CN103054869A - Application of amino dithio formic ester compound with triazolyl in preparing medicine taking LSD1 (Lysine Specificity Demethylase 1) as target - Google Patents
Application of amino dithio formic ester compound with triazolyl in preparing medicine taking LSD1 (Lysine Specificity Demethylase 1) as targetDownload PDFInfo
- Publication number
- CN103054869A CN103054869ACN2013100261986ACN201310026198ACN103054869ACN 103054869 ACN103054869 ACN 103054869ACN 2013100261986 ACN2013100261986 ACN 2013100261986ACN 201310026198 ACN201310026198 ACN 201310026198ACN 103054869 ACN103054869 ACN 103054869A
- Authority
- CN
- China
- Prior art keywords
- derivant
- reaction
- polysubstituted
- lsd1
- triazolyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images

Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Translated fromChineseDescription
Translated fromChinese技术领域technical field
本发明涉及药物化学领域,具体涉及一类含三唑基的氨基二硫代甲酸酯化合物作为新的组蛋白赖氨酸特异性去甲基化酶1(LSD1)抑制剂的应用。The invention relates to the field of medicinal chemistry, in particular to the application of a class of triazole-containing carbadithiocarbamate compounds as novel histone lysine-specific demethylase 1 (LSD1) inhibitors.
背景技术Background technique
组蛋白赖氨酸去甲基化酶1(Lysine Specific Demethylase 1,LSD1)是2004年由哈佛大学医学院ShiYang教授发现的第一个组蛋白去甲基化酶(YangShi. Cell. 2004, 119, 941–953),能够去除组蛋白H3K4、H3K9的单、双甲基化,从而调节组蛋白和其他蛋白的相互作用,并影响基因转录的激活和抑制,染色体失活等重要生命过程。Histone lysine demethylase 1 (Lysine Specific Demethylase 1, LSD1) is the first histone demethylase discovered by Professor Shi Yang of Harvard Medical School in 2004 (YangShi. Cell.2004, 119, 941–953 ), which can remove the single and double methylation of histone H3K4 and H3K9, thereby regulating the interaction between histone and other proteins, and affecting the activation and repression of gene transcription, chromosome inactivation and other important life processes.
LSD1是一个黄素腺嘌呤二核苷酸(FAD)依赖的去甲基化酶,在去甲基化的过程中能够生成一分子的过氧化氢和甲醛。LSD1还是氨基氧化酶的同源蛋白,和多胺氧化酶(Polyamine Oxidase, PAO)相似度为22.4%,和单胺氧化酶A和B(Monoamine Oxidases A and B)相似度为17.6%。LSD1 is a flavin adenine dinucleotide (FAD)-dependent demethylase that can generate a molecule of hydrogen peroxide and formaldehyde during the demethylation process. LSD1 is also a homologous protein of amino oxidase, with a similarity of 22.4% to polyamine oxidase (Polyamine Oxidase, PAO), and a similarity of 17.6% to monoamine oxidases A and B (Monoamine Oxidases A and B).
LSD1在多种肿瘤细胞中LSD1的表达量显著高于正常细胞,如神经母细胞瘤、眼癌、前列腺癌、乳腺癌、肺癌、膀胱癌等。而实验证明,通过RNAi技术或小分子抑制剂在细胞水平降低LSD1表达量或降低LSD1的活性能抑制细胞增殖并诱导一些细胞分化相关基因的表达;在小分子单胺氧化酶抑制剂PCPA的作用下亦能抑制多种肿瘤细胞和实体瘤的生长。因此,LSD1抑制剂不仅能作为表观遗传学的研究工具用于阐述生物学功能,而且能作为表观遗传学药物用于肿瘤的预防和治疗。The expression of LSD1 in various tumor cells is significantly higher than that in normal cells, such as neuroblastoma, eye cancer, prostate cancer, breast cancer, lung cancer, and bladder cancer. Experiments have shown that reducing the expression of LSD1 or reducing the activity of LSD1 at the cellular level through RNAi technology or small molecule inhibitors can inhibit cell proliferation and induce the expression of some cell differentiation-related genes; under the action of small molecule monoamine oxidase inhibitor PCPA, it can also Inhibits the growth of various tumor cells and solid tumors. Therefore, LSD1 inhibitors can not only be used as epigenetic research tools to elucidate biological functions, but also can be used as epigenetic drugs for the prevention and treatment of tumors.
点击化学由2001年诺贝尔化学奖获得者美国化学家Sharpless首次提出,目前主要指的是一类Cu(I)催化的叠氮化合物与炔基化合物反应生成1, 2, 3-三氮唑五元环化合物的反应。由于其反应条件温和、产率高,产物后处理简单等诸多优点而在药物先导化合物的优化、化合物库的组建中得到了广泛应用。1, 2, 3-三唑表现出了多种有趣的生物活性,例如抗菌,抗炎,抗过敏,抗结核,抗HIV活性等。许多上市的药物结构中都具有1, 2, 3-三唑结构片段,例如β-内酰胺酶抑制剂他唑巴坦(式.1)。最近,其在抗肿瘤药物先导化合物发现中的应用受到越来越多的关注。多个研究小组利用点击化学将1, 2, 3-三唑和其他药效基团进行连接,合成得到了一些具有优秀抗肿瘤活性和抑制MAO活性的化合物。例如Kamal小组在鬼臼毒素中引入1, 2, 3-三唑(式.2),合成得到了一系列抗肿瘤活性优于etoposide的化合物(H.M.Sampath Kumar. European Journal of Medicinal Chemistry. 2011, 46, 1983-1991);Marvin J. Miller将1,2,3-三唑和芳酰胺结合(式.3),发现了一类新型的具有优异抗肿瘤活性的化合物(Marvin J. Miller. J. Med. Chem. 2010, 53, 3389-3395);而Qing Zhu小组利用Click合成多个三唑基衍生物(式4)并具有较好的抗MAO-A活性(Qing Zhu. Bioorganic & Medicinal Chemistry Letters.2010, 20, 6222-6225)。 Click chemistry was first proposed by the American chemist Sharpless, who won the Nobel Prize in Chemistry in 2001. At present, it mainly refers to the reaction of azide compounds and alkyne compounds catalyzed by Cu(I) to generate 1, 2, 3-triazole penta Reactions of ring compounds. Due to its mild reaction conditions, high yield, simple product post-treatment and many other advantages, it has been widely used in the optimization of drug lead compounds and the construction of compound libraries. 1, 2, 3-triazoles exhibit a variety of interesting biological activities, such as antibacterial, anti-inflammatory, anti-allergic, anti-tuberculosis, anti-HIV activities, etc. Many marketed drugs have 1, 2, 3-triazole structural fragments in their structures, such as the β-lactamase inhibitor tazobactam (Formula 1). Recently, its application in the discovery of anticancer drug lead compounds has received more and more attention. A number of research groups used click chemistry to connect 1, 2, 3-triazoles and other pharmacophore groups, and synthesized some compounds with excellent anti-tumor activity and MAO-inhibitory activity. For example, the Kamal group introduced 1, 2, 3-triazole (Formula 2) into podophyllotoxin, and synthesized a series of compounds with better antitumor activity than etoposide (HMSampath Kumar. European Journal of Medicinal Chemistry.2011 , 46, 1983-1991); Marvin J. Miller combined 1,2,3-triazole and aromatic amide (Formula 3), and discovered a new class of compounds with excellent antitumor activity (Marvin J. Miller. J. Med . Chem.2010, 53, 3389-3395 ); and the Qing Zhu group used Click to synthesize multiple triazolyl derivatives (Formula 4) and had better anti-MAO-A activity (Qing Zhu. Bioorganic & Medicinal Chemistry Letters.2010 ,20, 6222-6225 ).


另外,氨基二硫代甲酸酯类化合物,由于其广泛的生物活性,如具有抗菌、抗炎、杀虫、抗肿瘤、防辐射以及螯合重金属等作用而引起药物化学领域的广泛关注。利用点击化学将1,2,3-三唑活性片段引入到氨基二硫代甲酸酯结构中,合成新型含三唑基的氨基二硫代甲酸酯化合物,研究新型含三唑基结构的氨基二硫代甲酸酯类化合物抑制LSD1的活性,对进一步研究以LSD1为靶点的新型抗肿瘤药物,开发自主知识产权药物具有重要意义。In addition, carbadithiocarbamate compounds have attracted extensive attention in the field of medicinal chemistry due to their wide range of biological activities, such as antibacterial, anti-inflammatory, insecticidal, anti-tumor, anti-radiation and chelating heavy metals. Use click chemistry to introduce 1,2,3-triazole active fragments into the carbamate structure, synthesize new triazole-containing carbamodithiocarbamate compounds, and study new triazole-containing carbamodithiocarbamate compounds Carbamate compounds inhibit the activity of LSD1, which is of great significance for the further study of new anti-tumor drugs targeting LSD1 and the development of drugs with independent intellectual property rights.
发明内容Contents of the invention
本发明的目的在于提供一类新型含三唑基的氨基二硫代甲酸酯化合物作为制备抑制LSD1活性先导药物的应用。The purpose of the present invention is to provide a new type of triazole group-containing aminodithiocarbamate compound as the application of the preparation of the lead drug for inhibiting LSD1 activity.
本发明所述一类含三唑基的氨基二硫代甲酸酯类化合物具有如下通式:A class of triazolyl-containing aminodithiocarbamate compounds of the present invention has the following general formula:

R1为H,不同位置的单取代C1-C5的烷基、甲氧基、氟、氯、溴、三氟甲基或多取代的氟、多取代的氯、多取代溴或多取代的甲氧基等。R1 is H, monosubstituted C1-C5 alkyl, methoxy, fluorine, chlorine, bromine, trifluoromethyl or multi-substituted fluorine, multi-substituted chlorine, multi-substituted bromine or multi-substituted methyl at different positions Oxygen etc.
通式I中优选:R1为不同位置的单取代C1-C3的烷基、甲氧基、氟、氯、三氟甲基或多取代的氟、多取代的氯、多取代的甲氧基等。Preferred in general formula I: R1 is a monosubstituted C1-C3 alkyl, methoxy, fluorine, chlorine, trifluoromethyl or polysubstituted fluorine, polysubstituted chlorine, polysubstituted methoxy group in different positions wait.
通式I中优选如下化合物:The following compounds are preferred in general formula I:
I-1:R1=o-F的衍生物;I-1: R1 = derivative ofo -F;
I-2:R1=p-F的衍生物;I-2: R1 = derivative ofp -F;
I-3:R1=p-Cl的衍生物;I-3: R1 = derivative ofp -Cl;
I-4:R1=o-Cl的衍生物;I-4: R1 = derivative ofo -Cl;
I-5:R1=p-CH3的衍生物;I-5: R1 = derivative ofp -CH3 ;
I-6:R1=p-OCH3的衍生物;I-6: R1 = derivative ofp -OCH3 ;
I-7: R1=o-CF3的衍生物;I-7: Derivatives of R1 =o -CF3 ;
I-8:R1=m, p-diCl的衍生物;I-8: Derivatives of R1 =m,p -diCl;
I-9:R1=m, p, m-triOCH3的衍生物;I-9: R1 = derivatives ofm, p, m -triOCH3 ;
I-10:R1=o, o-diF的衍生物。I-10 : a derivative of R1 =o, o -diF.
通式II中优选:R1为不同位置的单取代C1-C3的烷基、甲氧基、氟、氯或者多取代的氟、多取代的氯、多取代的甲氧基等。In the general formula II, it is preferred thatR1 is a monosubstituted C1-C3 alkyl, methoxy, fluorine, chlorine or multi-substituted fluorine, multi-substituted chlorine, multi-substituted methoxy group, etc. at different positions.
通式II中优选如下化合物:The following compounds are preferred in the general formula II:
II-1:R1=o-F的衍生物;II-1: R1 = derivative ofo -F;
II-2:R1=p-Cl的衍生物;II-2: R1 = derivative ofp -Cl;
II-3:R1=p-CH3的衍生物;II-3: R1 = derivative ofp -CH3 ;
II-4:R1=p-OCH3的衍生物;II-4: R1 = derivative ofp -OCH3 ;
II-5: R1=o-CF3的衍生物;II-5: Derivatives of R1 =o -CF3 ;
II-6:R1=m, p-diCl的衍生物;II-6: R1 = derivative ofm, p -diCl;
II-7:R1=m, p, m-triOCH3的衍生物;II-7: R1 = derivatives ofm, p, m -triOCH3 ;
II-8:R1=o, o-diF的衍生物。II-8 : R1 =o, o -diF derivative.
本发明所述的一类含三唑基的氨基二硫代甲酸酯类化合物主要通过下列方法制得:A class of triazolyl-containing aminodithiocarbamate compounds of the present invention is mainly prepared by the following methods:
1. 通式(I)的制备方法1. The preparation method of general formula (I)
有机溶剂中,化合物(IV)和通式(V)中的化合物在CuI/有机碱、CuSO4/抗坏血酸钠或Cu/CuSO4条件下发生1,3-环加成反应,所用的有机碱为三乙胺,二异丙基乙基胺;所用的有机溶剂为乙腈,叔丁醇/水,四氢呋喃/水,N, N-二甲基甲酰胺/水,乙醇/水等;反应温度在0-900C之间,通常在室温进行。所得产物经柱层析或者重结晶等提纯得到纯产品。重结晶所用溶剂为乙醇、甲醇、乙腈、丙酮、乙酸乙酯、四氢呋喃、二氯甲烷、氯仿中的一种或其中两种的混合物。 In an organic solvent, a 1,3-cycloaddition reaction occurs between the compound (IV ) and the compound of the general formula (V ) under the conditions of CuI/organic base, CuSO4 /sodium ascorbate or Cu/CuSO4 , and the organic base used is Triethylamine, diisopropylethylamine; The organic solvent used is acetonitrile, tert-butanol/water, tetrahydrofuran/water, N, N-dimethylformamide/water, ethanol/water, etc.; the reaction temperature is at 0 Between -900 C, usually at room temperature. The obtained product is purified by column chromatography or recrystallization to obtain a pure product. The solvent used for recrystallization is one or a mixture of two of ethanol, methanol, acetonitrile, acetone, ethyl acetate, tetrahydrofuran, dichloromethane, and chloroform.
R1为H,不同位置的单取代C1-C5的烷基、甲氧基、氟、氯、三氟甲基或多取代的氟、多取代的氯、多取代的甲氧基等。R1 is H, monosubstituted C1-C5 alkyl, methoxy, fluorine, chlorine, trifluoromethyl or multi-substituted fluorine, multi-substituted chlorine, multi-substituted methoxy, etc. at different positions.
2. 通式(IV)的制备方法:2. The preparation method of general formula (IV):
溶剂中,将商业可得的叔丁氧羰基单保护哌嗪在碱性条件下和二硫化碳、溴丙炔或者氯丙炔发生亲核反应,所用的碱是碳酸钠、碳酸钾、磷酸钠、十一水磷酸钠、磷酸钾、碳酸氢钾、碳酸氢钠、三乙胺等中的一种;所用的溶剂为丙酮、N, N-二甲基甲酰胺、乙腈、乙醇、甲醇、异丙醇、1,2-二氯乙烷、二氯甲烷、氯仿、四氢呋喃、二氧六环,蒸馏水其中之一或其中任意两种或三种的混合物;所得产物经柱层析或者重结晶等提纯得到纯产品。重结晶所用溶剂为乙醇、甲醇、乙腈、丙酮、乙酸乙酯、四氢呋喃、二氯甲烷、氯仿中的一种或其中两种的混合物。 In the solvent, the commercially available tert-butoxycarbonyl monoprotected piperazine reacts nucleophilically with carbon disulfide, propyne bromide or propyne chloride under alkaline conditions, and the base used is sodium carbonate, potassium carbonate, sodium phosphate, undecyl One of sodium phosphate, potassium phosphate, potassium bicarbonate, sodium bicarbonate, triethylamine, etc.; the solvent used is acetone, N, N-dimethylformamide, acetonitrile, ethanol, methanol, isopropanol, 1,2-dichloroethane, dichloromethane, chloroform, tetrahydrofuran, dioxane, distilled water, or a mixture of any two or three of them; the resulting product is purified by column chromatography or recrystallization to obtain pure product. The solvent used for recrystallization is one or a mixture of two of ethanol, methanol, acetonitrile, acetone, ethyl acetate, tetrahydrofuran, dichloromethane, and chloroform.
3. 通式(II)的制备方法:3. The preparation method of general formula (II):
通式(I)所示化合物在有机溶剂、酸性条件下脱去叔丁氧羰基,得通式(II)化合物。所用的酸为三氟乙酸、氯化氢、溴化氢等;所用的有机溶剂为二氯甲烷、1,4-二氧六环、乙酸乙酯、四氢呋喃等;反应温度在-10-600C之间,通常在室温进行。所得产物经柱层析或者重结晶等提纯得到纯产品。 The compound represented by the general formula (I) removes the tert-butoxycarbonyl group in an organic solvent under acidic conditions to obtain the compound of the general formula (II). The acid used is trifluoroacetic acid, hydrogen chloride, hydrogen bromide, etc.; the organic solvent used is dichloromethane, 1,4-dioxane, ethyl acetate, tetrahydrofuran, etc.; the reaction temperature is between -10-60° C , usually at room temperature. The obtained product is purified by column chromatography or recrystallization to obtain a pure product.

R1为H,不同位置的单取代C1-C5的烷基、甲氧基、氟、氯、三氟甲基或多取代的氟、多取代的氯、多取代的甲氧基等。R1 is H, monosubstituted C1-C5 alkyl, methoxy, fluorine, chlorine, trifluoromethyl or multi-substituted fluorine, multi-substituted chlorine, multi-substituted methoxy, etc. at different positions.
与现有的技术相比,本发明首次利用经典的点击化学将1,2,3-三唑活性单元与氨基二硫代甲酸酯结合,简单高效,绿色环保的合成含三唑结构的氨基二硫代甲酸酯类化合物。体外LSD1活性抑制试验表明,本发明所提供含三唑结构的氨基二硫代甲酸酯类化合物对LSD1活性具有明显的抑制作用,其中通式(I)中I-1:R1=o-F; I-3:R1=o-Cl;I-2:R1=p-F; I-5:R1=p-CH3; I-6:R1=p-OCH3的衍生物,通式(II)中II-4:R1=p-OCH3衍生物对LSD1的抑制活性明显优于苯环丙胺,可以将其做为活性成分用于制备组蛋白赖氨酸特异性去甲基化酶1抑制剂药物。Compared with the existing technology, the present invention combines the 1,2,3-triazole active unit with the carbamodithiocarbamate for the first time using the classic click chemistry, which is a simple, efficient, green and environmentally friendly synthesis of the amino group containing the triazole structure. Dithioformate compounds. The in vitro LSD1 activity inhibition test shows that the triazole-containing aminodithiocarbamate compounds provided by the present invention have obvious inhibitory effect on LSD1 activity, whereinI-1 in the general formula (I): R1 =o -F ;I-3:R1 =o -Cl;I-2:R1 =p -F;I-5:R1 =p -CH3 ;I-6: derivatives ofR1 =p -OCH3 ,II-4 in the general formula (II): R1 =p -OCH3 derivatives have significantly better inhibitory activity on LSD1 than phencypromine, and can be used as active ingredients for the preparation of histone lysine-specific demethylation Kase 1 inhibitor drugs.
附图说明Description of drawings
图 1为本发明合成的I-1、I-3、I-5、I-2、I-6、II-4化合物终浓度0.5μM时在胃癌细胞系MGC-803中对LSD1底物H3K9me2、H3K4me2 表达量的调节作用Western Blot定性图;Figure 1 shows the effects of compounds I-1, I-3, I-5, I-2, I-6, and II-4 synthesized by the present invention on LSD1 substrates H3K9me2, H3K9me2, Western Blot qualitative diagram of the regulatory effect of H3K4me2 expression;
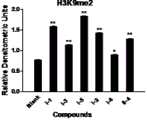
图 2为本发明合成的I-1、I-3、I-5、I-2、I-6、II-4化合物终浓度0.5μM时在胃癌细胞系MGC-803中对LSD1底物H3K9me2表达量的调节作用Western Blot图灰度分析半定量柱形图;Figure 2 shows the expression of the I-1, I-3, I-5, I-2, I-6, and II-4 compounds synthesized by the present invention on the expression of the LSD1 substrate H3K9me2 in the gastric cancer cell line MGC-803 at a final concentration of 0.5 μM Semi-quantitative histogram of quantitative adjustment Western Blot graph grayscale analysis;
图3为本发明合成的I-1、I-3、I-5、I-2、I-6、II-4化合物终浓度0.5μM时在胃癌细胞系MGC-803中对LSD1底物H3K4me2 表达量的调节作用Western Blot图灰度分析半定量柱形图。Figure 3 shows the expression of LSD1 substrate H3K4me2 in the gastric cancer cell line MGC-803 when the final concentration of compounds I-1, I-3, I-5, I-2, I-6 and II-4 synthesized by the present invention is 0.5 μM Quantitative effect Western Blot grayscale analysis semi-quantitative histogram.
具体实施方式Detailed ways
为了对本发明进行更好的说明,特举实施例如下:In order to better illustrate the present invention, special examples are as follows:
通式(V)的制备参考以下文献制得:The preparation of general formula (V) is obtained with reference to the following documents:
(a) Ina.Wilkening.; Giuseppe del Signore.; C.P.R.Hackenberger. Chem. Commun. 2011, 47, 349-351. (b) Mingyu Hu.; Junqi Li.; ShaoQ Yao. Org. Lett, 2008, 10, 5529-5531.。(a) Ina.Wilkening.; Giuseppe del Signore.; CPRHackenberger.Chem. Commun.2011 , 47, 349-351. (b ) Mingyu Hu.; -5531..
实施例 1 中间体(IV)的制备Preparation ofExample 1 Intermediate (IV )
将CS2(10mmol)逐滴加入到叔丁氧羰基单保护哌嗪(10mmol)和Na3PO4(6mmol)的丙酮溶液中,室温搅拌30分钟后加入溴丙炔(11mmol)搅拌反应,TLC跟踪检测。反应结束后,抽滤,滤液减压浓缩,浓缩物用二氯甲烷(3×50mL)和饱和食盐水萃取,合并有机相,无水硫酸钠干燥,减压浓缩,浓缩物重结晶或柱层析分离得化合物IV。收率92%,白色固体。1H NMR (400 MHz, Actone-d6, δ, ppm): 4.28 (br, 2H), 4.14 (d, 2H, J=2.68Hz), 4.00 (br, 2H), 3.58 (br, 4H), 2.78 (t, 1H, J=2.68Hz), 1.46 (s, 9H); HRMS (ESI) calcd for C13H21N2O2S2 [M+H]+:301.1044, found: 301.1046。Add CS2 (10 mmol) dropwise to the acetone solution of tert-butoxycarbonyl mono-protected piperazine (10 mmol) and Na3 PO4 (6 mmol), stir at room temperature for 30 minutes, then add propyne bromide (11 mmol) to stir the reaction, TLC Track detection. After the reaction, filter with suction, concentrate the filtrate under reduced pressure, extract the concentrate with dichloromethane (3×50mL) and saturated brine, combine the organic phases, dry over anhydrous sodium sulfate, concentrate under reduced pressure, and recrystallize the concentrate or column layer CompoundIV was isolated by analysis. Yield 92%, white solid.1 H NMR (400 MHz, Actone-d6 , δ, ppm): 4.28 (br, 2H), 4.14 (d, 2H, J=2.68Hz), 4.00 (br, 2H), 3.58 (br, 4H), 2.78 (t, 1H, J=2.68Hz), 1.46 (s, 9H); HRMS( ESI) calcd for C13 H21 N2 O 2 S2 [M+H]+ :301.1044, found: 301.1046.
实施例 2 通式(I)所示,R1 = o-F的衍生物(I-1)的制备Example 2 Preparation of the derivative (I-1 ) represented by the general formula (I), R1 =o -F
将化合物IV(5mmol)和2-氟苄基叠氮(5mmol)用THF-H2O (30-30mL)溶解,搅拌下加入五水硫酸铜(0.25mmol),抗坏血酸钠(0.5mmol),室温搅拌反应3-4小时,跟踪监测反应。反应结束后,向反应体系中加入H2O(40mL),反应体系用EtOAc(3×50mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-1。收率79.0%,白色固体,熔点:109-110oC。IR( KBr, cm-1) ν :3454, 2979, 1693, 1494, 1478, 1279, 1167, 1012, 986, 932, 791, 757, 695; 1H NMR (400 MHz, CDCl3, δ , ppm): 7.66 (s, 1H), 7.09-7.38 (m, 4H), 5.55 (s, 2H), 4.69 (s, 2H), 3.52 (t, 4H, J=5.20Hz), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3,δ, ppm): 196.42, 161.72, 159.26, 154.41, 144.00, 130.89, 130.81, 130.51, 130.48, 124.82, 124.78, 122.96, 121.98, 121.84, 115.92, 115.71, 80.63, 47.66, 47.61, 53.4, 31.84, 28.34; HRMS(ESI) calcd for C20H27FN5O2S2 [M+H]+: 452.1590, found: 452.1598.。Dissolve compoundIV (5mmol) and 2-fluorobenzyl azide (5mmol) in THF-H2 O (30-30mL), add copper sulfate pentahydrate (0.25mmol) and sodium ascorbate (0.5mmol) under stirring, room temperature The reaction was stirred for 3-4 hours, and the reaction was followed and monitored. After the reaction was completed, H2 O (40 mL) was added to the reaction system, the reaction system was extracted with EtOAc (3×50 mL), the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The concentrate was recrystallized from acetone to obtain productI-1 . Yield: 79.0%, white solid, melting point: 109-110o C. IR( KBr, cm-1 ) ν :3454, 2979, 1693, 1494, 1478, 1279, 1167, 1012, 986, 932, 791, 757, 695;1 H NMR (400 MHz, CDCl3 , δ , ppm) : 7.66 (s, 1H), 7.09-7.38 (m, 4H), 5.55 (s, 2H), 4.69 (s, 2H), 3.52 (t, 4H, J=5.20Hz), 1.47 (s, 9H);13 C NMR (100 MHz, CDCl3 ,δ, ppm): 196.42, 161.72, 159.26, 154.41, 144.00, 130.89, 130.81, 130.51, 130.48, 124.82, 124.78, 122.96, 121.98, 121.84, 115.92, 115.71, 80.63, 47.66 , 47.61, 53.4, 31.84, 28.34; HRMS(ESI) calcd for C20 H27 FN5 O2 S2 [M+H]+ : 452.1590, found: 452.1598..
实施例 3 通式(I)所示,R1 = p-F的衍生物(I-2)的制备Example 3 Preparation of the derivative (I-2 ) represented by the general formula (I), R1 =p -F
将化合物IV(5mmol)和4-氟苄基叠氮(5mmol)用THF-H2O(30-30mL)溶解,搅拌下加入五水硫酸铜(0.25mmol),抗坏血酸钠(0.5mmol),加毕室温搅拌反应1-2小时,跟踪监测反应。反应结束后,向反应体系中加入H2O(30mL),反应体系用EtOAc(3×50mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-2。收率78.7%,白色固体,熔点:105-106oC。IR ( KBr, cm-1) ν : 3447, 2975, 1691, 1460, 1423, 1219, 1167, 1039, 935, 788, 746; 1H NMR (400 MHz, CDCl3,δ, ppm): 7.59 (s, 1H), 7.34-7.87 (m, 3H), 5.55 (s, 2H), 4.72 (s, 2H), 4.26 (br, 2H), 3.92 (br, 2H), 3.56 (t, 4H, J=5.24Hz), 1.47 (s, 9H); 13C NMR (100 MHz, acetone-d6,δ, ppm): 195.7, 154.8, 141.9, 133.7, 133.1, 133.1, 130.2, 129.71, 127.7, 123.6, 78.4, 50.8, 31.8, 27.6; HRMS(ESI) calcd for C20H28N5O3S2 [M+H]+: 450.1634, found: 450.1638.。Dissolve compoundIV (5mmol) and 4-fluorobenzyl azide (5mmol) in THF-H2 O (30-30mL), add copper sulfate pentahydrate (0.25mmol) and sodium ascorbate (0.5mmol) under stirring, add After completion, the reaction was stirred at room temperature for 1-2 hours, and the reaction was tracked and monitored. After the reaction, H2 O (30 mL) was added to the reaction system, the reaction system was extracted with EtOAc (3×50 mL), the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The concentrate was recrystallized from acetone to obtain productI-2 . Yield: 78.7%, white solid, melting point: 105-106o C. IR ( KBr, cm-1 ) ν : 3447, 2975, 1691, 1460, 1423, 1219, 1167, 1039, 935, 788, 746;1 H NMR (400 MHz, CDCl3 ,δ, ppm): 7.59 (s , 1H), 7.34-7.87 (m, 3H), 5.55 (s, 2H), 4.72 (s, 2H), 4.26 (br, 2H), 3.92 (br, 2H), 3.56 (t, 4H, J=5.24 Hz), 1.47 (s, 9H);13 C NMR (100 MHz, acetone-d6 ,δ, ppm): 195.7, 154.8, 141.9, 133.7, 133.1, 133.1, 130.2, 129.71, 127.7, 123.6, 78.4, 50. , 31.8, 27.6; HRMS(ESI) calcd for C20 H28 N5 O3 S2 [M+H]+ : 450.1634, found: 450.1638..
实施例 4 通式(I)所示,R1 = p-Cl的衍生物(I-3)的制备Example 4 Preparation of the derivative (I-3 ) represented by the general formula (I), R1 =p -Cl
将化合物IV(3mmol)和4-氯苄基叠氮(3mmol)用THF-H2O(20-20mL)溶解,搅拌下加入五水硫酸铜(0.15mmol),抗坏血酸钠(0.3mmol),加毕室温搅拌反应2-3小时,跟踪监测反应。反应结束后,向反应体系中加入H2O(30mL),反应体系用EtOAc(3×40mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-3。收率85.5%,白色固体,熔点:177-178oC。IR ( KBr, cm-1) ν :3446, 3130, 2979, 2914, 1678, 1491, 1422, 1224, 1161, 1024, 994, 932, 777, 543, 499; 1H NMR (400 MHz, CDCl3,δ, ppm): 7.59 (s, 1H), 7.35 (d, 2H, J = 8.4Hz), 7.20 (d, 2H, J = 8.4Hz), 5.45 (s, 2H), 4.68 (s, 2H), 4.29 (br, 2H), 3.90 (br, 2H), 3.54 (t, 4H, J = 5.2Hz), 1.47(s, 9H);13C NMR (100 MHz, CDCl3, δ, ppm): 196.4, 154.4, 144.4, 134.8, 134.1, 129.4, 129.3, 122.8, 80.7, 53.4, 31.7, 28.4; HRMS(ESI) calcd for C20H27ClN5O2S2 [M+H]+: 468.1295, found: 468.1291.。 Dissolve compoundIV (3mmol) and 4-chlorobenzyl azide (3mmol) in THF-H2 O (20-20mL), add copper sulfate pentahydrate (0.15mmol) and sodium ascorbate (0.3mmol) under stirring, add After completion, the reaction was stirred at room temperature for 2-3 hours, and the reaction was tracked and monitored. After the reaction, H2 O (30 mL) was added to the reaction system, the reaction system was extracted with EtOAc (3×40 mL), the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The concentrate was recrystallized from acetone to obtain productI-3 . Yield: 85.5%, white solid, melting point: 177-178o C. IR ( KBr, cm-1 ) ν :3446, 3130, 2979, 2914, 1678, 1491, 1422, 1224, 1161, 1024, 994, 932, 777, 543, 499;1 H NMR (400 MHz, CDCl3 , δ, ppm): 7.59 (s, 1H), 7.35 (d, 2H,J = 8.4Hz), 7.20 (d, 2H,J = 8.4Hz), 5.45 (s, 2H), 4.68 (s, 2H), 4.29 (br, 2H), 3.90 (br, 2H), 3.54 (t, 4H,J = 5.2Hz), 1.47(s, 9H);13 C NMR (100 MHz, CDCl3 , δ, ppm): 196.4, 154.4, 144.4, 134.8, 134.1, 129.4, 129.3, 122.8, 80.7, 53.4, 31.7, 28.4; HRMS(ESI) calcd for C20 H27 ClN5 O2 S2 [M+H]+ : 468.1268.112 ..
实施例 5 通式(I)所示,R1 = o-Cl的衍生物(I-4)的制备Example 5 Preparation of the derivative (I-4 ) represented by the general formula (I), R1 =o -Cl
将化合物IV(3mmol)和4-氯苄基叠氮(3mmol)用THF-H2O(20-20mL)溶解,搅拌下加入五水硫酸铜(0.15mmol),抗坏血酸钠(0.3mmol),加毕室温搅拌反应2-3小时,跟踪监测反应。反应结束后,向反应体系中加入H2O(30mL),反应体系用EtOAc(3×40mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-4。收率85.5%,白色固体,熔点:177-178oC。IR ( KBr, cm-1) ν :3446, 3130, 2979, 2914, 1678, 1491, 1422, 1224, 1161, 1024, 994, 932, 777, 543, 499; 1H NMR (400 MHz, CDCl3,δ, ppm): 7.59 (s, 1H), 7.35 (d, 2H, J = 8.4Hz), 7.20 (d, 2H, J = 8.4Hz), 5.45 (s, 2H), 4.68 (s, 2H), 4.29 (br, 2H), 3.90 (br, 2H), 3.54 (t, 4H, J = 5.2 Hz), 1.47(s, 9H); 13C NMR (100 MHz, CDCl3, δ, ppm): 196.4, 154.4, 144.4, 134.8, 134.1, 129.4, 129.3, 122.8, 80.7, 53.4, 31.7, 28.4; HRMS(ESI) calcd for C20H27ClN5O2S2 [M+H]+: 468.1295, found: 468.1291.。 Dissolve compoundIV (3mmol) and 4-chlorobenzyl azide (3mmol) in THF-H2 O (20-20mL), add copper sulfate pentahydrate (0.15mmol) and sodium ascorbate (0.3mmol) under stirring, add After completion, the reaction was stirred at room temperature for 2-3 hours, and the reaction was tracked and monitored. After the reaction, H2 O (30 mL) was added to the reaction system, the reaction system was extracted with EtOAc (3×40 mL), the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The concentrate was recrystallized from acetone to obtain productI-4 . Yield: 85.5%, white solid, melting point: 177-178o C. IR ( KBr, cm-1 ) ν :3446, 3130, 2979, 2914, 1678, 1491, 1422, 1224, 1161, 1024, 994, 932, 777, 543, 499;1 H NMR (400 MHz, CDCl3 , δ, ppm): 7.59 (s, 1H), 7.35 (d, 2H,J = 8.4Hz), 7.20 (d, 2H,J = 8.4Hz), 5.45 (s, 2H), 4.68 (s, 2H), 4.29 (br, 2H), 3.90 (br, 2H), 3.54 (t, 4H,J = 5.2 Hz), 1.47(s, 9H);13 C NMR (100 MHz, CDCl3 , δ, ppm): 196.4, 154.4, 144.4, 134.8, 134.1, 129.4, 129.3, 122.8, 80.7, 53.4, 31.7, 28.4; HRMS(ESI) calcd for C20 H27 ClN5 O2 S2 [M+H]+ : 468.1268.112 ..
实施例 6 通式(I)所示,R1 = p-CH3的衍生物(I-5)的制备Example 6 Preparation of the derivative (I-5 ) represented by the general formula (I), R1 =p -CH3
将化合物IV(5mmol)和4-甲基苄基叠氮(5mmol)用乙腈(40mL)溶解,搅拌下加入碘化亚铜(0.5mmol),三乙胺(10mmol),加毕室温搅拌反应4-6小时,跟踪监测反应。反应结束后,将反应体系真空浓缩,浓缩物用EtOAc(50mL)溶解,用稀盐酸,饱和碳酸氢钠水溶液,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-5。收率79.9%,白色固体,熔点:182-183oC。IR ( KBr, cm-1) ν:3454, 2975, 1682, 1457, 1422, 1224, 1078, 994, 933, 867, 772, 524; 1H NMR (400 MHz, CDCl3,δ, ppm): 7.55 (s, 1H), 7.16(s, 4H), 5.43 (s, 2H), 4.67 (s, 2H), 4.29 (br, 2H), 3.91 (br, 2H), 3.51 (t, 4H, J=5.16Hz), 2.35 (s, 3H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3,δ, ppm): 196.5, 154.4, 138.7, 131.6, 129.8, 128.1, 80.6, 54.01, 31.9, 28.4, 21.2; HRMS(ESI) calcd for C21H30N5O2S2 [M+H]+: 448.1841, found: 448.1840.。Dissolve compoundIV (5mmol) and 4-methylbenzyl azide (5mmol) in acetonitrile (40mL), add cuprous iodide (0.5mmol) and triethylamine (10mmol) under stirring, and stir at room temperature to complete reaction 4 -6 hours, follow up and monitor the reaction. After the reaction, the reaction system was concentrated in vacuo, the concentrate was dissolved in EtOAc (50mL), washed with dilute hydrochloric acid, saturated aqueous sodium bicarbonate, saturated brine, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the concentrate was used Acetone was recrystallized to obtain productI-5 . Yield: 79.9%, white solid, melting point: 182-183o C. IR ( KBr, cm-1 ) ν: 3454, 2975, 1682, 1457, 1422, 1224, 1078, 994, 933, 867, 772, 524;1 H NMR (400 MHz, CDCl3 ,δ, ppm): 7.55 (s, 1H), 7.16(s, 4H), 5.43 (s, 2H), 4.67 (s, 2H), 4.29 (br, 2H), 3.91 (br, 2H), 3.51 (t, 4H, J=5.16 Hz), 2.35 (s, 3H), 1.47 (s, 9H);13 C NMR (100 MHz, CDCl3 ,δ, ppm): 196.5, 154.4, 138.7, 131.6, 129.8, 128.1, 80.6, 54.01, 31.9, 28.4, 21.2; HRMS(ESI) calcd for C21 H30 N5 O2 S2 [M+H]+ : 448.1841, found: 448.1840..
实施例 7 通式(I)所示,R1 = p-OCH3的衍生物(I-6)的制备Example 7 Preparation of derivative (I-6 ) represented by general formula (I), R1 =p -OCH3
将化合物IV(3mmol)和4-甲氧基苄基叠氮(3mmol)用乙腈(30mL)溶解,搅拌下加入碘化亚铜(0.3mmol),三乙胺(6mmol),加毕室温搅拌反应2-3小时,跟踪监测反应。反应结束后,将反应体系真空浓缩,浓缩物用EtOAc(50mL)溶解,用稀盐酸,饱和碳酸氢钠水溶液,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用乙酸乙酯重结晶,得产物I-6。收率85.8%,白色固体,熔点:129-130oC。IR ( KBr, cm-1) ν :3502, 3125, 2975,1686, 1542, 1453, 1281, 1173, 1016, 982, 932, 775, 698 ; 1H NMR (400 MHz, CDCl3,δ, ppm): 7.54 (s, 2H), 7.21 (d, 2H, J=8.0), 6.88 (d, 2H, J=8.0), 5.41 (s, 2H), 4.67 (s, 2H), 4.29 (br, 2H), 3.90 (br, 2H),3.80 (s, 3H), 3.51 (t, 4H, J=5.2Hz), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, δ, ppm): 196.4, 164.0, 161.6, 154.4, 130.5, 130.5, 130.0, 129.9, 116.2, 116.0, 80.7, 53.4, 31.8, 28.3; HRMS (ESI) calcd for C21H30N5O3S2 [M+H]+:464.1790, found:464.1794.。Dissolve compoundIV (3mmol) and 4-methoxybenzyl azide (3mmol) in acetonitrile (30mL), add cuprous iodide (0.3mmol) and triethylamine (6mmol) under stirring, and stir the reaction at room temperature 2-3 hours, follow up and monitor the reaction. After the reaction, the reaction system was concentrated in vacuo, the concentrate was dissolved in EtOAc (50mL), washed with dilute hydrochloric acid, saturated aqueous sodium bicarbonate solution, saturated brine, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the concentrate was used Ethyl acetate was recrystallized to obtain productI-6 . Yield: 85.8%, white solid, melting point: 129-130o C. IR ( KBr, cm-1 ) ν :3502, 3125, 2975,1686, 1542, 1453, 1281, 1173, 1016, 982, 932, 775, 698 ;1 H NMR (400 MHz, CDCl3 ,δ, ppm) : 7.54 (s, 2H), 7.21 (d, 2H, J=8.0), 6.88 (d, 2H, J=8.0), 5.41 (s, 2H), 4.67 (s, 2H), 4.29 (br, 2H) , 3.90 (br, 2H), 3.80 (s, 3H), 3.51 (t, 4H, J=5.2Hz), 1.47 (s, 9H);13 C NMR (100 MHz, CDCl3 , δ, ppm): 196.4 , 164.0, 161.6, 154.4, 130.5, 130.5, 130.0, 129.9, 116.2, 116.0, 80.7,53.4 , 31.8, 28.3; HRMS (ESI) calcd forC21H30N5O3S2 [M+H]+: 464.1790, found: 464.1794..
实施例 8 通式(I)所示,R1 = o-CF3的衍生物(I-7)的制备Example 8 Preparation of derivative (I-7 ) represented by general formula (I), R1 =o -CF3
将化合物IV(5mmol)和2-三氟甲基苄基叠氮(5mmol)用乙腈(40mL)溶解,搅拌下加入碘化亚铜(0.5mmol),二异丙基乙基胺(7.5mmol),加毕室温搅拌反应2-3小时,跟踪监测反应。反应结束后,将反应体系真空浓缩,浓缩物用EtOAc(50mL)溶解,用稀盐酸,饱和碳酸氢钠水溶液,饱和食盐水洗涤,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-7。收率81.5%,白色固体,熔点:138-139oC。IR( KBr, cm-1) ν:3454, 2979, 1693, 1494, 1478, 1279, 1167, 1012, 986, 932, 791, 757, 695; 1H NMR (400 MHz, CDCl3, δ , ppm): 8.15 (s, 1H), 7.04-7.39 (m, 4H), 5.54 (s, 2H), 4.83 (s, 2H), 4.30 (br, 2H), 3.91(br, 2H) 3.52 (t, 4H, J=5.24Hz), 1.47(s, 9H); HRMS (ESI) calcd for C21H27F3N5O2S2 [M+H]+: 502.1558, found:502.1559.。Dissolve compoundIV (5mmol) and 2-trifluoromethylbenzyl azide (5mmol) in acetonitrile (40mL), add cuprous iodide (0.5mmol) and diisopropylethylamine (7.5mmol) under stirring After the addition, the reaction was stirred at room temperature for 2-3 hours, and the reaction was tracked and monitored. After the reaction, the reaction system was concentrated in vacuo, and the concentrate was dissolved in EtOAc (50mL), washed with dilute hydrochloric acid, saturated aqueous sodium bicarbonate, saturated brine, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was reduced to Concentrate under reduced pressure, and the concentrate is recrystallized from acetone to obtain productI-7 . Yield: 81.5%, white solid, melting point: 138-139o C. IR( KBr, cm-1 ) ν:3454, 2979, 1693, 1494, 1478, 1279, 1167, 1012, 986, 932, 791, 757, 695;1 H NMR (400 MHz, CDCl3 , δ , ppm) : 8.15 (s, 1H), 7.04-7.39 (m, 4H), 5.54 (s, 2H), 4.83 (s, 2H), 4.30 (br, 2H), 3.91(br, 2H) 3.52 (t, 4H, J=5.24Hz), 1.47(s, 9H); HRMS (ESI) calcd for C21 H27 F3 N5 O2 S2 [M+H]+ : 502.1558, found: 502.1559..
实施例9通式(I)所示R1 = m, p-diCl的衍生物(I-8)的制备Example 9 Preparation of R1 =m, p -diCl derivative (I-8 ) represented by general formula (I)
将化合物IV(4mmol)和3,4-二氯苄基叠氮(4mmol)用叔丁醇-水(30-30mL)溶解,搅拌下加入五水硫酸铜(0.8mmol),铜粉(4mmol),加毕室温搅拌反应3-5小时,跟踪监测反应。反应结束后,向反应体系中加入H2O(30mL),反应体系用EtOAc(3×40mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-8。收率90.2%,白色固体,熔点:153-154oC。IR ( KBr, cm-1) ν:3129, 2984, 1693, 1470, 1347, 1159, 1131, 990, 963, 798, 740, 698, 542 ; 1H NMR (400 MHz, CDCl3, δ, ppm): 7.64 (s, 1H), 7.43 (d, 1H, J = 8.3 Hz), 7.35 (d, 1H, J = 2.0 Hz), 7.08 (dd, 1H, J1=2.0 Hz, J2=8.2 Hz), 5.44 (s, 2H), 4.70 (s, 2H), 4.30 (br, 2H), 3.91 (br, 2H), 3.52 (t, 4H, J = 5.1Hz), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, δ, ppm): 196.3, 154.4, 144.6, 134.8, 133.3, 133.1, 131.1, 129.9, 127.2, 122.9, 80.7, 52.8, 31.7, 28.3; HRMS (ESI) calcd for C20H26Cl2N5O2S2 [M+H]+: 502.0905, found: 502.0900.。Dissolve compoundIV (4mmol) and 3,4-dichlorobenzyl azide (4mmol) in tert-butanol-water (30-30mL), add copper sulfate pentahydrate (0.8mmol) and copper powder (4mmol) under stirring After the addition, the reaction was stirred at room temperature for 3-5 hours, and the reaction was tracked and monitored. After the reaction, H2 O (30 mL) was added to the reaction system, the reaction system was extracted with EtOAc (3×40 mL), the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The concentrate was recrystallized from acetone to obtain productI-8 . Yield: 90.2%, white solid, melting point: 153-154o C. IR ( KBr, cm-1 ) ν:3129, 2984, 1693, 1470, 1347, 1159, 1131, 990, 963, 798, 740, 698, 542 ;1 H NMR (400 MHz, CDCl3 , δ, ppm) : 7.64 (s, 1H), 7.43 (d, 1H,J = 8.3 Hz), 7.35 (d, 1H,J = 2.0 Hz), 7.08 (dd, 1H,J1 =2.0 Hz,J2 =8.2 Hz) , 5.44 (s, 2H), 4.70 (s, 2H), 4.30 (br, 2H), 3.91 (br, 2H), 3.52 (t, 4H,J = 5.1Hz), 1.47 (s, 9H);13 C NMR (100 MHz, CDCl3 , δ, ppm): 196.3, 154.4, 144.6, 134.8, 133.3, 133.1, 131.1, 129.9, 127.2, 122.9, 80.7, 52.8, 31.7, 28.3; HRMS (ESI C) for H20 calc26 Cl2 N5 O2 S2 [M+H]+ : 502.0905, found: 502.0900.
实施例10通式(I)所示R1 = m, p, m-triOCH3的衍生物(I-9)的制备Example 10 Preparation of R1 =m, p, m -triOCH3 derivative (I-9 ) represented by general formula (I)
将化合物IV(5mmol)和3,4,5-三甲氧基苄基叠氮(5mmol)用叔丁醇-水(40-40mL)溶解,搅拌下加入五水硫酸铜(1mmol),铜粉(5mmol),加毕室温搅拌反应3-5小时,跟踪监测反应。反应结束后,向反应体系中加入H2O(30mL),反应体系用EtOAc(3×40mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-9。收率83.4%,白色固体, 熔点:134-135oC。IR( KBr, cm-1) ν:3453, 2979, 1693, 1494, 1478, 1279, 1167, 1012, 986, 932, 791, 757, 695 ; 1H NMR (400 MHz, CDCl3,δ, ppm): 7.63 (s, 1H), 6.48 (s, 2H), 5.41 (s, 2H), 4.70 (s, 2H), 4.30 (br, 2H), 3.91 (br, 2H), 3.85 (s, 3H), 3.84 (s, 6H), 3.55 (t, 4H, J=5.24Hz), 1.48 (s, 9H);HRMS (ESI) calcd for C23H34N5O2S2 [M+H]+: 524.2001, found: 524.2005.。Dissolve compoundIV (5mmol) and 3,4,5-trimethoxybenzyl azide (5mmol) in tert-butanol-water (40-40mL), add copper sulfate pentahydrate (1mmol) under stirring, copper powder ( 5mmol), after the addition was completed, the reaction was stirred at room temperature for 3-5 hours, and the reaction was tracked and monitored. After the reaction, H2 O (30 mL) was added to the reaction system, the reaction system was extracted with EtOAc (3×40 mL), the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The concentrate was recrystallized from acetone to obtain productI-9 . Yield: 83.4%, white solid, melting point: 134-135o C. IR( KBr, cm-1 ) ν:3453, 2979, 1693, 1494, 1478, 1279, 1167, 1012, 986, 932, 791, 757, 695 ;1 H NMR (400 MHz, CDCl3 ,δ, ppm) : 7.63 (s, 1H), 6.48 (s, 2H), 5.41 (s, 2H), 4.70 (s, 2H), 4.30 (br, 2H), 3.91 (br, 2H), 3.85 (s, 3H), 3.84 (s, 6H), 3.55 (t, 4H, J=5.24Hz), 1.48 (s, 9H); HRMS (ESI) calcd for C23 H34 N 5O2 S2 [M+H]+ : 524.2001 , found: 524.2005..
实施例 11 通式(I)所示, R1 =o, o-diF的衍生物(I-10)的制备Example 11 Preparation of the derivative (I-10 ) represented by the general formula (I), R1 =o, o -diF
将化合物IV(5mmol)和2,6-二氟苄基叠氮(5mmol)用THF-H2O(30-30mL)溶解,搅拌下加入五水硫酸铜(1mmol),铜粉(5mmol),加毕室温搅拌反应2-3小时,跟踪监测反应。反应结束后,向反应体系中加入H2O(30mL),反应体系用EtOAc(3×40mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,浓缩物用丙酮重结晶,得产物I-10。收率78.4%,白色固体,熔点:141-142oC。1H NMR (400 MHz, CDCl3, δ, ppm): 7.71 (s, 1H), 7.32-7.41 (m, 1H), 6.94-6.99 (m, 2H), 5.58 (s, 2H), 4.67 (s, 2H), 4.30 (br, 2H), 3.90 (br, 2H), 3.53 (s, 4H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, δ, ppm): 196.43, 162.66, 162.59, 160.16, 160.10, 154.41, 131.51, 131.40, 131.30, 111.94, 111.88, 111.75, 111.69, 110.95, 110.76, 110.57, 80.62, 41.39, 31.85, 28.35; HRMS (ESI) calcd for C20H26F2N5O2S2 [M+H]+:470.1496, found:470.1498.。Dissolve compoundIV (5mmol) and 2,6-difluorobenzyl azide (5mmol) in THF-H2 O (30-30mL), add copper sulfate pentahydrate (1mmol) and copper powder (5mmol) under stirring, After the addition was completed, the reaction was stirred at room temperature for 2-3 hours, and the reaction was tracked and monitored. After the reaction, H2 O (30 mL) was added to the reaction system, the reaction system was extracted with EtOAc (3×40 mL), the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The concentrate was recrystallized from acetone to obtain productI-10 . Yield: 78.4%, white solid, melting point: 141-142o C.1 H NMR (400 MHz, CDCl3 , δ, ppm): 7.71 (s, 1H), 7.32-7.41 (m, 1H), 6.94-6.99 (m, 2H), 5.58 (s, 2H), 4.67 (s , 2H), 4.30 (br, 2H), 3.90 (br, 2H), 3.53 (s, 4H), 1.47 (s, 9H);13 C NMR (100 MHz, CDCl3 , δ, ppm): 196.43, 162.66 , 162.59, 160.16, 160.10, 154.41, 131.51, 131.40, 131.30, 111.94, 111.88, 111.69, 110.95, 110.57, 80.62,31.85 ,28.35 ;5 O2 S2 [M+H]+ :470.1496, found: 470.1498..
实施例 12 通式(II)所示, R1 = o-F的衍生物(II-1)的制备 Example 12 Preparation of the derivative (II-1 ) represented by the general formula (II), R1 =o -F
将化合物I-1(2mmol)用二氯甲烷(20mL)溶解,冰浴下加入CF3COOH (40mmol),加毕改为室温搅拌反应,跟踪监测反应。反应结束后,将反应体系减压浓缩,浓缩物用二氯甲烷(30mL)溶解,分别用饱和碳酸氢钠,水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得产物II-1。收率96.7%,淡黄色固体,熔点:93-94oC。IR ( KBr, cm-1) ν :3138, 2910, 1693, 1474, 1425, 1219, 1132, 1031, 996, 933, 785, 763, 731, 694; 1H NMR (400 MHz, CDCl3, δ, ppm): 7.67 (s, 1H), 7.09-7.34 (m, 4H), 5.55 (s, 2H), 4.69 (s, 2H), 4.30 (br, 2H), 3.93 (br, 2H), 2.94 (t, 4H, J = 4.8 Hz); HRMS (ESI) calcd for C15H19FN5S2 [M+H]+:352.1066, found: 352.1064.。CompoundI-1 (2mmol) was dissolved in dichloromethane (20mL), and CF3 COOH (40mmol) was added under ice-cooling. After the addition was completed, the reaction was stirred at room temperature, and the reaction was tracked and monitored. After the reaction, the reaction system was concentrated under reduced pressure, and the concentrate was dissolved in dichloromethane (30 mL), washed with saturated sodium bicarbonate, water, and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the productII-1 . Yield: 96.7%, pale yellow solid, melting point: 93-94o C. IR ( KBr, cm-1 ) ν :3138, 2910, 1693, 1474, 1425, 1219, 1132, 1031, 996, 933, 785, 763, 731, 694;1 H NMR (400 MHz, CDCl3 , δ, ppm): 7.67 (s, 1H), 7.09-7.34 (m, 4H), 5.55 (s, 2H), 4.69 (s, 2H), 4.30 (br, 2H), 3.93 (br, 2H), 2.94 (t , 4H,J = 4.8 Hz); HRMS (ESI) calcd for C15 H19 FN5 S2 [M+H]+ :352.1066, found: 352.1064..
实施例 13 通式(II)所示, R1 = p-Cl的衍生物(II-2)的制备Example 13 Preparation of the derivative (II-2 ) represented by the general formula (II), R1 =p -Cl
将化合物I-3(2mmol)用二氯甲烷(20mL)溶解,冰浴下加入CF3COOH (40mmol),加毕改为室温搅拌反应,跟踪监测反应。反应结束后,将反应体系减压浓缩,浓缩物用二氯甲烷(30mL)溶解,分别用饱和碳酸氢钠,水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得产物II-2。收率97.9%,淡黄色固体熔点:138-139oC。IR ( KBr, cm-1) ν:3138, 2910, 1693, 1474, 1425, 1219, 1031, 996, 933, 785, 763, 694; 1H NMR (400 MHz, CDCl3, δ, ppm): 7.60 (s, 1H), 7.35 (d, 2H, J = 8.1 Hz ), 7.20 (d, 2H, J = 8.1 Hz), 5.45 (s, 2H), 4.69 (s, 2H), 3.90 (br, 2H), 2.94 (s, 4H); 13C NMR (100 MHz, CDCl3, δ, ppm):196.03, 164.07, 161.60, 144.33, 130.47, 130.44, 129.97, 129.88, 122.74, 116.21, 116.00, 53.41, 45.15, 31.7; HRMS (ESI) calcd for C15H19FN5S2 [M+H]+:352.1066, found: 352.1063.。CompoundI-3 (2mmol) was dissolved in dichloromethane (20mL), and CF3 COOH (40mmol) was added under ice-cooling. After the addition was completed, the reaction was stirred at room temperature, and the reaction was tracked and monitored. After the reaction, the reaction system was concentrated under reduced pressure, and the concentrate was dissolved in dichloromethane (30 mL), washed with saturated sodium bicarbonate, water, and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the productII-2 . Yield 97.9%, light yellow solid melting point: 138-139o C. IR ( KBr, cm-1 ) ν: 3138, 2910, 1693, 1474, 1425, 1219, 1031, 996, 933, 785, 763, 694;1 H NMR (400 MHz, CDCl3 , δ, ppm): 7.60 (s, 1H), 7.35 (d, 2H,J = 8.1 Hz ), 7.20 (d, 2H,J = 8.1 Hz), 5.45 (s, 2H), 4.69 (s, 2H), 3.90 (br, 2H) , 2.94 (s, 4H);13 C NMR (100 MHz, CDCl3 , δ, ppm): 196.03, 164.07, 161.60, 144.33, 130.47, 130.44, 129.97, 129.88, 122.74, 5, 116.21, 5, 3.0. ; HRMS (ESI) calcd for C15 H19 FN5 S2 [M+H]+ :352.1066, found: 352.1063.
实施例 14 通式(II)所示, R1 = p-CH3的衍生物(II-3)的制备Example 14 Preparation of derivative (II-3 ) represented by general formula (II), R1 =p -CH3
将化合物I-5(2.5mmol)用二氯甲烷(20mL)溶解,冰浴下加入CF3COOH(50mmol),加毕改为室温搅拌反应,跟踪监测反应。反应结束后,将反应体系减压浓缩,浓缩物用二氯甲烷(30mL)溶解,分别用饱和碳酸氢钠,水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得产物II-3。收率94.0%,淡黄色固体,熔点:74-75oC。IR ( KBr, cm-1) ν:3135, 2910, 1697, 1474, 1425, 1219, 1031, 996, 933, 785, 763, 731, 695; 1H NMR (400 MHz, CDCl3, δ, ppm): 7.56 (s, 1H), 7.16-7.33 (m, 4H), 5.43 (s, 2H), 4.67 (s, 2H), 4.31 (br, 2H), 3.92 (br, 2H), 2.96 (br, 4H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3, δ, ppm):195.82, 138.61, 131.59, 129.74, 128.08, 122.72, 53.96, 31.80, 29.68, 21.17; HRMS (ESI) calcd for C16H22N5S2 [M+H]+:348.1317, found: 348.1319.。CompoundI-5 (2.5mmol) was dissolved in dichloromethane (20mL), and CF3 COOH (50mmol) was added under ice-cooling. After the addition was completed, the reaction was stirred at room temperature, and the reaction was tracked and monitored. After the reaction, the reaction system was concentrated under reduced pressure, and the concentrate was dissolved in dichloromethane (30 mL), washed with saturated sodium bicarbonate, water, and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the productII-3 . Yield 94.0%, light yellow solid, melting point: 74-75o C. IR ( KBr, cm-1 ) ν:3135, 2910, 1697, 1474, 1425, 1219, 1031, 996, 933, 785, 763, 731, 695;1 H NMR (400 MHz, CDCl3 , δ, ppm) : 7.56 (s, 1H), 7.16-7.33 (m, 4H), 5.43 (s, 2H), 4.67 (s, 2H), 4.31 (br, 2H), 3.92 (br, 2H), 2.96 (br, 4H ), 2.35 (s, 3H);13 C NMR (100 MHz, CDCl3 , δ, ppm):195.82, 138.61, 131.59, 129.74, 128.08, 122.72, 53.96, 31.80, 29.68, 21.17; HRMS (ESI) for cal C16 H22 N5 S2 [M+H]+ :348.1317, found: 348.1319.
实施例 15 通式(II)所示, R1=p-OCH3的衍生物(II-4)的制备 Example 15 Preparation of derivative (II-4 ) represented by general formula (II), R1 =p -OCH3
将化合物I-6(4mmol)冰浴下用氯化氢的1,4-二氧六环溶液(4mol/L, 30mL)溶解,加毕保持冰浴搅拌反应,跟踪监测反应。反应结束后,将反应体系减压浓缩,浓缩物用二氯甲烷(30mL)溶解,分别用饱和碳酸氢钠,水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得产物II-4。收率98.7%, 淡黄色固体,熔点:95-96oC。IR ( KBr, cm-1) ν :3138, 2910, 1693, 1474, 1425, 1383, 1219, 1031, 996, 933, 785, 763, 694; 1H NMR (400 MHz, CDCl3, δ, ppm): 7.55 (s, 1H), 7.24 (d, 2H, J = 8.7 Hz), 6.91 (d, 2H, J = 8.7 Hz), 5.42 (s, 2H), 4.68 (s, 2H), 4.31 (br, 2H), 3.95 (br, 2H), 3.80 (s, 3H), 2.96 (t, 4H, J = 4.6 Hz); 13C NMR (100 MHz, CDCl3, δ, ppm):195.75, 159.89, 144.12, 129.61, 126.61, 122.53, 114.45, 55.34, 53.69, 45.63, 31.79; HRMS (ESI) calcd for C16H22N5OS2 [M+H]+:364.1266, found: 364.1263。Dissolve compoundI-6 (4 mmol) in 1,4-dioxane solution of hydrogen chloride (4 mol/L, 30 mL) under ice bath, keep stirring in ice bath for reaction after addition, and track and monitor the reaction. After the reaction, the reaction system was concentrated under reduced pressure, and the concentrate was dissolved in dichloromethane (30 mL), washed with saturated sodium bicarbonate, water, and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the productII-4 . Yield: 98.7%, light yellow solid, melting point: 95-96o C. IR ( KBr, cm-1 ) ν :3138, 2910, 1693, 1474, 1425, 1383, 1219, 1031, 996, 933, 785, 763, 694;1 H NMR (400 MHz, CDCl3 , δ, ppm) : 7.55 (s, 1H), 7.24 (d, 2H,J = 8.7 Hz), 6.91 (d, 2H,J = 8.7 Hz), 5.42 (s, 2H), 4.68 (s, 2H), 4.31 (br, 2H), 3.95 (br, 2H), 3.80 (s, 3H), 2.96 (t, 4H,J = 4.6 Hz);13 C NMR (100 MHz, CDCl3 , δ, ppm): 195.75, 159.89, 144.12, 129.61, 126.61, 122.53, 114.45, 55.34, 53.69, 45.63, 31.79; HRMS (ESI) calcd for C16 H22 N5 OS2 [M+H]+ :364.1266, found: 364.1263.
实施例 16通式(II)所示,R1=o-CF3的衍生物(II-5)的制备Example 16 Preparation of the derivative (II-5 ) represented by the general formula (II) with R1 =o -CF3
将化合物I-7(2.00g, 4mmol)冰浴下用氯化氢的1,4-二氧六环溶液(4mol/L, 30mL)溶解,加毕保持冰浴搅拌反应,跟踪监测反应。反应结束后,将反应体系减压浓缩,浓缩物用二氯甲烷(30mL)溶解,分别用饱和碳酸氢钠,水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得产物II-7 1.52g,收率94.5%,淡黄色固体,熔点:151-152oC。1H NMR (400 MHz, CDCl3, δ , ppm): 7.66 (s, 1H), 7.65 (d, 2H, J = 8.2 Hz), 7.36 (d, 2H, J = 8.2 Hz), 5.57 (s, 2H), 4.71 (s, 2H), 4.33 (br, 2H), 3.91(br, 2H), 2.98(s, 4H,); HRMS (ESI) calcd for C16H19F3N5S2 [M+H]+: 402.1034, found: 402.1035.。CompoundI-7 (2.00g, 4mmol) was dissolved in 1,4-dioxane solution of hydrogen chloride (4mol/L, 30mL) under ice bath, and the reaction was kept stirring in ice bath after addition, and the reaction was tracked and monitored. After the reaction, the reaction system was concentrated under reduced pressure, and the concentrate was dissolved in dichloromethane (30 mL), washed with saturated sodium bicarbonate, water, and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the productII-7 1.52g, yield 94.5%, light yellow solid, melting point: 151-152o C.1 H NMR (400 MHz, CDCl3 , δ , ppm): 7.66 (s, 1H), 7.65 (d, 2H,J = 8.2 Hz), 7.36 (d, 2H,J = 8.2 Hz), 5.57 (s, 2H), 4.71 (s, 2H), 4.33 (br, 2H), 3.91(br, 2H), 2.98(s, 4H,); HRMS (ESI) calcd for C16 H19 F3 N5 S2 [M +H]+ : 402.1034, found: 402.1035..
实施例 17通式(II)所示,R1=m, p-diCl的衍生物(II-6)的制备Example 17 Preparation of the derivative (II-6 ) represented by the general formula (II), R1 =m, p -diCl
将化合物I-8(5mmol)冰浴下用氯化氢的1,4-二氧六环溶液(4mol/L, 40mL)溶解,加毕保持冰浴搅拌反应,跟踪监测反应。反应结束后,将反应体系减压浓缩,浓缩物用二氯甲烷(30mL)溶解,分别用饱和碳酸氢钠,水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得产物II-5。收率95.5%,淡黄色固体,熔点:122-123oC。IR ( KBr, cm-1) ν:3138, 2910, 1693, 1474, 1425, 1219, 1163, 1031, 996, 933, 785, 763, 694; 1H NMR (400 MHz, CDCl3, δ, ppm): 7.65 (s, 1H), 7.45 (d, 1H, J = 8.3 Hz), 7.35(d, 1H, J = 1.9 Hz), 7.10 (dd, 1H, J1 = 2.0 Hz, J2 = 8.3 Hz), 5.44 (s, 2H), 4.70 (s, 2H), 4.31 (br, 2H), 3.92 (br, 2H), 2.95 (s, 4H); 13C NMR (100 MHz, CDCl3, δ, ppm):195.62, 144.87, 134.79, 133.27, 133.07, 131.09, 129.86, 127.17, 122.92, 52.81, 45.63, 31.56; HRMS (ESI) calcd for C15H18Cl2N5S2 [M+H]+:402.0381, found: 402.0369.。Dissolve compoundI-8 (5 mmol) in 1,4-dioxane solution of hydrogen chloride (4 mol/L, 40 mL) under ice bath, keep stirring in ice bath for reaction after addition, and track and monitor the reaction. After the reaction, the reaction system was concentrated under reduced pressure, and the concentrate was dissolved in dichloromethane (30 mL), washed with saturated sodium bicarbonate, water, and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the productII-5 . Yield: 95.5%, pale yellow solid, melting point: 122-123o C. IR ( KBr, cm-1 ) ν:3138, 2910, 1693, 1474, 1425, 1219, 1163, 1031, 996, 933, 785, 763, 694;1 H NMR (400 MHz, CDCl3 , δ, ppm) : 7.65 (s, 1H), 7.45 (d, 1H,J = 8.3 Hz), 7.35 (d, 1H,J = 1.9 Hz), 7.10 (dd, 1H,J1 = 2.0 Hz,J2 = 8.3 Hz) , 5.44 (s, 2H), 4.70 (s, 2H), 4.31 (br, 2H), 3.92 (br, 2H), 2.95 (s, 4H);13 C NMR (100 MHz, CDCl3 , δ, ppm) :195.62, 144.87, 134.79, 133.27, 133.07, 131.09, 129.86, 127.17, 122.92, 52.81, 45.63, 31.56; HRMS (ESI) calcd for C15 H18 Cl +2 N5 S2 [M+ found: 402.0369..
实施例 18 通式(II)所示,R1=m, p, m-triOCH3, n=1的衍生物(II-7)的制备Example 18 Preparation of derivative (II-7 ) represented by general formula (II), R1 =m, p, m -triOCH3 , n=1
将化合物I-9(2mmol)冰浴下用氯化氢饱和乙酸乙酯溶液(30mL)溶解,加毕改为室温搅拌反应,跟踪监测反应。反应结束后,将反应体系分别用饱和碳酸氢钠,水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得产物II-6。收率96.0%,淡黄色固体,熔点:120-121oC。IR ( KBr, cm-1) ν:3138, 2910, 1693, 1474, 1425, 1219, 1031, 996, 933, 785, 763, 694; 1H NMR (400 MHz, CDCl3, δ, ppm): 7.64 (s, 1H), 6.47 (s, 2H), 5.40 (s, 2H), 4.69 (s, 2H), 4.30 (br, 2H), 3.91 (br, 2H), 3.83(s, 3H), 3.82(s, 6H), 2.93(s, 4H); 13C NMR (100 MHz, CDCl3, δ, ppm):195.63, 153.66, 144.42, 138.23, 130.15, 122.83, 105.16, 60.85, 56.23, 54.35, 45.72, 31.68; HRMS (ESI) calcd for C18H26N5O3S2 [M+H]+:424.1477, found: 424.1472.。CompoundI-9 (2 mmol) was dissolved in hydrogen chloride saturated ethyl acetate solution (30 mL) under ice bath, and after the addition was completed, the reaction was stirred at room temperature, and the reaction was tracked and monitored. After the reaction, the reaction system was washed with saturated sodium bicarbonate, water, and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the productII-6 . Yield: 96.0%, pale yellow solid, melting point: 120-121o C. IR ( KBr, cm-1 ) ν: 3138, 2910, 1693, 1474, 1425, 1219, 1031, 996, 933, 785, 763, 694;1 H NMR (400 MHz, CDCl3 , δ, ppm): 7.64 (s, 1H), 6.47 (s, 2H), 5.40 (s, 2H), 4.69 (s, 2H), 4.30 (br, 2H), 3.91 (br, 2H), 3.83(s, 3H), 3.82( s, 6H), 2.93(s, 4H);13 C NMR (100 MHz, CDCl3 , δ, ppm): 195.63, 153.66, 144.42, 138.23, 130.15, 122.83, 105.16, 60.85, 56.263, 54.375, 435 ; HRMS (ESI) calcd for C18 H26 N5 O3 S2 [M+H]+ :424.1477, found: 424.1472.
实施例 19 通式(II)所示,R1=o, o-diF, n=1的衍生物(II-8)的制备Example 19 Preparation of derivative (II-8 ) represented by general formula (II), R1 =o, o -diF, n=1
将化合物I-10(3mmol)用氯化氢饱和乙酸乙酯溶液(40mL)溶解,加毕改为室温搅拌反应,跟踪监测反应。反应结束后,将反应体系分别用饱和碳酸氢钠,水,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得产物II-7。收率93.3%,淡黄色固体,熔点:144-145oC。1H NMR (400 MHz, CDCl3, δ, ppm): 8.10 (s, 1H), 7.50-7.86(m, 3H), 4.89 (s, 2H), 4.37 (br, 2H), 3.93 (br, 2H), 3.05(s, 4H); HRMS (ESI) calcd for C15H18F2N5S2 [M+H]+:370.0792, found: 370.0795.。 CompoundI-10 (3mmol) was dissolved in hydrogen chloride saturated ethyl acetate solution (40mL), and after the addition was completed, the reaction was changed to stirring at room temperature, and the reaction was tracked and monitored. After the reaction, the reaction system was washed with saturated sodium bicarbonate, water, and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the productII-7 . Yield: 93.3%, pale yellow solid, melting point: 144-145o C.1 H NMR (400 MHz, CDCl3 , δ, ppm): 8.10 (s, 1H), 7.50-7.86(m, 3H), 4.89 (s, 2H), 4.37 (br, 2H), 3.93 (br, 2H ), 3.05(s, 4H); HRMS (ESI) calcd for C15 H18 F2 N5 S2 [M+H]+ :370.0792, found: 370.0795..
实施例20上述化合物的LSD1抑制活性测定:The LSD1 inhibitory activity assay of the above-mentioned compoundof embodiment 20 :
1.实验方法:1. Experimental method:
样品为实施例所合成的上述化合物、纯化而得;样品储备液:称取3-5 mg样品置于1.5 mL EP管中,然后用DMSO配制成浓度是20 mM的溶液,4 °C保存放置,实验时根据所需浓度用DMSO稀释。将待测样品与LSD1蛋白于室温孵育后,加入LSD1反应底物H3K4me2并孵育反应,最后加入荧光染料Amplex和辣根过氧化酶HRP室温孵育,在酶标仪上激发光530 nm,发射光590 nm检测荧光数值:The sample is obtained by purification of the above compound synthesized in the example; sample stock solution: Weigh 3-5 mg sample and place it in a 1.5 mL EP tube, then prepare a solution with a concentration of 20 mM with DMSO, and store it at 4 °C , diluted with DMSO according to the required concentration during the experiment. After incubating the sample to be tested with LSD1 protein at room temperature, add the LSD1 reaction substrate H3K4me2 and incubate the reaction, and finally add the fluorescent dye Amplex and horseradish peroxidase HRP to incubate at room temperature. nm detection fluorescence value:

试验结果采用SPSS软件计算IC50值。The test results were calculated by using SPSS software to calculate the IC50 value.
2.实验结果2. Experimental results
实验结果如下例表1所示。The experimental results are shown in Table1 below.
表 1 上述化合物对LSD1及对人胃癌细胞MGC-803,乳腺癌细胞MCF-7,前列腺癌细胞PC3,食管癌细胞EC-109抗肿瘤活性评价数据:Table1 Evaluation data of the anti-tumor activity of the above compounds on LSD1 and human gastric cancer cell MGC-803, breast cancer cell MCF-7, prostate cancer cell PC3, and esophageal cancer cell EC-109:
实施例21已筛选的LSD1抑制剂在细胞水平对LSD1活性的抑制作用Example 21 Inhibitory effect of screened LSD1 inhibitors on LSD1 activity at the cellular level
1. 实验方法:1. Experimental method:
样品为实施例20所筛选出的化合物I-1、I-3、I-5、I-2、I-6、II-4。样品储备液:称取3-5 mg样品置于1.5 mL EP管中,然后用DMSO配制成浓度是20 mM的溶液,4 °C保存放置,实验时根据所需浓度用DMSO稀释。取对数生长期的胃癌细胞系MGC-803,消化接种于100mL细胞培养瓶中培养至80%满,加入终浓度0.5μM的化合物I-1、I-3、I-5、I-2、I-6、II-4,24小时候用酸沉淀法提取组蛋白并用BCA法定量。加入6×Loading Buffer后蛋白变性,根据蛋白浓度计算上样体积,加1.5μg蛋白于配置好的12%SDS-PAGE胶中电泳,电用完后用硝酸纤维素膜进行半干转,5%牛奶封闭电转完的硝酸纤维素膜2h后,加入抗体H3K4me2或H3K4me9进行一抗过夜孵育。次日用PBST洗膜3次并加入二抗体显影曝光。根据曝光出的条带颜色分析检测抑制剂在细胞水平对LSD1活性的抑制作用。The samples are compounds I-1, I-3, I-5, I-2, I-6, and II-4 screened out in Example 20. Sample stock solution: Weigh 3-5 mg sample and place it in a 1.5 mL EP tube, then prepare a solution with a concentration of 20 mM with DMSO, store it at 4 °C, and dilute it with DMSO according to the required concentration during the experiment. Gastric cancer cell line MGC-803 in the logarithmic growth phase was taken, digested and inoculated in a 100mL cell culture flask and cultured to 80% full, and compound I-1, I-3, I-5, I-2, I-6, II-4, histones were extracted by acid precipitation method and quantified by BCA method after 24 hours. After adding 6×Loading Buffer, the protein is denatured, and the loading volume is calculated according to the protein concentration, and 1.5 μg of protein is added to the prepared 12% SDS-PAGE gel for electrophoresis. After blocking the electroporated nitrocellulose membrane with milk for 2 hours, the antibody H3K4me2 or H3K4me9 was added for overnight incubation with the primary antibody. The next day, the membrane was washed 3 times with PBST, and the secondary antibody was added for development and exposure. According to the color analysis of the exposed bands, the inhibitory effect of the inhibitor on the activity of LSD1 at the cellular level was detected.
2.实验结果2. Experimental results
实验结果见附图1-3所示。结果表明:本发明合成的化合物对LSD1活性有抑制作用,明显优于苯环丙胺,可以将其做为活性成分用于制备组蛋白赖氨酸特异性去甲基化酶1抑制剂药物。The experimental results are shown in Figures1-3 . The results show that the compound synthesized by the invention has inhibitory effect on LSD1 activity, which is obviously better than phencypromine, and it can be used as an active ingredient to prepare histone lysine specific demethylase 1 inhibitor medicine.
Claims (6)


Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2013100261986ACN103054869A (en) | 2013-01-18 | 2013-01-18 | Application of amino dithio formic ester compound with triazolyl in preparing medicine taking LSD1 (Lysine Specificity Demethylase 1) as target |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2013100261986ACN103054869A (en) | 2013-01-18 | 2013-01-18 | Application of amino dithio formic ester compound with triazolyl in preparing medicine taking LSD1 (Lysine Specificity Demethylase 1) as target |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103054869Atrue CN103054869A (en) | 2013-04-24 |
Family
ID=48097966
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2013100261986APendingCN103054869A (en) | 2013-01-18 | 2013-01-18 | Application of amino dithio formic ester compound with triazolyl in preparing medicine taking LSD1 (Lysine Specificity Demethylase 1) as target |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN103054869A (en) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106045881A (en)* | 2016-05-26 | 2016-10-26 | 新乡医学院 | Resveratrol derivative, preparation method thereof and application of resveratrol derivative serving as LSD1 inhibitor |
| US9493450B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493442B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| WO2016198649A1 (en) | 2015-06-12 | 2016-12-15 | Oryzon Genomics, S.A. | Biomarkers associated with lsd1 inhibitors and uses thereof |
| US9527835B2 (en) | 2014-02-13 | 2016-12-27 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| WO2017013061A1 (en) | 2015-07-17 | 2017-01-26 | Oryzon Genomics, S.A. | Biomarkers associated with lsd1 inhibitors and uses thereof |
| US9670210B2 (en) | 2014-02-13 | 2017-06-06 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9695180B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US9695167B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted triazolo[1,5-a]pyridines and triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US9695168B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,5-α]pyridines and imidazo[1,5-α]pyrazines as LSD1 inhibitors |
| US9758523B2 (en) | 2014-07-10 | 2017-09-12 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| WO2017158136A1 (en) | 2016-03-16 | 2017-09-21 | Oryzon Genomics, S.A. | Methods to determine kdm1a target engagement and chemoprobes useful therefor |
| US9944647B2 (en) | 2015-04-03 | 2018-04-17 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| WO2018083189A1 (en) | 2016-11-03 | 2018-05-11 | Oryzon Genomics, S.A. | Biomarkers for determining responsiveness to lsd1 inhibitors |
| WO2018106984A1 (en) | 2016-12-09 | 2018-06-14 | Constellation Pharmaceuticals, Inc. | Markers for personalized cancer treatment with lsd1 inhibitors |
| US10166221B2 (en) | 2016-04-22 | 2019-01-01 | Incyte Corporation | Formulations of an LSD1 inhibitor |
| WO2019025588A1 (en) | 2017-08-03 | 2019-02-07 | Oryzon Genomics, S.A. | Methods of treating behavior alterations |
| US10329255B2 (en) | 2015-08-12 | 2019-06-25 | Incyte Corporation | Salts of an LSD1 inhibitor |
| WO2020188089A1 (en) | 2019-03-20 | 2020-09-24 | Oryzon Genomics, S.A. | Methods of treating attention deficit hyperactivity disorder using kdm1a inhibitors such as the compound vafidemstat |
| WO2020188090A1 (en) | 2019-03-20 | 2020-09-24 | Oryzon Genomics, S.A. | Methods of treating borderline personality disorder |
| WO2021004610A1 (en) | 2019-07-05 | 2021-01-14 | Oryzon Genomics, S.A. | Biomarkers and methods for personalized treatment of small cell lung cancer using kdm1a inhibitors |
| US10968200B2 (en) | 2018-08-31 | 2021-04-06 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| WO2022214303A1 (en) | 2021-04-08 | 2022-10-13 | Oryzon Genomics, S.A. | Combinations of lsd1 inhibitors for treating myeloid cancers |
| WO2023217784A1 (en) | 2022-05-09 | 2023-11-16 | Oryzon Genomics, S.A. | Methods of treating nf1-mutant tumors using lsd1 inhibitors |
| WO2023217758A1 (en) | 2022-05-09 | 2023-11-16 | Oryzon Genomics, S.A. | Methods of treating malignant peripheral nerve sheath tumor (mpnst) using lsd1 inhibitors |
| WO2024110649A1 (en) | 2022-11-24 | 2024-05-30 | Oryzon Genomics, S.A. | Combinations of lsd1 inhibitors and menin inhibitors for treating cancer |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102180845A (en)* | 2011-03-29 | 2011-09-14 | 郑州大学 | Butenolide structure-containing benzyl amino dithio formiate compounds, and preparation method and application thereof |
| CN102850283A (en)* | 2012-10-18 | 2013-01-02 | 郑州大学 | Triazolyl-containing amino-dithio formic ether compound as well as preparation method and application of compound |
- 2013
- 2013-01-18CNCN2013100261986Apatent/CN103054869A/enactivePending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102180845A (en)* | 2011-03-29 | 2011-09-14 | 郑州大学 | Butenolide structure-containing benzyl amino dithio formiate compounds, and preparation method and application thereof |
| CN102850283A (en)* | 2012-10-18 | 2013-01-02 | 郑州大学 | Triazolyl-containing amino-dithio formic ether compound as well as preparation method and application of compound |
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10174030B2 (en) | 2014-02-13 | 2019-01-08 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493450B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493442B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10717737B2 (en) | 2014-02-13 | 2020-07-21 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9527835B2 (en) | 2014-02-13 | 2016-12-27 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10676457B2 (en) | 2014-02-13 | 2020-06-09 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9670210B2 (en) | 2014-02-13 | 2017-06-06 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10513493B2 (en) | 2014-02-13 | 2019-12-24 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10300051B2 (en) | 2014-02-13 | 2019-05-28 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US11155532B2 (en) | 2014-02-13 | 2021-10-26 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US11247992B2 (en) | 2014-02-13 | 2022-02-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9994546B2 (en) | 2014-02-13 | 2018-06-12 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10047086B2 (en) | 2014-07-10 | 2018-08-14 | Incyte Corporation | Imidazopyridines and imidazopyrazines as LSD1 inhibitors |
| US9695180B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US10640503B2 (en) | 2014-07-10 | 2020-05-05 | Incyte Corporation | Imidazopyridines and imidazopyrazines as LSD1 inhibitors |
| US10556908B2 (en) | 2014-07-10 | 2020-02-11 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US10125133B2 (en) | 2014-07-10 | 2018-11-13 | Incyte Corporation | Substituted [1,2,4]triazolo[1,5-a]pyridines and substituted [1,2,4]triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US10112950B2 (en) | 2014-07-10 | 2018-10-30 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US9758523B2 (en) | 2014-07-10 | 2017-09-12 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| US10138249B2 (en) | 2014-07-10 | 2018-11-27 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| US9695168B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,5-α]pyridines and imidazo[1,5-α]pyrazines as LSD1 inhibitors |
| US9695167B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted triazolo[1,5-a]pyridines and triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US10968221B2 (en) | 2014-07-10 | 2021-04-06 | Incyte Corporation | Substituted [1,2,4]triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US10800779B2 (en) | 2015-04-03 | 2020-10-13 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US11401272B2 (en) | 2015-04-03 | 2022-08-02 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US9944647B2 (en) | 2015-04-03 | 2018-04-17 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| WO2016198649A1 (en) | 2015-06-12 | 2016-12-15 | Oryzon Genomics, S.A. | Biomarkers associated with lsd1 inhibitors and uses thereof |
| WO2017013061A1 (en) | 2015-07-17 | 2017-01-26 | Oryzon Genomics, S.A. | Biomarkers associated with lsd1 inhibitors and uses thereof |
| US10329255B2 (en) | 2015-08-12 | 2019-06-25 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US11498900B2 (en) | 2015-08-12 | 2022-11-15 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US10723700B2 (en) | 2015-08-12 | 2020-07-28 | Incyte Corporation | Salts of an LSD1 inhibitor |
| WO2017158136A1 (en) | 2016-03-16 | 2017-09-21 | Oryzon Genomics, S.A. | Methods to determine kdm1a target engagement and chemoprobes useful therefor |
| US10166221B2 (en) | 2016-04-22 | 2019-01-01 | Incyte Corporation | Formulations of an LSD1 inhibitor |
| CN106045881A (en)* | 2016-05-26 | 2016-10-26 | 新乡医学院 | Resveratrol derivative, preparation method thereof and application of resveratrol derivative serving as LSD1 inhibitor |
| WO2018083189A1 (en) | 2016-11-03 | 2018-05-11 | Oryzon Genomics, S.A. | Biomarkers for determining responsiveness to lsd1 inhibitors |
| WO2018106984A1 (en) | 2016-12-09 | 2018-06-14 | Constellation Pharmaceuticals, Inc. | Markers for personalized cancer treatment with lsd1 inhibitors |
| WO2019025588A1 (en) | 2017-08-03 | 2019-02-07 | Oryzon Genomics, S.A. | Methods of treating behavior alterations |
| EP4512473A2 (en) | 2017-08-03 | 2025-02-26 | Oryzon Genomics, S.A. | Methods of treating behavior alterations |
| US10968200B2 (en) | 2018-08-31 | 2021-04-06 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| US11512064B2 (en) | 2018-08-31 | 2022-11-29 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| WO2020188090A1 (en) | 2019-03-20 | 2020-09-24 | Oryzon Genomics, S.A. | Methods of treating borderline personality disorder |
| WO2020188089A1 (en) | 2019-03-20 | 2020-09-24 | Oryzon Genomics, S.A. | Methods of treating attention deficit hyperactivity disorder using kdm1a inhibitors such as the compound vafidemstat |
| WO2021004610A1 (en) | 2019-07-05 | 2021-01-14 | Oryzon Genomics, S.A. | Biomarkers and methods for personalized treatment of small cell lung cancer using kdm1a inhibitors |
| WO2022214303A1 (en) | 2021-04-08 | 2022-10-13 | Oryzon Genomics, S.A. | Combinations of lsd1 inhibitors for treating myeloid cancers |
| WO2023217784A1 (en) | 2022-05-09 | 2023-11-16 | Oryzon Genomics, S.A. | Methods of treating nf1-mutant tumors using lsd1 inhibitors |
| WO2023217758A1 (en) | 2022-05-09 | 2023-11-16 | Oryzon Genomics, S.A. | Methods of treating malignant peripheral nerve sheath tumor (mpnst) using lsd1 inhibitors |
| WO2024110649A1 (en) | 2022-11-24 | 2024-05-30 | Oryzon Genomics, S.A. | Combinations of lsd1 inhibitors and menin inhibitors for treating cancer |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103054869A (en) | Application of amino dithio formic ester compound with triazolyl in preparing medicine taking LSD1 (Lysine Specificity Demethylase 1) as target | |
| CN103319466B (en) | Containing the 1,2,3-triazoles-dithiocarbamates compound of tonka bean camphor parent nucleus, preparation method and application thereof | |
| Wang et al. | Design, synthesis and biological evaluation of [1, 2, 4] triazolo [1, 5-a] pyrimidines as potent lysine specific demethylase 1 (LSD1/KDM1A) inhibitors | |
| CN106831489B (en) | Tranylcypromine acylhydrazone, preparation method and applications | |
| Yang et al. | 4, 6-Substituted-1H-Indazoles as potent IDO1/TDO dual inhibitors | |
| CN106045881B (en) | Resveratrol derivative, its preparation method and the application as LSD1 inhibitor | |
| Horiuchi et al. | Discovery of novel thieno [2, 3-d] pyrimidin-4-yl hydrazone-based inhibitors of Cyclin D1-CDK4: Synthesis, biological evaluation and structure–activity relationships. Part 2 | |
| CN107174584B (en) | Application of piperazine structure-containing compound in preparation of LSD1 inhibitor | |
| CN113527195B (en) | 5-aryl nicotinamide LSD1/HDAC double-target inhibitor, preparation method and application thereof | |
| Liu et al. | Discovery and synthesis of novel indole derivatives-containing 3-methylenedihydrofuran-2 (3H)-one as irreversible LSD1 inhibitors | |
| CN112110936B (en) | Tetrahydroquinoline derivative and preparation method and application thereof | |
| EP2168952B1 (en) | Morpholine derivatives as renin inhibitors | |
| CN103992336A (en) | Oxa- or thio-evodiamine anti-tumor derivatives and preparation method thereof | |
| CN111592487B (en) | A class of diarylethene-based LSD1/HDACs dual-target inhibitors containing hydroxamic acid group, preparation method and application thereof | |
| WO2019141131A1 (en) | Bromodomain inhibitor compound and use thereof | |
| CN105254635A (en) | Imidazo pyrazine compound, medicine composition of imidazo pyrazine compound and purpose of imidazo pyrazine compound | |
| CN102850283B (en) | Triazolyl-containing amino-dithio formic ether compound as well as preparation method and application of compound | |
| CN103467359A (en) | Cinnamon amides histone deacetylase inhibitor with benzpyrole and preparation method and application of same | |
| CN107501169A (en) | A kind of trans diarylethene LSD1 inhibitor, its preparation method and application | |
| CN102295640A (en) | 3-heterocyclic Schiff base-5-fluoro-indole-2-ketone compound and preparation method and application thereof | |
| CN109293664B (en) | Pyrimido 1,2,4-triazole hydrazine compounds and preparation method and application thereof | |
| WO2018015788A1 (en) | ANTIMETASTATIC 2H-SELENOPHENO[3,2-h]CHROMENES, SYNTHESIS THEREOF, AND METHODS OF USING SAME AGENTS | |
| CN101717397B (en) | Substituted pyrido [2',1':2,3] imidazo [4,5-c ] isoquinolinone compounds, synthetic method and application thereof, and pharmaceutical composition containing the compounds | |
| KR20100132553A (en) | Novel N- (2-amino-phenyl) -acrylamide | |
| CN114539267A (en) | Evodiamine derivative and application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C12 | Rejection of a patent application after its publication | ||
| RJ01 | Rejection of invention patent application after publication | Application publication date:20130424 |