissue is cut, the cartilage tissue pieces are cut into 1-2mm by sterile ophthalmic scissors3Size pieces, immersed in PBS throughout the procedure. The fragments were transferred to 50ml centrifuge tubes, digested for 30 minutes in 37 ℃ incubator with the addition of fresh 0.25% trypsin, and occasionally mixed to remove any fibroblasts that may have adhered. After centrifugation at 1000rpm for 4 minutes, cartilage pieces were collected and washed once with PBS, 0.2% collagenase type II, 37 ℃ and 5% CO were added2Digesting for 4-6 hours in an incubator, shaking the centrifuge tube once every 20 minutes after 2 hours, observing whether the cartilage pieces are basically digested and whether the suspension is turbid, after the cartilage pieces are basically digested, filtering with a sterilized 120-mesh filter, transferring the filtrate into a 50ml centrifuge tube with a pipette, washing the cells attached to the bottom of the filter and the bottom of the culture dish with PBS, collecting, centrifuging for 10 minutes at 1000rpm, discarding the supernatant, adding a fresh culture medium (10% FBS + α -MEM), gently blowing and beating the cells to fully suspend, adding the cells into a 10cm culture dish, culturing, changing the culture medium once after 3 days, and obtaining primary chondrocytes4/cm2Subculture at the density of (2). All other experiments were taken from 3-6 passages of cells as primary cells.
MTT cytotoxicity assay
Reagent:
α-MEM(Hyclone);
DMEM(Hyclone);
fetal bovine serum (Hyclone);
PBS buffer: 8g NaCl, 0.2g KCl, 3.5g Na2HPO4·12H2O,0.24g KH2PO4Dissolving with 1L of ultrapure water, adjusting pH to 7.2-7.4, and autoclaving;
0.25% Trypsin-EDTA solution (Gibco)TM,Invitrogen);
Double resistance: cyan-streptomycin solution (100 ×), containing penicillin sodium salt 5,000 units/ml, streptomycin 5,000 μ g/ml (hyclone);
MTT(Sigma);
DMSO (Shanghai chemical Co.).
MTT cytotoxicity assay:
after the cells (BMSC, synoviocytes, chondrocytes 3-6 passages as primary cells) were confluent, the supernatant was discarded, the cells were washed once with PBS, then digested with 0.25% trypsin at 37 ℃ for 2 minutes, after most of the cells had contracted and rounded, the digestion was stopped by adding the medium, and after centrifugation at 1000rpm for 4 minutes, the cells were collected. Adding culture medium, gently blowing to make cells sufficiently suspend, taking 10 μ l, performing cell counting with cell counter, and diluting cell concentration with culture medium to 5 × 104After one/ml, 200. mu.l of the cell suspension, i.e.1X 10, was added to the 96-well plate4Per well. At 37℃ 5% CO2After culturing for 24 hours in an incubator, respectively adding the non-viral vector PEI600CyD and PEI-25kDa, 5. mu.l per well, ten concentration gradients, the highest gradient having a final concentration of 0.25. mu.g/. mu.l, whereby the concentrations are diluted down-by-one and the lowest concentration being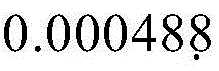 μ g/. mu.l. After 4 hours of action of the non-viral vector, the medium was changed, 200. mu.l of fresh medium was added to each well, and 20. mu.l (5mg/ml) of MTT was added thereto, and after 4 hours of action, the presence of purple crystals in the cells was observed under a microscope using an inverted microscope. The supernatant was discarded, 200. mu.l DMSO was added to each well, and the mixture was placed inThe shaker was incubated at 37 ℃ and 100rpm for 30 minutes to dissolve the purple crystals. And after the purple crystals are dissolved, putting the purple crystals into an enzyme-labeling instrument for absorbance measurement, wherein the test wavelength is set to 570nm, and the reference wavelength is set to 630 nm. The difference value obtained by subtracting the absorbance value of the reference wavelength from the absorbance value of the test wavelength is the final absorbance value, and then the absorbance value of the control group (i.e. the group without the non-viral vector treatment) is removed by the absorbance value of the sample group, so that the percentage value of the cell viability can be obtained.
μ g/. mu.l. After 4 hours of action of the non-viral vector, the medium was changed, 200. mu.l of fresh medium was added to each well, and 20. mu.l (5mg/ml) of MTT was added thereto, and after 4 hours of action, the presence of purple crystals in the cells was observed under a microscope using an inverted microscope. The supernatant was discarded, 200. mu.l DMSO was added to each well, and the mixture was placed inThe shaker was incubated at 37 ℃ and 100rpm for 30 minutes to dissolve the purple crystals. And after the purple crystals are dissolved, putting the purple crystals into an enzyme-labeling instrument for absorbance measurement, wherein the test wavelength is set to 570nm, and the reference wavelength is set to 630 nm. The difference value obtained by subtracting the absorbance value of the reference wavelength from the absorbance value of the test wavelength is the final absorbance value, and then the absorbance value of the control group (i.e. the group without the non-viral vector treatment) is removed by the absorbance value of the sample group, so that the percentage value of the cell viability can be obtained.

Cell transfection of complexes and quantitative analysis by flow cytometry
Reagent:
α-MEM(Hyclone);
DMEM(Hyclone);
fetal bovine serum (Hyclone);
PBS buffer: 8g NaCl, 0.2g KCl, 3.5g Na2HPO4·12H2O,0.24g KH2PO4Dissolving with 1L of ultrapure water, adjusting pH to 7.2-7.4, and autoclaving;
0.25% Trypsin-EDTA solution (Gibco)TM,Invitrogen);
Double resistance: cyan-streptomycin solution (100 ×), containing penicillin sodium salt 5,000 units/ml, streptomycin 5,000 μ g/ml (hyclone);
LipofectamineTM2000Reagent, Lipo2000(Invitrogen) for short.
PEI600-complexation of CyD and PEI-25kDa with pEGFP, transfection and quantitative analysis by flow cytometry:
BMSC, primary synoviocytes, primary chondrocytes at1X 105Perwell, 1ml seed per well in 12 well plates. After 24 hours of plating, the cell density was close to above 90%, the medium was discarded, washed once with PBS and then transfectant was added. Preparing a transfection solution: firstly, preparing PEI-25kDa into 1 mu g/mu l stock solution by using ultrapure water, and preparing PEI600CyD was prepared as a stock solution with ultrapure water at 2.7. mu.g/. mu.l, and the plasmid was diluted to 0.05. mu.g/. mu.l for use. Then based on 4.5. mu.g/. mu.l PEI-25kDa ═ 100 nmol/. mu.l effective "N", 13.5. mu.g/. mu.l PEI600-CyD ═ 100 nmol/. mu.l effective "N", 1. mu.g/. mu.l massThe particle is calculated as 3 nmol/. mu.l effective "P". Then 40. mu.l, 0.05. mu.g/. mu.l of pEGFP and 40. mu.l of PEI-25kDa or PEI at the corresponding concentration (different concentrations calculated from different N/P)600And (3) carrying out equal-volume mixing on the-CyD, slowly dripping, shaking on a shaker for 20 seconds at the highest speed, standing for 30 minutes at room temperature, adding 1ml of serum-free medium DMEM, mixing uniformly, and standing for 5 minutes for use. The groups established by the inventors herein are: for BMSC cells and primary synoviocytes (PEI-25 kDa: N/P ═ 10, 8, 4, 2; PEI600-CyD: N/P ═ 30, 25, 20, 15), for primary chondrocytes (PEI-25 kDa: N/P ═ 10, 8, 4, 2; PEI600CyD N/P25, 20, 15, 10 for the positive control group, see Invitrogen for instructions for use of Lipo 2000. briefly, 4. mu.l Lipo2000 was added to 100. mu.l serum-free DMEM (α -MEM for chondrocyte experiments), 2. mu.g pEGFP was added to 100. mu.l serum-free DMEM (α -MEM for chondrocyte experiments) and then shaken for 20s, Lipo2000 was added to pEGFP, and after shaking for 20s, it was left to stand at room temperature for 25min for use, after 4 hours of transfection, the transfection solution was discarded, fresh DMEM 1ml containing 10% (v/v) fetal bovine serum and 1% volume diabody (α -MEM for chondrocyte experiments), after 48 hours, observation was performed under a fluorescence microscope, after completion of observation, the cells in 12-well plates were digested with 0.25% Tryp-EDTA solution, collected once by flow tube (1000rpm, collected for 4 minutes, and then resuspended in PBS, and then collected by flow cytometry.
Statistical significance was determined by random-factor analysis of variance (Student ANOVA) using the SAS statistical package and compared between groups using the SNK test.
Quantum dot labeling of plasmid pEGFP
Reagent:
Psoralen-PEO3-Biotin(PIERCE);
potassium acetate (Shanghai chemical Co.);
absolute ethanol (Shanghai chemical Co.);
6% (w/v) Bovine Serum Albumin (BSA): 300mgBSA was dissolved in 5ml PBS;
DMSO (shanghai city chemicals);
quantum dots Qdot Streptavidin Conjugates-605 (Invitrogen);
quantum dot labeling of plasmid pEGFP:
plasmid pEGFP was diluted to 0.1. mu.g/. mu.l with ultrapure water, and a volume of plasmid solution 1/100, 20mM Psoralen-PEO, was taken out of the dark3Biotin was mixed and reacted for 30 minutes under an ultraviolet lamp with 365nm excitation light, which was performed on ice. After completion of the reaction, the reacted plasmid was precipitated by centrifugation at 12000rpm for 10 minutes using a potassium acetate solution having a final concentration of 0.2M and 2 volumes of anhydrous ethanol, and the supernatant (containing therein unreacted plasmid and Psoralen-PEO) was discarded3-Biotin). And cleaning the precipitate with 70% ethanol, drying the precipitate, diluting with water, and measuring the concentration with an ultraviolet spectrophotometer for later use.
mu.M quantum dots were diluted to 20nM with 6% BSA. Simultaneously adding Psoralen-PEO3-Biotin-linked plasmids were diluted to 5nM with the same quantum dot volume and mixed, incubated at room temperature in the dark for 4 hours, centrifuged at 12000rpm for 10 minutes with potassium acetate solution of final concentration 0.2M and 2 times the volume of absolute ethanol to precipitate reacted plasmids, at the same time the supernatant was discarded, the precipitate was washed with 70% ethanol, dried, diluted with water and its concentration was measured with UV spectrophotometer for future use.
Example II
Example 1 PEI600Cytotoxicity of CyD
(1)PEI600Cytotoxicity of CyD on BMSC
As can be seen from FIG. 1, PEI was used for either 4 hours or 24 hours of action600The cytotoxicity of-CyD on BMSC is obviously less than that of PEI-25 kDa. At 4 hours, when PEI600The final concentration of CyD reaches 0.25 mug/ul, and the survival rate of BMSC cells measured by MTT method can reach 97.63%. Whereas the PEI-25kDa group showed only 10.10% cell viability at this concentration. At 24 hours, when PEI600Final concentration of CyDWhen the concentration of the BMSC is 0.25 mu g/mu l, the survival rate of the BMSC cells measured by an MTT method can reach 77.90 percent. Whereas the PEI-25kDa group showed only 8.06% cell viability at this concentration.
(2)PEI600Cytotoxicity of CyD on Primary synoviocytes
As can be seen from FIG. 2, PEI was used for either 4 hours or 24 hours of action600CyD is significantly less cytotoxic to primary synoviocytes than PEI-25 kDa. At 4 hours, when PEI600The final concentration of CyD reaches 0.25 mu g/mu l, and the survival rate of primary synovial cells measured by an MTT method can reach 72.02%. Whereas the PEI-25kDa group showed only 11.94% cell viability at this concentration. At 24 hours, when PEI600The final concentration of CyD reaches 0.25 mu g/mu l, and the survival rate of primary synovial cells measured by an MTT method can reach 48.93 percent. Whereas the PEI-25kDa group showed only 7.65% cell viability at this concentration.
(3)PEI600Cytotoxicity of CyD on Primary chondrocytes
As can be seen from FIG. 3, PEI was observed to be present for either 4 hours or 24 hours of action600CyD is significantly less cytotoxic to primary chondrocytes than PEI-25 kDa. At 4 hours, when PEI600The final concentration of CyD reaches 0.03125 mug/ul, and the survival rate of primary chondrocytes measured by MTT method can reach 76.93%. Whereas the PEI-25kDa group showed only 12.04% cell viability at this concentration. At 24 hours, when PEI600The final concentration of CyD reaches 0.03125 mug/ul, and the survival rate of primary chondrocytes measured by MTT method can reach 63.84%. Whereas the PEI-25kDa group showed only 4.19% cell viability at this concentration.
Combining the above results, it can be seen that each cell has a large difference in cytotoxicity to the material, primary chondrocytes are sensitive to non-viral vectors, and PEI is relatively speaking600CyD and PEI-25kDa are much less cytotoxic for BMSC and primary synoviocytes. Nevertheless, one and the same phenomenon can be found on these three cells, namely PEI600The cytotoxicity of-CyD on all three cells is significantly less than that of PEI-25 kDa.
Example 2 PEI600Analysis of transfection efficiency of CyD
(1)PEI600Analysis of transfection efficiency of CyD for BMSC
FIG. 4A is a photograph of the PEI-25kDa panel taken at an N/P of 10 and PEI600The pictures of the CyD group were taken at an N/P of 30. The Lipo2000 group was a positive Control group, and the Control group was a negative Control group (naked plasmid group). The left side is a photograph taken under fluorescence and the right side is a photograph taken under white light for the same field of view, all photographs being taken under a 20 x objective. Combining FIG. 4A and FIG. 4B, PEI was discovered600The transfection efficiencies of-CyD (N/P ═ 30) and PEI-25kDa (N/P ═ 10) were 11.20% and 10.20%, respectively, higher than those of positive control Lipo2000 group (3.53%), and were statistically different, with P representing P<0.01. With reference to FIG. 4C, PEI can also be found600The transfection efficiency of CyD for BMSC increases with increasing N/P.
(2)PEI600Analysis of the transfection efficiency of CyD on Primary synoviocytes
FIG. 5A is a photograph of the PEI-25kDa panel taken at an N/P of 10 and PEI600The pictures of the CyD group were taken at an N/P of 30. The Lipo2000 group was a positive Control group, and the Control group was a negative Control group (naked plasmid group). The left side is a photograph taken under fluorescence and the right side is a photograph taken under white light for the same field of view, all photographs being taken under a 20 x objective. Combining FIGS. 5A and 5B, PEI was discovered600The transfection efficiencies of-CyD (N/P ═ 30) and PEI-25kDa (N/P ═ 10) were 16.37% and 10.50%, respectively, which were lower than those of positive control Lipo2000 group (61.00%), and were statistically different, and represent P<0.01. And PEI600There is also a statistical difference between-CyD (N/P ═ 30) and PEI-25kDa (N/P ═ 10). With reference to FIG. 5C, PEI can also be found600The transfection efficiency of CyD for primary synoviocytes increased with increasing N/P.
(3)PEI600Analysis of transfection efficiency of CyD on Primary chondrocytes
FIG. 6A is a photograph of the PEI-25kDa panel taken at an N/P of 10 and PEI600Photographs of the CyD group areShot when the N/P is 25. The Lipo2000 group was a positive Control group, and the Control group was a negative Control group (naked plasmid group). The left side is a photograph taken under fluorescence and the right side is a photograph taken under white light for the same field of view, all photographs being taken under a 20 x objective. Combining FIGS. 6A and 6B, PEI was discovered600The transfection efficiencies of-CyD (N/P ═ 25) and PEI-25kDa (N/P ═ 10) were 13.37% and 12.10%, respectively, which were higher than those of positive control Lipo2000 group (5.30%), and were statistically different, with P representing P<0.05. And PEI600There was no statistical difference between-CyD (N/P ═ 25) and PEI-25kDa (N/P ═ 10). With reference to FIG. 6C, PEI can also be found600The transfection efficiency of CyD on primary chondrocytes increases with increasing N/P.
Example 3 tracer analysis in cells after pEGFP transfection in complexes
(1) Tracing distribution condition of pEGFP transfected in cell by quantum dot marking method
The distribution of pEGFP in BMSC cells after transfection is traced by using a quantum dot labeling method is shown in FIG. 7. The upper row of the figure is a superposition effect graph of the quantum dot marked plasmid under 4 times of objective lens and the cell under white light, and the lower row is a superposition effect graph of the quantum dot marked plasmid and the cell nucleus stained by Hochests under 40 objective lenses. By observing the upper row of photographs, it was found that most of the red color appeared as a cluster in the Control group (naked plasmid group), indicating that the plasmid was aggregated in the Control group and that the plasmid was difficult to enter the cells. In the Lipo2000 group, the appearance of red spots of gradually fine particles was found, indicating that a part of the plasmids did not aggregate under the packaging of Lipo2000, and that the part of the plasmids entered the cells relatively easily. Continued observation of the PEI-25kDa group and PEI600CyD group, the fine red spots are found more and more, in PEI600Almost all plasmids of the CyD group are well packaged, and only a small fraction of the plasmids undergo agglomeration. Moreover, it was found that PEI600CyD group, plasmid in diffuse state, distributed in the whole cell. By observing the photographs in the lower row, it was found that there were few plasmids in the Control group and that the plasmids appeared to be substantially all represented coagulumThe state of aggregation. In the Lipo2000 group, the number of plasmids was found to be significantly increased, but still, a state of aggregation was substantially exhibited. While in the PEI25-kDa group (N/P ═ 10) and PEI600In the CyD group (N/P-30), the carrier material can be found to be able to complex plasmids well, without agglomeration of the plasmids occurring, but rather with a more homogeneous distribution within the cells. After careful observation, it was found that the number of plasmids in the nucleus was in the PEI25-kDa group (N/P ═ 10) and PEI600There is also significantly more in the CyD group (N/P ═ 30) than in the Control and Lipo2000 groups. This indicates that the support material PEI600CyD is better able to help the plasmid enter the nucleus of BMSC.
Example 4 PEI600Efficiency of transfection of CyD into other cells
Currently, the most classical non-viral transfection reagent on the market is Lipofectamine from InvitrogenTM2000, after extensive research, the inventors found that PEI was present in spite of its presence in the 293T cell line, the Hela cell line600The transfection efficiency of-CyD is significantly less than that of Lipofecatemine2000 on both cell lines. However, in primary chondrocytes and primary mesenchymal stem cells, PEI was found600The transfection efficiency of-CyD is obviously better than that of lipofectamine 2000, and can reach 3-4 times of that of lipofectamine 2000. See fig. 8.
And after comparison research with another cationic polymer Chitosan (Chitosan) and its derivative PCL-Chitosan (grafted Polycaprolactone (PCL) which improves the capability of releasing plasmid from carrier to a certain extent), PCL-PEG-Chitosan (grafted PCL and polyethylene glycol (PEG), which can also reduce the particle size of the compound and improve the capability of releasing plasmid from carrier) (all from Shanghai university of transportation), PEI is found, compared with the existing mainstream non-viral gene carrier600CyD has the highest transfection efficiency for osteoarticular-related cells, especially primary bone marrow mesenchymal stem cells and primary chondrocytes, see FIGS. 9-11.
Discussion of the related Art
The invention researches a non-viral vector PEI for the first time600-use of CyD in bone joint-related cells. By MTT experiment, the non-viral vector PEI25-kDa and PEI600Cytotoxicity of CyD was investigated and PEI was found600CyD is significantly less cytotoxic to BMSC, primary synoviocytes and primary cartilage than PEI 25-kDa. This is mainly due to the fact that when the non-viral vector PEI600After entering lysosome, CyD is degraded to form PEI600And CyD, PEI with a molecular weight of 600 has relatively small cytotoxicity per se, while CyD, a natural macromolecular compound, has relatively small cytotoxicity per se, so that PEI600The cytotoxicity of-CyD is less. Conversely, PEI-25kDa, a synthetic polymer compound, is very difficult to degrade in cells and therefore can cause great cytotoxicity.
Through cell transfection experiments, the plasmid pEGFP capable of expressing green fluorescent protein and non-viral vector PEI are utilized600-CyD and PEI25-kDa were complexed separately and then BMSC, primary synoviocytes and primary chondrocytes were transfected separately, quantitative analysis of transfection efficiency was performed using flow cytometry, and Lipo2000 and naked plasmid groups were set as positive and negative control groups, respectively.
Through quantitative analysis, the inventor finds that for BMSC and PEI600CyD at the N/P of 30 is basically consistent with the transfection efficiency of PEI-25kDa at the N/P of 10, and the obvious difference is larger than that of a positive control group and a negative control group, which indicates that the transfection efficiency of the PEI-based non-viral vector is better than that of the liposome-based non-viral vector on the BMSC cells.
Further, the inventors of the present invention have further analyzed the reason why the difference is caused, and have studied the distribution of pEGFP in BMSC cells after transfection with different non-viral vectors by quantum dot labeling of pEGFP, and found that the pEGFP is relatively easy to aggregate in the naked plasmid group and hardly penetrates through the cell membrane to enter the cytoplasm, whereas with the liposome type non-viral vector Lipo2000, the aggregation of plasmid is slowed down, but the aggregation still cannot effectively penetrate through the cell membrane of BMSC, indicating that Lipo2000 is not sufficient in the ability to rupture the cell membrane of BMSC cells. And for PEI600CyD, when N/P equals 30, the plasmid in the complex can cross more efficientlyThe cell membrane and into the cytoplasm, and the inventors were also able to find the presence of many plasmids in the nucleus, demonstrating that PEI600The low molecular weight PEI in CyD can exert certain 'proton sponge effect' without being retained in lysosomes and degraded so as not to enter cell nucleus.
It is worth mentioning here that the inventors have performed gel electrophoresis on the compounded transfection solution, and found that Lipo2000 and pEGFP are not compounded at all, and the principle that Lipo2000 functions is probably to break the membrane or increase the permeability of the cell membrane first, and then facilitate the entry of pEGFP. Interestingly, the transfection efficiency of Lipo2000 group on primary synoviocytes increased dramatically under the same conditions, suggesting that it is meaningful to study the cell membrane composition of BMSCs and the cell membrane composition of primary synoviocytes for an in-depth discussion of the mechanism of Lipo2000 to cross cell membranes.
To sum up, PEI600CyD can be used as a low-toxicity and effective non-viral gene vector to transfect BMSC, primary synoviocytes and primary chondrocytes in vitro. And the next experiment can be carried out in the animal body to evaluate the biocompatibility and the transfection efficiency of the animal body, and the metabolic pathways and the organ distribution of the animal body can be deeply observed by using a marking technology and a living body imager. And an animal model of osteoarticular diseases can be constructed, plasmid DNA with therapeutic genes is utilized to carry out gene therapy on the animal model, and the therapeutic effect is observed.
All documents referred to herein are incorporated by reference into this application as if each were individually incorporated by reference. Furthermore, it should be understood that various changes and modifications of the present invention can be made by those skilled in the art after reading the above teachings of the present invention, and these equivalents also fall within the scope of the present invention as defined by the appended claims.
Claims (2)
Translated fromChinese
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610023604.7ACN105671078B (en) | 2010-09-27 | 2010-09-27 | Nano non-viral gene delivery system and application thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610023604.7ACN105671078B (en) | 2010-09-27 | 2010-09-27 | Nano non-viral gene delivery system and application thereof |
| CN2010102927786ACN102417914A (en) | 2010-09-27 | 2010-09-27 | Nano non-viral gene delivery system and application thereof |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2010102927786ADivisionCN102417914A (en) | 2010-09-27 | 2010-09-27 | Nano non-viral gene delivery system and application thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN105671078A CN105671078A (en) | 2016-06-15 |
| CN105671078Btrue CN105671078B (en) | 2020-04-07 |
Family
ID=56300667
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201610023604.7AActiveCN105671078B (en) | 2010-09-27 | 2010-09-27 | Nano non-viral gene delivery system and application thereof |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN105671078B (en) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1639228A (en)* | 2002-02-22 | 2005-07-13 | 植入疗法公司 | Carbohydrate-modified polymers, compositions and uses related thereto |
| CN101338318A (en)* | 2008-01-25 | 2009-01-07 | 林李家宓 | Polycation transgenic vector and synthetic method thereof |
- 2010
- 2010-09-27CNCN201610023604.7Apatent/CN105671078B/enactiveActive
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1639228A (en)* | 2002-02-22 | 2005-07-13 | 植入疗法公司 | Carbohydrate-modified polymers, compositions and uses related thereto |
| CN101338318A (en)* | 2008-01-25 | 2009-01-07 | 林李家宓 | Polycation transgenic vector and synthetic method thereof |
Non-Patent Citations (2)
| Title |
|---|
| Construction of Polyethyleneimine-β- cyclodextrin/pDNA Multilayer Structure for Improved In Situ Gene Transfection;Yan Hu;《Advanced Biomaterials》;20100218;第12卷;摘要,第819页左栏,第820页右栏-824页左栏* |
| 环糊精偶联低分子量聚乙烯亚胺构建新型非病毒转基因载体及其应用;黄宏靓;《中国博士学位论文全文数据库(医药卫生科技辑)》;20070815(第2期);摘要、第29,43-51页* |
Also Published As
| Publication number | Publication date |
|---|---|
| CN105671078A (en) | 2016-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2883235T3 (en) | Injectable macroporous hydrogels | |
| Zhang et al. | Nuclear entry of hyperbranched polylysine nanoparticles into cochlear cells | |
| Hosseinkhani et al. | Development of 3D in vitro platform technology to engineer mesenchymal stem cells | |
| CN108743620A (en) | Bioactive materials promote source of human stem cell excretion body to treat corneal injury | |
| CN109054377A (en) | A kind of dendroid daiamid combination graphene oxide advanced composite material (ACM) and preparation method and application | |
| CN111467373A (en) | A kind of dental pulp stem cell exosome preparation, preparation method and application thereof | |
| CN114225044B (en) | A reagent for modifying extracellular vesicles and its preparation method | |
| CN115581810B (en) | Hydrogel rich in exosomes as well as preparation method and application thereof | |
| CN106492194A (en) | A kind of stem cell excretion body preparation and its preparation method and application | |
| CN110917400A (en) | Nano-hybrid silk fibroin hydrogel and preparation method and application thereof | |
| Ruan et al. | Biofunctionalized self-assembly of peptide amphiphile induces the differentiation of bone marrow mesenchymal stem cells into neural cells | |
| CN113616810A (en) | P-selectin-targeted engineered extracellular vesicle composition and preparation method and application thereof | |
| CN109528653A (en) | Film property vesica with gene editing function and preparation method thereof, pharmaceutical composition and purposes | |
| CN111116904A (en) | Phenylboronic acid modified fluorine-containing high polymer material and application thereof in intracellular delivery of protein | |
| CN108904468A (en) | A method of building targets extracellular vesica | |
| CN104974343B (en) | Modified polyethyleneimine and its application in Gene transfer vector reagent is prepared | |
| Li et al. | ScreenFect A: an efficient and low toxic liposome for gene delivery to mesenchymal stem cells | |
| CN104119451B (en) | A kind of cationic silk fibroin and preparation method thereof | |
| Zhu et al. | Chorionic villi-derived nanofibers enhanced mesenchymal stem cell extracellular vesicle secretion and bioactivity for endometrium regeneration toward intrauterine adhesion treatment | |
| CN101445806B (en) | Calcium-ionized mesoporous silica nanoparticle gene delivery system and its preparation method | |
| ES2763864T3 (en) | Preparation of a dental type structure using stem cells | |
| CN110151726A (en) | Use of resveratrol-loaded human pluripotent stem cell exosomes in the preparation of drugs for the treatment of bone and joint degenerative diseases | |
| CN105671078B (en) | Nano non-viral gene delivery system and application thereof | |
| CN113846064A (en) | FGF18 gene modified mesenchymal stem cell and preparation method and application thereof | |
| CN113425853B (en) | Glutathione-modified brain-targeted zinc oxide quantum dot gene transport carrier and preparation method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| CP03 | Change of name, title or address | Address after:200031 Yueyang Road, Shanghai, No. 319, No. Patentee after:Shanghai Institute of nutrition and health, Chinese Academy of Sciences Address before:200031 No. 320, Yueyang Road, Shanghai, Xuhui District Patentee before:SHANGHAI INSTITUTES FOR BIOLOGICAL SCIENCES, CHINESE ACADEMY OF SCIENCES | |
| CP03 | Change of name, title or address |