CN101537007A - Application of N-(thiofuran-2) pyrazolo (1, 5-a) pyridine-3-formanides compounds for preparing antineoplastic - Google Patents
Application of N-(thiofuran-2) pyrazolo (1, 5-a) pyridine-3-formanides compounds for preparing antineoplasticDownload PDFInfo
- Publication number
- CN101537007A CN101537007ACN 200910068159CN200910068159ACN101537007ACN 101537007 ACN101537007 ACN 101537007ACN 200910068159CN200910068159CN 200910068159CN 200910068159 ACN200910068159 ACN 200910068159ACN 101537007 ACN101537007 ACN 101537007A
- Authority
- CN
- China
- Prior art keywords
- pyrazolo
- cdk9
- pyrimidine
- thiophene
- activity
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images



Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Translated fromChineseDescription
Translated fromChinese技术领域technical field
本发明涉及一种抗恶性肿瘤的化合物的应用,属于抗肿瘤药物的研究领域。具体的说,本发明涉及一种N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物及其取代物在抑制肺癌、骨肉瘤、卵巢癌、宫颈癌、乳腺癌等多种恶性肿瘤中的用途。The invention relates to the application of an anti-malignant tumor compound and belongs to the research field of anti-tumor drugs. Specifically, the present invention relates to a kind of N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide compound and its substitution in inhibiting lung cancer, osteosarcoma, ovarian cancer, cervical cancer , breast cancer and other malignant tumors.
背景技术Background technique
恶性肿瘤是严重威胁人类健康的一大类疾病,其形成主要原因是细胞基因水平的变异所导致的细胞异常增生。真核细胞的增殖分裂是一个精确而复杂的调控过程。增殖过程是通过细胞周期来完成的,而细胞周期的有序进行,有其严格的分子调控机制。随着分子生物学的发展,这些复杂的调控分子及其作用机制慢慢为人类所发现和阐明。目前已发现的参与细胞周期调控主要有三大类分子:细胞周期依赖性激酶(cyclin-dependent kinases,CDKs),细胞周期蛋白(cyclins),细胞周期蛋白依赖性激酶抑制剂(cyclin-dependent kinase inhibitors,CKIs),其中CDK处于中心地位。cyclin可以与对应的CDK以1∶1方式结合形成复合物,作为该复合物的调节亚基对相应的CDK的活性进行正性调节,研究表明,未结合cyclin的CDK及CDK单体均无活性。而与cyclin作用相反的CKI则是一类负性调节CDK的核蛋白。Malignant tumors are a large class of diseases that seriously threaten human health. The main cause of their formation is the abnormal proliferation of cells caused by mutations at the level of cell genes. The proliferation and division of eukaryotic cells is a precise and complex regulatory process. The proliferation process is completed through the cell cycle, and the orderly progress of the cell cycle has its strict molecular regulation mechanism. With the development of molecular biology, these complex regulatory molecules and their mechanism of action are gradually discovered and elucidated by humans. There are three main types of molecules that have been found to participate in cell cycle regulation: cell cycle-dependent kinases (cyclin-dependent kinases, CDKs), cyclins, and cyclin-dependent kinase inhibitors (cyclin-dependent kinase inhibitors, CKIs), with CDK at the center. Cyclin can combine with the corresponding CDK in a 1:1 manner to form a complex, and as the regulatory subunit of the complex, positively regulate the activity of the corresponding CDK. Studies have shown that CDK and CDK monomers that are not bound to cyclin are inactive. . CKI, which is opposite to cyclin, is a kind of nuclear protein that negatively regulates CDK.
据研究统计显示,有超过90%的人类癌症中cyclin,CDK,CKI和Rb途径中相关基因发生了变异,其中CDK和其相应的cyclin的失常最为频繁,由此可见其重要性。CDK家族目前已经发现十三个成员(CDK1-CDK13)按其胞内功能不同分为两类:控制细胞周期的CDKs和控制细胞转录的CDKs。CDK9属于后者。CDK9属于丝氨酸类激酶,它与对应cyclin(s)结合形成的复合物叫做正性转录延长因子b(P-TEFb),该复合物能够磷酸化RNA聚合酶II(RNA polymerase II)和一些负性转录延长因子(NELF和N-TEFs)从而使转录从起始部位得以延伸,是转录得以延长的关键点。研究发现CDK9的表达水平或(和)活性的异常会引起细胞内多种蛋白表达或(和)其mRNA异常,进而导致细胞的生长异常、性状异常、周期异常、增殖异常。According to research statistics, more than 90% of human cancers have mutations in genes related to cyclin, CDK, CKI and Rb pathways, among which the abnormality of CDK and its corresponding cyclin is the most frequent, which shows its importance. Thirteen members of the CDK family (CDK1-CDK13) have been found so far and are divided into two categories according to their intracellular functions: CDKs that control the cell cycle and CDKs that control cell transcription. CDK9 belongs to the latter group. CDK9 belongs to serine kinase, and the complex formed by binding to corresponding cyclin(s) is called positive transcription elongation factor b (P-TEFb), which can phosphorylate RNA polymerase II (RNA polymerase II) and some negative Transcription elongation factors (NELF and N-TEFs) are key points for transcription elongation, thereby extending transcription from the initiation site. Studies have found that the abnormal expression level or (and) activity of CDK9 will cause the abnormal expression of various proteins in cells or (and) their mRNA, which in turn will lead to abnormal growth, abnormal traits, abnormal cycle and abnormal proliferation of cells.
近年来有许多针对分子靶点的抗癌药物出现,这类药物的针对性强,效果显著,被称为“靶标药物”。早在100年前Paul Ehrlich就提出了靶向治疗(targeting therapy)的概念,随着分子生物学的发展及人们对肿瘤发生机制研究的深入,针对特定致癌机制的靶标药物在近些年得到了很快的发展,这些药物直接攻击致癌病因,选择性强,临床试验效果显著,且副作用轻,多药联合使用时常能增强传统化疗药物的作用。在过去的十多年里,抗肿瘤药物的研究普遍针对肿瘤细胞失控的分子基础,集中在恢复肿瘤细胞周期的调控机制上,因而产生了许多针对不同CDK的直接或间接的抑制剂,以及针对细胞周期检控点的抑制剂。In recent years, many anti-cancer drugs targeting molecular targets have emerged. These drugs are highly targeted and effective, and are called "targeted drugs". As early as 100 years ago, Paul Ehrlich proposed the concept of targeting therapy. With the development of molecular biology and the deepening of people's research on the mechanism of tumorigenesis, targeted drugs targeting specific carcinogenic mechanisms have been obtained in recent years. Rapidly developing, these drugs directly attack the cause of cancer, have strong selectivity, have remarkable effects in clinical trials, and have mild side effects. The combined use of multiple drugs can often enhance the effect of traditional chemotherapy drugs. In the past ten years, the research on anti-tumor drugs has generally focused on the molecular basis of tumor cell out-of-control, focusing on restoring the regulatory mechanism of tumor cell cycle, thus producing many direct or indirect inhibitors against different CDKs, and targeting Inhibitors of cell cycle checkpoints.
CDKs是肿瘤治疗颇具潜力的的靶点。目前报道过50种以上的CDK抑制剂,其中一些具有潜在的抗肿瘤活性,某些多CDK抑制剂已经被开发为抗肿瘤药物,另有一些正在进行临床前或临床实验,而新的CDK抑制剂也在不断被研发。目前针对细胞周期的抑制剂主要有以下几大类,Flavopiridol(夫拉平度),第一代CDK抑制剂的代表,已经被广泛的用于抗肿瘤临床治疗研究中。它是以印度乡土植物中分离得到的一种生物碱为原型,经过半合成得到的一种人工半合成黄酮类衍生物。它具有多种抗癌机理,它对EGFR(表皮生长因子受体)酪氨酸激酶和蛋白激酶A(PKA)有抑制作用。同时还具有广谱CDK抑制作用,能够有效抑制CDK1,2,4,6和7在细胞周期中所起的作用,其IC50为40-200nM之间。后来研究又发现它对CDK9/cyclinT的活性也有抑制作用,能够抑制转录。另一主要大类是嘌呤类化合物,包括二甲氨基嘌呤,异戊烯腺嘌呤,Olomoucine(奥罗莫星),Roscovitine(一种CDK激酶抑制剂)等。他们对CDK1和CDK2分别有不同程度的抑制作用,使细胞停滞在G1/S期或G2/M期,从而诱导细胞凋亡。其中的代表Roscovitine的分子机制是它能竞争性结合ATP位点,对人CDK2/cyclinE,CDK7/cyclinH,CDK9/cyclinT1有极高的选择抑制作用。分子药理学研究显示,经Roscovitine处理过的直肠癌细胞可导致pRb(视网膜细胞瘤蛋白)磷酸化水平降低。但在临床实验中表现出作用毒副作用大,单独用药抑癌特性并不明显等特点。其他CDK抑制剂,如丁内酯、Staurosporine类(星形孢菌素)、氯吲哚磺胺类、2-氨基噻唑类均针对CDK1,2或4等分子靶点所研发。最近有报道有另一种新型的咪唑[1,2-a]吡啶类CDKs抑制剂也可以有效抑制CDK1,2和9,具有很好的开发前景。除此以外,对新靶点CDK9的特异性抑制剂研究成果的报道很少。究其原因,除了因为CDK9靶点较新,且以前对它的研究偏重于艾滋病治疗以外,另一个重要原因是由于目前还没有CDK9晶体结构,这些都极大地制约了利用CDK9作为靶点来研制抗肿瘤药物的发展。CDKs are potential targets for tumor therapy. At present, more than 50 CDK inhibitors have been reported, some of which have potential anti-tumor activity, some multi-CDK inhibitors have been developed as anti-tumor drugs, and others are undergoing preclinical or clinical trials, and new CDK inhibitors Agents are also being developed continuously. At present, there are mainly the following categories of inhibitors targeting the cell cycle. Flavopiridol (flavopiridol), a representative of the first generation of CDK inhibitors, has been widely used in anti-tumor clinical treatment research. It is an artificial semi-synthetic flavonoid derivative obtained through semi-synthesis based on an alkaloid isolated from native plants in India. It has multiple anti-cancer mechanisms, and it has inhibitory effects on EGFR (epidermal growth factor receptor) tyrosine kinase and protein kinase A (PKA). At the same time, it also has a broad-spectrum CDK inhibitory effect, which can effectively inhibit the role of CDK1, 2, 4, 6 and 7 in the cell cycle, and its IC50 is between 40-200nM. Later studies found that it also inhibited the activity of CDK9/cyclinT and could inhibit transcription. Another major category is purine compounds, including dimethylaminopurine, isopentenyl adenine, Olomoucine (oromucine), Roscovitine (a CDK kinase inhibitor) and so on. They have different degrees of inhibition on CDK1 and CDK2, respectively, so that cells are arrested in G1/S phase or G2/M phase, thereby inducing apoptosis. The representative molecular mechanism of Roscovitine is that it can competitively bind to the ATP site, and has a very high selective inhibitory effect on human CDK2/cyclinE, CDK7/cyclinH, and CDK9/cyclinT1. Molecular pharmacology studies have shown that Roscovitine-treated rectal cancer cells can lead to a decrease in the phosphorylation level of pRb (retinoblastoma protein). However, in clinical trials, it has shown the characteristics of high toxicity and side effects, and the tumor suppressor property is not obvious when used alone. Other CDK inhibitors, such as butyrolactone, Staurosporine (staurosporine), chloroindosulfonamides, and 2-aminothiazoles are all developed for molecular targets such as CDK1, 2, or 4. Recently, it has been reported that another novel imidazo[1,2-a]pyridine CDKs inhibitor can also effectively inhibit CDK1, 2 and 9, and has a good development prospect. In addition, there are few reports on the research results of specific inhibitors of the new target CDK9. The reason is that, in addition to the relatively new target of CDK9, and the previous research on it focused on AIDS treatment, another important reason is that there is no crystal structure of CDK9, which greatly restricts the use of CDK9 as a target to develop Development of antineoplastic drugs.
因此,目前针对CDK9为靶点,需要开发一种能抑制恶性肿瘤,尤其是肺癌、骨肉瘤、卵巢癌、宫颈癌、乳腺癌等多种恶性肿瘤的药物及其制备方法。Therefore, currently targeting CDK9, it is necessary to develop a drug capable of inhibiting malignant tumors, especially lung cancer, osteosarcoma, ovarian cancer, cervical cancer, breast cancer, and a preparation method thereof.
发明内容Contents of the invention
本发明基于以下原理:从靶点构建开始采用同源模建的方法得到CDK9的三维晶体结构,并以此为基础,采用DOCK方法与Specs公司化合物库小分子进行对接,通过计算机虚拟筛选、生物活性筛选和小分子化合物与靶点之间相互作用的分子机制研究,寻找能高效特异抑制CDK9激酶活性的小分子抑制剂。通过阐明并验证了N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物在抑制肿瘤细胞生长的分子机制。我们首次发现,本发明的N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物在抑制肺癌、骨肉瘤、卵巢癌、宫颈癌、乳腺癌等多种恶性肿瘤细胞生长中显示出高效药理活性,为寻找新的有效的细胞周期类抗肿瘤药物开辟了一条新的途径。The present invention is based on the following principle: starting from the construction of the target point, the three-dimensional crystal structure of CDK9 is obtained by using the method of homology modeling, and based on this, the DOCK method is used to dock the small molecule in the compound library of Specs Company, and through computer virtual screening, biological Activity screening and molecular mechanism research on the interaction between small molecule compounds and targets, looking for small molecule inhibitors that can efficiently and specifically inhibit CDK9 kinase activity. By elucidating and verifying the molecular mechanism of N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide compounds in inhibiting tumor cell growth. We have found for the first time that the N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamides of the present invention are effective in inhibiting lung cancer, osteosarcoma, ovarian cancer, cervical cancer, breast cancer, etc. The high-efficiency pharmacological activity is shown in the growth of malignant tumor cells, which opens up a new way for finding new and effective cell cycle anti-tumor drugs.
在本发明以前,N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类特别是本次购买的化合物仅仅只是一类普通的化合物,但是我们研究了其在抗肿瘤方面的特性,并且深入到分子水平研究其与CDK9的相互作用。另外,N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺及其类似物都可以从市场上买到,来源比较方便。Before the present invention, N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamides, especially the compound purchased this time, was only a common compound, but we studied its Anti-tumor properties, and in-depth study of its interaction with CDK9 at the molecular level. In addition, N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide and its analogues can be purchased from the market, and the source is relatively convenient.
因此,本发明的第一个目的在于提供一种N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物在抑制肺癌、骨肉瘤、卵巢癌、宫颈癌、乳腺癌等多种恶性肿瘤中的用途。Therefore, the first object of the present invention is to provide a kind of N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamides in inhibiting lung cancer, osteosarcoma, ovarian cancer, cervical cancer , breast cancer and other malignant tumors.
在一个具体实施方案中,本发明提供了如下式(I)的化合物(即N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺)在抗恶性肿瘤的用途:In a specific embodiment, the present invention provides the following compounds of formula (I) (i.e. N-(thiophene-2) pyrazolo [1,5-a] pyrimidine-3-carboxamide) in anti-cancer purposes :

N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide
其中:in:
R1、R2、R3为C1-C6的直链或支链烷基、C1-C4全氟烷基、C1-C4烷酰基、C1-C4酯基、C1-C4羧基、C1-C4酰胺基、C1-C4硫醚、C1-C4肼基、C1-C4酰肼、卤素、磺酸基、磺胺、-NO2、-NH2、-CHO、-OH、-H、苯基及取代苯基、各种杂环及取代杂环;R1 , R2 , R3 are C1 -C6 linear or branched chain alkyl, C1 -C4 perfluoroalkyl, C1 -C4 alkanoyl, C1 -C4 ester, C1 -C4 carboxyl, C1 -C4 amido, C1 -C4 sulfide, C1 -C4 hydrazine, C1 -C4 hydrazide, halogen, sulfonic acid, sulfonamide, -NO2 , -NH2 , -CHO, -OH, -H, phenyl and substituted phenyl, various heterocycles and substituted heterocycles;
R1’R2’:(CH2)4、CH2=CH2-CH2=CH2;R1 'R2 ': (CH2 )4 , CH2 =CH2 -CH2 =CH2 ;
R2’R3’:(CH2)4、CH2=CH2-CH2=CH2;R2 'R3 ': (CH2 )4 , CH2 =CH2 -CH2 =CH2 ;
R1’、R2’、R3’为-H、C1-C6的直链或支链烷基、C1-C4全氟烷基、C1-C4烷酰基、C1-C4酯基、C1-C4羧基、C1-C4酰胺基、C1-C4硫醚、C1-C4肼基、C1-C4酰肼、卤素、磺酸基、磺胺、-NO2、-NH2、-CHO、-OH、苯基及取代苯基、各种杂环及取代杂环;R1 ', R2 ', R3 ' are -H, C1 -C6 straight or branched chain alkyl, C1 -C4 perfluoroalkyl, C1-C4 alkanoyl, C1 -C4 Ester group, C1 -C4 carboxyl group, C1 -C4 amido group, C1 -C4 sulfide group, C1 -C4 hydrazine group, C1 -C4 hydrazide group, halogen, sulfonic acid group, sulfonamide, -NO2 , -NH2 , -CHO, -OH, phenyl and substituted phenyl, various heterocycles and substituted heterocycles;
R1、R2、R3、R1’、R2’、R3’杂环包括呋喃、噻吩、吡咯、恶唑、噻唑、咪唑、异恶唑、异噻唑、吡唑、吡啶、吡喃、嘧啶、哒嗪、吡嗪、三嗪、苯并呋喃、苯并噻吩、吲哚、喹啉、异喹啉、苯并吡喃、嘌呤及其它嘧啶并咪唑环系、喋啶及其它嘧啶吡嗪环系;R1 , R2 , R3 , R1 ', R2 ', R3 ' heterocycles include furan, thiophene, pyrrole, oxazole, thiazole, imidazole, isoxazole, isothiazole, pyrazole, pyridine, pyran , pyrimidine, pyridazine, pyrazine, triazine, benzofuran, benzothiophene, indole, quinoline, isoquinoline, benzopyran, purine and other pyrimidimidazole ring systems, pteridine and other pyrimidine pyrimidine Azine ring system;
R1、R2、R3、R1’、R2’、R3’苯基和杂环上的取代基可以是烷基、烯基、炔基、烷氧基、醚基、硫醚基、氟烷基、羰基、酯基、羧基、酰胺基、肼基、酰肼、卤素、磺酸基、磺胺、-NO2、-NH2、-CHO、-OH,可以是单取代也可以是多取代;The substituents on R1 , R2 , R3 , R1 ', R2 ', R3 'phenyl and heterocycle can be alkyl, alkenyl, alkynyl, alkoxy, ether, thioether , fluoroalkyl, carbonyl, ester, carboxyl, amido, hydrazine, hydrazide, halogen, sulfonic acid, sulfonamide, -NO2 , -NH2 , -CHO, -OH, can be monosubstituted or multiple substitutions;
通式I化合物药学上可接受的盐,包括盐酸盐、磷酸盐、硫酸盐、醋酸盐、马来酸盐、枸橼酸盐、苯磺酸盐、甲基苯磺酸盐、富马酸盐、酒石酸盐。Pharmaceutically acceptable salts of compounds of general formula I, including hydrochloride, phosphate, sulfate, acetate, maleate, citrate, benzenesulfonate, toluenesulfonate, fumarate salts, tartrates.
在另一个具体实施方案中,所述的用途可以是利用N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物来抑制或治疗恶性肿瘤(优选肺癌、骨肉瘤、卵巢癌、宫颈癌、乳腺癌等)中的用途;或所述的用途可以是利用N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物来制备抑制或治疗恶性肿瘤(优选肺癌、骨肉瘤、卵巢癌、宫颈癌、乳腺癌等)的药物中的用途。In another specific embodiment, the use can be the use of N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamides to inhibit or treat malignant tumors (preferably lung cancer, osteosarcoma, ovarian cancer, cervical cancer, breast cancer, etc.); or the use can be the use of N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamides To prepare the purposes in the medicine of suppressing or treating malignant tumor (preferably lung cancer, osteosarcoma, ovarian cancer, cervical cancer, breast cancer etc.).
本发明的第二个目的在于提供上述化合物的取代物在抑制恶性肿瘤中的用途。The second object of the present invention is to provide the use of the substitutes of the above compounds in inhibiting malignant tumors.
在一个具体实施方案中,提供一种N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物的取代物在抑制肿瘤增殖方面的应用。In a specific embodiment, an application of a substitute of N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide compounds in inhibiting tumor growth is provided.
在另一个具体实施方案中,提供一种取代N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物在抑制肺癌、骨肉瘤、卵巢癌、宫颈癌、乳腺癌等多种恶性肿瘤体外活性方面的应用。In another specific embodiment, a substituted N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide compound is provided for inhibiting lung cancer, osteosarcoma, ovarian cancer, cervical cancer, Application in in vitro activity of various malignant tumors such as breast cancer.
还在另一具体实施方案中,提供一种取代N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物在抑制细胞周期依赖性激酶CDK9激酶活性方面的应用。In another specific embodiment, there is provided a substituted N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide compound in inhibiting the activity of the cell cycle-dependent kinase CDK9 kinase application.
本发明第三个发明目的是提供一种治疗恶性肿瘤或抑制细胞周期依赖性激酶CDK9激酶活性的药物组合物。The third object of the present invention is to provide a pharmaceutical composition for treating malignant tumors or inhibiting the activity of the cell cycle-dependent kinase CDK9 kinase.
在一个具体实施方案中,它包含上述的化合物以及一种或多种药学上可接受的载体、赋形剂或稀释剂。In a specific embodiment, it comprises a compound as described above and one or more pharmaceutically acceptable carriers, excipients or diluents.
本发明第四个发明目的是提供筛选CDK9的新型小分子抑制剂N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物的方法。The fourth object of the present invention is to provide a method for screening N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamides, novel small molecule inhibitors of CDK9.
在一个具体实施方案中,所述的方法是采用同源模建方法得到细胞周期依赖性激酶家族成员CDK9的三维晶体构象,用DOCK(分子对接)对小分子三维数据库进行筛选;通过MTT法肿瘤细胞生长抑制实验对挑选出的化合物进行生物活性测定,然后挑选高活性的化合物N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类进行分子机制研究,验证化合物对CDK9激酶活性的抑制作用。In a specific embodiment, the method is to use the homology modeling method to obtain the three-dimensional crystal conformation of CDK9, a member of the cell cycle-dependent kinase family, and use DOCK (molecular docking) to screen the three-dimensional database of small molecules; The cell growth inhibition experiment is used to measure the biological activity of the selected compounds, and then select the highly active compound N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamides for molecular mechanism research and verification Inhibition of CDK9 kinase activity by compounds.
在一个具体实施方案中,所述的同源模建及DOCK筛选步骤如下:In a specific embodiment, the homology modeling and DOCK screening steps are as follows:
为了得到CDK9三维结构,本实验室根据已知CDK9的氨基酸序列,在SGI图形工作站上,利用软件Insight II的同源模建(MODELLER/HOMOLOGY)模块,对CDK9进行三维结构的同源模建。主要步骤如下:以CDK9的一级序列为探针,用BlastP程序在PDB库中搜索其同源蛋白,选择同源性高并且蛋白类型已知的同源蛋白作为模板。用同源蛋白叠加,确定SCR(结构保守区)和LOOP区。将模建蛋白和同源蛋白序列联配,能与同源蛋白SCR区相匹配的序列为模建蛋白的SCR区,然后赋予模建蛋白SCR区空间坐标,接着利用Modeler软件包,采用数据库查询方法,根据LOOP的长度及两端的SCR的坐标,搜索出合适的LOOP结构,同样采用数据库查询的方法,在模建过程中直接从参考蛋白考博模建蛋白的侧链,对不适侧链通过分子力学方法模建为较为合理的结构。In order to obtain the three-dimensional structure of CDK9, based on the known amino acid sequence of CDK9, our laboratory used the homology modeling (MODELLER/HOMOLOGY) module of the software Insight II on the SGI graphics workstation to perform homology modeling on the three-dimensional structure of CDK9. The main steps are as follows: use the primary sequence of CDK9 as a probe, use the BlastP program to search for homologous proteins in the PDB library, and select homologous proteins with high homology and known protein types as templates. Superimposed with homologous proteins, the SCR (structurally conserved region) and LOOP regions were determined. Align the modeled protein and homologous protein sequences, the sequence that can match the SCR region of the homologous protein is the SCR region of the modeled protein, and then assign the spatial coordinates of the SCR region of the modeled protein, and then use the Modeler software package to query the database method, according to the length of the LOOP and the coordinates of the SCRs at both ends, a suitable LOOP structure is searched, and the method of database query is also used to directly model the side chain of the protein from the reference protein Kobo in the modeling process. The mechanical method is modeled as a more reasonable structure.
模建后结构的动力学优化:利用Soak/Assembly程序,对整个蛋白加上两层厚度为5

将CDK9的三维结构中的二级结构显示出来,观察其中的beta折叠和alpha螺旋的分布,对蛋白质的表面进行活性区搜索,把搜索到的活性区逐一显示进行比较,同时结合文献和活性区特征选出可能的结合活性区。将CDK9动力学优化后的PDB文件将所有的组氨酸His改为Hid,对生成二硫键的半胱氨酸Cys改为Cyx,经Insight II模拟以及归属力场之后,存储为receptor.mol2文件,供DOCK使用。受体分子表面有DMS程序生成,再用SPHGEN生成活性位点的负像,负像由不同大小的圆球堆积而成,作为活性区域特征的代表。然后用基于分子锚点片段出发的构象搜索方法搜索数据库中配体可能的构象,根据负像区的特点自动模拟配体的作用方式,并根据能量打分记录理论上最佳的作用方式。最后撰写dock.in文件,确定各个参数文件的位置,在LINUX下运行DOCK模拟和筛选。Display the secondary structure in the three-dimensional structure of CDK9, observe the distribution of the beta fold and alpha helix, search the active region on the surface of the protein, and compare the searched active regions one by one, combining the literature and the active region Characterization selects possible binding activity regions. Change all histidine His to Hid in the PDB file after CDK9 kinetic optimization, and change the cysteine Cys that generates disulfide bonds to Cyx, and store it as receptor.mol2 after Insight II simulation and attribution force field file, for use by DOCK. The surface of the receptor molecule is generated by DMS program, and then SPHGEN is used to generate the negative image of the active site. The negative image is formed by stacking spheres of different sizes, which are representative of the characteristics of the active area. Then use the conformational search method based on the molecular anchor fragment to search the possible conformation of the ligand in the database, automatically simulate the mode of action of the ligand according to the characteristics of the negative image area, and record the theoretically optimal mode of action according to the energy score. Finally, write the dock.in file, determine the location of each parameter file, and run DOCK simulation and screening under LINUX.
本发明第五个发明目的是提供由上述方法所获得的一种CDK9三维晶体结构及其模建方法。The fifth object of the present invention is to provide a three-dimensional crystal structure of CDK9 obtained by the above method and its modeling method.
本发明第六个发明目的是提供一种用于治疗恶性肿瘤的药物组合物,它包含作为活性成分的通式I中任一的化合物以及一种或多种药学上可接受的载体、赋形剂或稀释剂;或者,包含作为活性成分的由上述同源模建的筛选方法所制备的化合物以及一种或多种药学上可接受的载体、赋形剂或稀释剂。The sixth object of the present invention is to provide a pharmaceutical composition for treating malignant tumors, which comprises any compound of the general formula I as an active ingredient and one or more pharmaceutically acceptable carriers, excipients or diluent; or, as an active ingredient, the compound prepared by the above homology modeling screening method and one or more pharmaceutically acceptable carriers, excipients or diluents.
附图说明Description of drawings
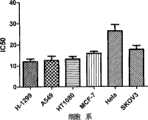
图1:高活性的N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类取代衍生物的具体代表C-21化合物进行了生物学活性测定,扩大其体外实验应用的范围。横坐标表示的是我室所保存的各种肿瘤细胞系H-1299为肺鳞癌细胞系,A549为另一种肺鳞癌细胞系,HT1080为骨肉瘤细胞系,MCF-7乳腺癌细胞系,Hela为宫颈癌细胞系,SKOV3为卵巢癌细胞系;纵坐标表示采用C-21对上述肿瘤细胞系处理后所得出的IC50值(肿瘤细胞被半数杀伤时的药物浓度值)Figure 1: Specific representative C-21 compound of highly active N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide substituted derivatives was tested for biological activity and expanded its in vitro range of experimental applications. The abscissa indicates the various tumor cell lines preserved in our laboratory. H-1299 is a lung squamous cell line, A549 is another lung squamous cell line, HT1080 is an osteosarcoma cell line, MCF-7 breast cancer cell line , Hela is the cervical cancer cell line, SKOV3 is the ovarian cancer cell line; the ordinate indicates the IC50 value (drug concentration value when the tumor cells are half-killed) obtained after treating the above tumor cell lines with C-21
图2:C-21化合物结构式,它是通式I在R1三氟甲基取代,R2苯二甲氧基取代,R2’R3’:(CH2)4取代,R1’乙酯基取代后得到的N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类一种具体取代衍生物Figure 2: The structural formula of C-21 compound, it is general formula I in which R1 is substituted with trifluoromethyl, R2 is substituted with phenylenedimethoxy, R2'R3 ': (CH2 )4 substituted, R1'B A specific substituted derivative of N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide obtained after ester group substitution
图3:C-21对CDK9的激酶作用的抑制实验的免疫印迹图,其中PBS为阴性对照处理,并分别用2,5,10,20,40μM的C-21处理细胞裂解液。pSer2CTD表示经药物处理后RNA聚合酶II大亚基C末端2位丝氨酸(也即是CDK9的磷酸化作用位点)的磷酸化程度,可由图中黑色斑点的深浅看出。图4:化合物C-21与CDK9晶体结构结合作用模式,其中彩色棒状图为C-21的分子结构式与其余墨绿色结构为CDK9的三维晶体结构模建图,其中实体墨绿色部分为其激酶活性区。图中三根亮绿色线表示C-21与CDK9激酶活性区形成的三个氢键。Figure 3: Western blot of the inhibitory effect of C-21 on CDK9 kinase, in which PBS was treated as a negative control, and cell lysates were treated with 2, 5, 10, 20, 40 μM C-21, respectively. pSer2CTD indicates the degree of phosphorylation of
具体实施方式:Detailed ways:
下面结合实施例对本发明做进一步的描述。The present invention will be further described below in conjunction with the examples.
实施例1:MMT测化合物的对肿瘤细胞的抑制作用Embodiment 1: MMT detects the inhibitory effect of compound on tumor cells
采用常规MTT法测定各化合物对肿瘤细胞系的增殖抑制活性。初步活性实验取对数生长期的H1299细胞,用含有10%血清的RPMI1640培养液调整细胞浓度为1×105/ml,接种于96孔培养板中,每孔100μl,在37℃,5%CO2条件下培养24h。分组加药,每个浓度设三个平行孔,处理组加不同浓度的药物,阴性对照组加等体积的PBS,培养48h后,每孔加入5mg/ml的MTT 20μl,37℃继续培养4h后弃上清,每孔加入150μl DMSO,振荡至沉淀完全溶解,在酶标仪上检测570nm光密度(OD)值。计算肿瘤细胞生长抑制率。在以同一药物的不同浓度对肿瘤细胞生长抑制率作图可得到剂量反应曲线,求出该药物的半数抑制浓度IC50。The proliferation inhibitory activity of each compound on tumor cell lines was determined by conventional MTT method. For the preliminary activity experiment, H1299 cells in the logarithmic growth phase were taken, and the cell concentration was adjusted to 1×105 /ml with RPMI1640 culture medium containing 10% serum, and seeded in 96-well culture plates, 100 μl per well, at 37°C, 5% Cultivate for 24 hours under CO2 conditions. Add drugs in groups, set three parallel wells for each concentration, add different concentrations of drugs to the treatment group, and add equal volume of PBS to the negative control group, after 48 hours of incubation, add 20 μl of 5 mg/ml MTT to each well, and continue to incubate at 37°C for 4 hours Discard the supernatant, add 150 μl DMSO to each well, shake until the precipitate is completely dissolved, and measure the optical density (OD) value at 570 nm on a microplate reader. Calculate the tumor cell growth inhibition rate. The dose-response curve can be obtained by plotting the tumor cell growth inhibition rate with different concentrations of the same drug, and the half inhibitory concentration IC50 of the drug can be calculated.
进一步活性实验仍然采用MTT法,选择上步结果中活性最高的C-21化合物用于本室保存的各类实体瘤细胞系,测定其抑制率,求出该药对各个肿瘤细胞系的IC50。For further activity experiments, the MTT method was still used, and the C-21 compound with the highest activity in the previous step was selected to be used in various solid tumor cell lines preserved in our laboratory, and its inhibition rate was measured to obtain the IC50 of the drug on each tumor cell line. .
实验结果:Experimental results:
27个化合物中12个化合物显著抑制肿瘤细胞的增殖(IC50<50μmoL),其中1个化合物的半数抑制浓度(IC50)在20μmoL以下,各个化合物的IC50见下表。选择其中活性最高的C-21化合物进一步进行研究。C-21对细胞系A459、Hela、HT1080、SKOV3、MCF-7等各类实体瘤细胞系的增殖抑制率如下表Among the 27 compounds, 12 compounds significantly inhibited the proliferation of tumor cells (IC50 <50 μmoL), and the half inhibitory concentration (IC50 ) of one compound was below 20 μmoL. The IC50 of each compound is shown in the table below. The C-21 compound with the highest activity was selected for further research. The growth inhibition rate of C-21 on cell lines A459, Hela, HT1080, SKOV3, MCF-7 and other solid tumor cell lines is as follows


上表中“*”出现浑浊现象"*" in the above table appears turbid
我们对高活性的C-21化合物进一步进行了生物学活性测定,扩大其体外实验应用的范围。结果显示其对肺癌、骨肉瘤、卵巢癌、宫颈癌、乳腺癌等多种恶性肿瘤细胞有生长明显的抑制作用,其IC50在10-30μM之间,且多数IC50>20μM。如图1所示。We further tested the biological activity of the highly active C-21 compound to expand the scope of its application in vitro. The results show that ithas obvious inhibitory effect on the growth of lung cancer, osteosarcoma, ovarian cancer, cervical cancer, breast cancer and other malignant tumor cells, and its IC50 is between 10-30 μM, and most of them are >20 μM. As shown in Figure 1.
进一步给出C-21的结构式如图2Further give the structural formula of C-21 as shown in Figure 2
实施例2:C-21对CDK9的激酶作用的抑制实验Example 2: Inhibition experiment of C-21 on the kinase action of CDK9
本研究选择抑制活性最高的C-21号化合物(一种N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类具体取代衍生物)进一步进行分子机制的研究。In this study, compound C-21 (a specific substituted derivative of N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamide) with the highest inhibitory activity was selected for further molecular mechanism research .
取对数生长期的H1299细胞,按RIPA细胞裂解液说明书操作,再经超声数下,得到新鲜细胞裂解液,BCA试剂盒定量。调节细胞裂解液和CTD蛋白液浓度至合适,加入不同浓度的药物C-21,震荡混匀(可同时加入一定量的ATP液),37℃反应4h,取适量进行10%SDS-PAGE电泳。电泳完成后,根据凝胶面积按0.65mA/cm2接通电源,电转移3小时。转移结束后,PBST洗PVFD膜15min,室温温和摇动,放入封闭液(5%脱脂奶)中4℃轻摇过夜。将膜封于塑料膜中按0.1ml/cm2加入抗磷酸化的RNA聚合酶II的单克隆抗体的稀释液,室温下温和摇动2h。取膜PBST洗3次,每次10min,再封于另一干净塑料膜中按0.1ml/cm2加入羊抗鼠IgG稀释液,室温下温和摇动2h。弃去二抗,PBST洗3遍,每次10min。最后按BeyoECL Plus说明书于暗室显影。H1299 cells in the logarithmic growth phase were taken, operated according to the instructions of RIPA cell lysate, and then subjected to ultrasound to obtain fresh cell lysate, which was quantified by BCA kit. Adjust the concentration of cell lysate and CTD protein solution to an appropriate level, add different concentrations of drug C-21, shake and mix (a certain amount of ATP solution can be added at the same time), react at 37°C for 4 hours, and take an appropriate amount for 10% SDS-PAGE electrophoresis. After the electrophoresis is completed, the power is turned on at 0.65 mA/cm2 according to the gel area, and the electrotransfer is performed for 3 hours. After the transfer, the PVFD membrane was washed with PBST for 15 min, gently shaken at room temperature, and placed in blocking solution (5% skimmed milk) at 4°C overnight. Seal the membrane in a plastic film and add the diluent of anti-phosphorylated RNA polymerase II monoclonal antibody at 0.1ml/cm2 , and shake gently at room temperature for 2h. Take the membrane and wash it three times with PBST, each time for 10 minutes, and then seal it in another clean plastic film, add goat anti-mouse IgG dilution solution at 0.1ml/cm2 , and shake gently at room temperature for 2 hours. Discard the secondary antibody, wash 3 times with PBST, 10 min each time. Finally, according to the BeyoECL Plus instructions, develop in the dark room.
其结果如图3。其中PBS(细胞等渗液)为阴性对照,分别用等体积的PBS,2,5,10,20,40μM的C-21处理细胞裂解液。pSer2CTD表示RNA聚合酶II大亚基C末端2位丝氨酸的磷酸化程度,可由图中免疫印记条带的深浅看出。由图可知RNA聚合酶II的磷酸化程度随药物剂量的加大而减弱,由此说明CDK9的激酶活性的下降程度与药物浓度之间呈一定的剂量依赖关系。The result is shown in Figure 3. Among them, PBS (cell isotonic solution) was used as a negative control, and the cell lysate was treated with equal volumes of PBS, 2, 5, 10, 20, and 40 μM C-21, respectively. pSer2CTD indicates the degree of phosphorylation of
实施例3:化合物C-21与CDK9晶体结构结合作用模式Example 3: Binding mode of compound C-21 and CDK9 crystal structure
根据已知CDK9的氨基酸序列,在SGI图形工作站上,利用软件Insight II的同源模建(MODELLER/HOMOLOGY)模块,对CDK9进行三维结构的同源模建。主要步骤如下:以CDK9的一级序列为探针,用BlastP程序在PDB库中搜索其同源蛋白,选择同源性高并且蛋白类型已知的同源蛋白作为模板。用同源蛋白叠加,确定SCR(结构保守区)和LOOP区。将模建蛋白和同源蛋白序列联配,能与同源蛋白SCR区相匹配的序列为模建蛋白的SCR区,然后赋予模建蛋白SCR区空间坐标,接着利用Modeler软件包,采用数据库查询方法,根据LOOP的长度及两端的SCR的坐标,搜索出合适的LOOP结构,同样采用数据库查询的方法,在模建过程中直接从参考蛋白考博模建蛋白的侧链,对不适侧链通过分子力学方法模建为较为合理的结构。According to the known amino acid sequence of CDK9, on the SGI graphics workstation, the homology modeling (MODELLER/HOMOLOGY) module of the software Insight II was used to carry out the homology modeling of the three-dimensional structure of CDK9. The main steps are as follows: use the primary sequence of CDK9 as a probe, use the BlastP program to search for homologous proteins in the PDB library, and select homologous proteins with high homology and known protein types as templates. Superimposed with homologous proteins, the SCR (structurally conserved region) and LOOP regions were determined. Align the modeled protein and homologous protein sequences, the sequence that can match the SCR region of the homologous protein is the SCR region of the modeled protein, and then assign the spatial coordinates of the SCR region of the modeled protein, and then use the Modeler software package to query the database method, according to the length of the LOOP and the coordinates of the SCRs at both ends, a suitable LOOP structure is searched, and the method of database query is also used to directly model the side chain of the protein from the reference protein Kobo in the modeling process. The mechanical method is modeled as a more reasonable structure.
将CDK9的二级结构显示出来,观察其中的beta折叠和alpha螺旋的分布,对蛋白质的表面进行活性区搜索,把搜索到的活性区逐一显示进行比较,同时结合文献和活性区特征选出可能的结合活性区。将CDK9动力学优化后的PDB文件将所有的组氨酸His改为Hid,对生成二硫键的半胱氨酸Cys改为Cyx,经Insight II模拟以及归属力场之后,存储为receptor.mol2文件,供DOCK使用。受体分子表面有DMS程序生成,再用SPHGEN生成活性位点的负像,负像由不同大小的圆球堆积而成,作为活性区域特征的代表。然后用基于分子锚点片段出发的构象搜索方法搜索数据库中配体可能的构象,根据负像区的特点自动模拟配体的作用方式,并根据能量打分记录理论上最佳的作用方式。最后撰写dock.in文件,确定各个参数文件的位置,在LINUX下运行DOCK模拟和筛选。Display the secondary structure of CDK9, observe the distribution of beta folds and alpha helices, search the active region on the surface of the protein, display and compare the searched active regions one by one, and select possible ones based on the literature and the characteristics of the active region the binding active region. Change all histidine His to Hid in the PDB file after CDK9 kinetic optimization, and change the cysteine Cys that generates disulfide bonds to Cyx, and store it as receptor.mol2 after Insight II simulation and attribution force field file, for use by DOCK. The surface of the receptor molecule is generated by DMS program, and then SPHGEN is used to generate the negative image of the active site. The negative image is formed by stacking spheres of different sizes, which are representative of the characteristics of the active area. Then use the conformational search method based on the molecular anchor fragment to search the possible conformation of the ligand in the database, automatically simulate the mode of action of the ligand according to the characteristics of the negative image area, and record the theoretically optimal mode of action according to the energy score. Finally, write the dock.in file, determine the location of each parameter file, and run DOCK simulation and screening under LINUX.
图4为小分子化合物C-21与CDK9三维晶体结构DOCK时的相互结合作用模式图。由图4可知C-21与CDK9的激酶活性区共形成了三个氢键,吡唑并[1,5-a]嘧啶结构以舒展的方式完全伸入活性区内部,其氮原子与103位苯丙氨酸形成一个氢键。另外106位半胱氨酸分别与酰胺的氧原子和噻吩的硫原子各形成一个氢键,从图上分析C-21与CDK9在能量和结构上匹配很好,确实能有效的阻止CDK9活性区与其下游底物的结合,从而使RNA聚合酶II的CTD段磷酸化水平下降。Figure 4 is a schematic diagram of the interaction between the small molecule compound C-21 and the three-dimensional crystal structure DOCK of CDK9. It can be seen from Figure 4 that three hydrogen bonds have been formed between C-21 and the kinase active region of CDK9, and the pyrazolo[1,5-a]pyrimidine structure completely extends into the active region in a stretched manner. Phenylalanine forms a hydrogen bond. In addition, cysteine at
综上所述,通过分子药理学实验的可以进一步的证明:本发明的N-(噻吩-2)吡唑并[1,5-a]嘧啶-3-甲酰胺类化合物显示出明显的抑制多种肿瘤细胞生长作用,其分子机制研究验证了模建及DOCK结果的正确性,体现出高效,分子靶标确定及针对性极强等特点,为进一步开发其优化后的类似物成为候选药物作出了机理上的准备,也为与其他类型抗肿瘤药物联合用药提供了新思路。In summary, it can be further proved by molecular pharmacology experiments that N-(thiophene-2)pyrazolo[1,5-a]pyrimidine-3-carboxamides of the present invention show significant inhibition of multiple The growth of tumor cells, its molecular mechanism research verified the correctness of the modeling and DOCK results, reflecting the characteristics of high efficiency, molecular target determination and strong pertinence, and made a contribution for the further development of its optimized analogues to become candidate drugs The preparation of the mechanism also provides a new idea for the combination with other types of anti-tumor drugs.
Claims (7)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 200910068159CN101537007A (en) | 2009-03-18 | 2009-03-18 | Application of N-(thiofuran-2) pyrazolo (1, 5-a) pyridine-3-formanides compounds for preparing antineoplastic |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 200910068159CN101537007A (en) | 2009-03-18 | 2009-03-18 | Application of N-(thiofuran-2) pyrazolo (1, 5-a) pyridine-3-formanides compounds for preparing antineoplastic |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101537007Atrue CN101537007A (en) | 2009-09-23 |
Family
ID=41120548
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 200910068159PendingCN101537007A (en) | 2009-03-18 | 2009-03-18 | Application of N-(thiofuran-2) pyrazolo (1, 5-a) pyridine-3-formanides compounds for preparing antineoplastic |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN101537007A (en) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102798708A (en)* | 2012-08-23 | 2012-11-28 | 中国科学院长春应用化学研究所 | Method for detecting binding specificity between ligand and target and drug screening method |
| JP2013531018A (en)* | 2010-07-13 | 2013-08-01 | エフ.ホフマン−ラ ロシュ アーゲー | Pyrazolo [1,5a] pyrimidine and thieno [3,2b] pyrimidine derivatives as IRAK4 modulators |
| US8912198B2 (en) | 2012-10-16 | 2014-12-16 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8957078B2 (en) | 2013-03-15 | 2015-02-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8969360B2 (en) | 2013-03-15 | 2015-03-03 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9309250B2 (en) | 2011-06-22 | 2016-04-12 | Vertex Pharmaceuticals Incorporated | Substituted pyrrolo[2,3-b]pyrazines as ATR kinase inhibitors |
| US9334244B2 (en) | 2010-05-12 | 2016-05-10 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9340546B2 (en) | 2012-12-07 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9365557B2 (en) | 2008-12-19 | 2016-06-14 | Vertex Pharmaceuticals Incorporated | Substituted pyrazin-2-amines as inhibitors of ATR kinase |
| US9630956B2 (en) | 2010-05-12 | 2017-04-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9663519B2 (en) | 2013-03-15 | 2017-05-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9670215B2 (en) | 2014-06-05 | 2017-06-06 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9791456B2 (en) | 2012-10-04 | 2017-10-17 | Vertex Pharmaceuticals Incorporated | Method for measuring ATR inhibition mediated increases in DNA damage |
| US9862709B2 (en) | 2011-09-30 | 2018-01-09 | Vertex Pharmaceuticals Incorporated | Processes for making compounds useful as inhibitors of ATR kinase |
| US10160760B2 (en) | 2013-12-06 | 2018-12-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10478430B2 (en) | 2012-04-05 | 2019-11-19 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase and combination therapies thereof |
| CN110538188A (en)* | 2019-08-08 | 2019-12-06 | 云南农业大学 | Novel use of astragaloside as CDK2 kinase inhibitor |
| US10813929B2 (en) | 2011-09-30 | 2020-10-27 | Vertex Pharmaceuticals Incorporated | Treating cancer with ATR inhibitors |
| US11179394B2 (en) | 2014-06-17 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of Chk1 and ATR inhibitors |
| CN113975271A (en)* | 2021-09-02 | 2022-01-28 | 苏州大学附属第一医院 | Application of small molecular compound in preparing medicine for treating tumor |
| US11464774B2 (en) | 2015-09-30 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA damaging agents and ATR inhibitors |
- 2009
- 2009-03-18CNCN 200910068159patent/CN101537007A/enactivePending
Cited By (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10961232B2 (en) | 2008-12-19 | 2021-03-30 | Vertex Pharmaceuticals Incorporated | Substituted pyrazines as ATR kinase inhibitors |
| US10479784B2 (en) | 2008-12-19 | 2019-11-19 | Vertex Pharmaceuticals Incorporated | Substituted pyrazin-2-amines as inhibitors of ATR kinase |
| US9701674B2 (en) | 2008-12-19 | 2017-07-11 | Vertex Pharmaceuticals Incorporated | Substituted pyrazines as ATR kinase inhibitors |
| US9365557B2 (en) | 2008-12-19 | 2016-06-14 | Vertex Pharmaceuticals Incorporated | Substituted pyrazin-2-amines as inhibitors of ATR kinase |
| US9630956B2 (en) | 2010-05-12 | 2017-04-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9334244B2 (en) | 2010-05-12 | 2016-05-10 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| JP2013531018A (en)* | 2010-07-13 | 2013-08-01 | エフ.ホフマン−ラ ロシュ アーゲー | Pyrazolo [1,5a] pyrimidine and thieno [3,2b] pyrimidine derivatives as IRAK4 modulators |
| US10023589B2 (en) | 2010-07-13 | 2018-07-17 | Hoffmann-La Roche Inc. | Pyrazolo[1,5a]pyrimidine derivatives as IRAK4 modulators |
| US9255110B2 (en) | 2010-07-13 | 2016-02-09 | Roche Palo Alto Llc | Pyrazolo[1,5a]pyrimidine derivatives as IRAK4 modulators |
| US9309250B2 (en) | 2011-06-22 | 2016-04-12 | Vertex Pharmaceuticals Incorporated | Substituted pyrrolo[2,3-b]pyrazines as ATR kinase inhibitors |
| US10822331B2 (en) | 2011-09-30 | 2020-11-03 | Vertex Pharmaceuticals Incorporated | Processes for preparing ATR inhibitors |
| US10813929B2 (en) | 2011-09-30 | 2020-10-27 | Vertex Pharmaceuticals Incorporated | Treating cancer with ATR inhibitors |
| US9862709B2 (en) | 2011-09-30 | 2018-01-09 | Vertex Pharmaceuticals Incorporated | Processes for making compounds useful as inhibitors of ATR kinase |
| US10208027B2 (en) | 2011-09-30 | 2019-02-19 | Vertex Pharmaceuticals Incorporated | Processes for preparing ATR inhibitors |
| US11110086B2 (en) | 2012-04-05 | 2021-09-07 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase and combination therapies thereof |
| US10478430B2 (en) | 2012-04-05 | 2019-11-19 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase and combination therapies thereof |
| CN102798708A (en)* | 2012-08-23 | 2012-11-28 | 中国科学院长春应用化学研究所 | Method for detecting binding specificity between ligand and target and drug screening method |
| US9791456B2 (en) | 2012-10-04 | 2017-10-17 | Vertex Pharmaceuticals Incorporated | Method for measuring ATR inhibition mediated increases in DNA damage |
| US8912198B2 (en) | 2012-10-16 | 2014-12-16 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9718827B2 (en) | 2012-12-07 | 2017-08-01 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11117900B2 (en) | 2012-12-07 | 2021-09-14 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9650381B2 (en) | 2012-12-07 | 2017-05-16 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10392391B2 (en) | 2012-12-07 | 2019-08-27 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9340546B2 (en) | 2012-12-07 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11370798B2 (en) | 2012-12-07 | 2022-06-28 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US12187731B2 (en) | 2012-12-07 | 2025-01-07 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10787452B2 (en) | 2012-12-07 | 2020-09-29 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8969360B2 (en) | 2013-03-15 | 2015-03-03 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8957078B2 (en) | 2013-03-15 | 2015-02-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9663519B2 (en) | 2013-03-15 | 2017-05-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10815239B2 (en) | 2013-12-06 | 2020-10-27 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10160760B2 (en) | 2013-12-06 | 2018-12-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11485739B2 (en) | 2013-12-06 | 2022-11-01 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10800781B2 (en) | 2014-06-05 | 2020-10-13 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10093676B2 (en) | 2014-06-05 | 2018-10-09 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9670215B2 (en) | 2014-06-05 | 2017-06-06 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11179394B2 (en) | 2014-06-17 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of Chk1 and ATR inhibitors |
| US11464774B2 (en) | 2015-09-30 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA damaging agents and ATR inhibitors |
| CN110538188A (en)* | 2019-08-08 | 2019-12-06 | 云南农业大学 | Novel use of astragaloside as CDK2 kinase inhibitor |
| CN113975271A (en)* | 2021-09-02 | 2022-01-28 | 苏州大学附属第一医院 | Application of small molecular compound in preparing medicine for treating tumor |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101537007A (en) | Application of N-(thiofuran-2) pyrazolo (1, 5-a) pyridine-3-formanides compounds for preparing antineoplastic | |
| Zhang et al. | Polo-like kinase 1 inhibitors in human cancer therapy: development and therapeutic potential | |
| Wu et al. | Recent advances in dual PI3K/mTOR inhibitors for tumour treatment | |
| Buckley et al. | 6-Substituted hexamethylene amiloride (HMA) derivatives as potent and selective inhibitors of the human urokinase plasminogen activator for use in cancer | |
| Maira et al. | Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity | |
| Akinleye et al. | Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics | |
| Saurat et al. | Design, synthesis, and biological activity of pyridopyrimidine scaffolds as novel PI3K/mTOR dual inhibitors | |
| Tintori et al. | Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo [3, 4-d] pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma | |
| Gehringer et al. | c-Jun N-terminal kinase inhibitors: A patent review (2010–2014) | |
| Liu et al. | Hybridization-based discovery of novel quinazoline-2-indolinone derivatives as potent and selective PI3Kα inhibitors | |
| Zhang et al. | Design, synthesis, and biological evaluation of substituted pyrimidines as potential phosphatidylinositol 3-kinase (PI3K) inhibitors | |
| Zhan et al. | Design, synthesis, and biological evaluation of dimorpholine substituted thienopyrimidines as potential class I PI3K/mTOR dual inhibitors | |
| Brasca et al. | Optimization of 6, 6-dimethyl pyrrolo [3, 4-c] pyrazoles: Identification of PHA-793887, a potent CDK inhibitor suitable for intravenous dosing | |
| Bata et al. | Inhibitors of the Hippo pathway kinases STK3/MST2 and STK4/MST1 have utility for the treatment of acute myeloid leukemia | |
| Rehan et al. | Virtual screening of naphthoquinone analogs for potent inhibitors against the cancer‐signaling PI3K/AKT/mTOR pathway | |
| Chen et al. | Synthesis and SAR of novel 4-morpholinopyrrolopyrimidine derivatives as potent phosphatidylinositol 3-kinase inhibitors | |
| JP2016523892A (en) | PI3K protein kinase inhibitors, in particular delta and / or gamma inhibitors | |
| Du et al. | Structure-based design of a potent and selective covalent inhibitor for SRC kinase that targets a P-loop cysteine | |
| Lv et al. | Furthering the design and the discovery of small molecule ATP-competitive mTOR inhibitors as an effective cancer treatment | |
| Liu et al. | Synthesis and biological evaluation of novel dasatinib analogues as potent DDR 1 and DDR 2 kinase inhibitors | |
| Shi et al. | Anti-angiogenic therapy: Strategies to develop potent VEGFR-2 tyrosine kinase inhibitors and future prospect | |
| Sharma et al. | Designing of kinase hinge binders: A medicinal chemistry perspective | |
| Poulsen et al. | Structure and ligand-based design of mTOR and PI3-kinase inhibitors leading to the clinical candidates VS-5584 (SB2343) and SB2602 | |
| Marzaro et al. | Discovery of biarylaminoquinazolines as novel tubulin polymerization inhibitors | |
| Manetti | Recent advances in the rational design and development of LIM kinase inhibitors are not enough to enter clinical trials |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication | Open date:20090923 |