
Read the methods paperhere
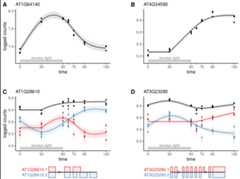
Application ofcpam to RNA-seq time series ofArabidopsis plants treated with excess-light.
Our new packagecpam provides a comprehensiveframework for analysing time series omics data. The method uses modernstatistical approaches while remaining user-friendly, through sensibledefaults and an interactive interface. Researchers can directly addresskey questions in time series analysis—when changes occur, what patternsthey follow, and how responses are related. While we have focused ontranscriptomics, the framework is applicable to other high-dimensionaltime series measurements.
If you encounter issues or have suggestions for improvements, pleaseopen anissue. Wewelcome questions and discussion about usingcpam foryour research throughDiscussions.Our goal is to work with users to makecpam a robustand valuable tool for time series omics analysis. We can also becontacted via the email addresses listed in our paperhere.
The package is available on CRAN and can be installed using thefollowing command:
install.packages("cpam")library(cpam)In thisArabidopsis thaliana time series example, we usedthe softwarekallisto togenerate counts from RNA-seq data. To load the counts, we provide thefile path for each kallisto output file (alternatively you can providethe counts directly as count matrix, or use other quantificationsoftware)
# load example dataload(system.file("extdata","exp_design_path.rda",package ="cpam"))head(exp_design_path)#> sample time path condition#> 1 JHSS01 0 output/kallisto/JHSS01/abundance.h5 treatment#> 2 JHSS02 0 output/kallisto/JHSS02/abundance.h5 treatment#> 3 JHSS03 0 output/kallisto/JHSS03/abundance.h5 treatment#> 4 JHSS04 0 output/kallisto/JHSS04/abundance.h5 treatment#> 5 JHSS05 0 output/kallisto/JHSS05/abundance.h5 treatment#> 6 JHSS06 5 output/kallisto/JHSS06/abundance.h5 treatmentN.B. This is not needed if your counts are aggregated at the genelevel, but transcript-level analysis with aggregation of
# load example dataload(system.file("extdata","t2g_arabidopsis.rda",package ="cpam"))head(t2g_arabidopsis)#> target_id gene_id#> 1 AT1G01010.1 AT1G01010#> 2 AT1G01020.2 AT1G01020#> 3 AT1G01020.6 AT1G01020#> 4 AT1G01020.1 AT1G01020#> 5 AT1G01020.4 AT1G01020#> 6 AT1G01020.5 AT1G01020 cpo<-prepare_cpam(exp_design = exp_design_path,count_matrix =NULL,t2g = t2g_arabidopsis,model ="case-only",import_type ="kallisto",num_cores =5) cpo<-compute_p_values(cpo) cpo<-estimate_changepoint(cpo) cpo<-select_shape(cpo)Load the shiny app for an interactive visualisation of theresults:
visualise(cpo)# not shown in vignetteOr plot one gene at a time:
plot_cpam(cpo,gene_id ="AT3G23280")
Isoform 1 (AT3G23280.1) has a changepoint at 67.5 min and has amonotonic increasing concave (micv) shape. Isoform 2 (AT3G23280.2) hasno changepoint and has an unconstrained thin-plate (tp) shape.
We can generate a results table which has
results(cpo)#> # A tibble: 15,279 × 25#> target_id gene_id p cp shape lfc.0 lfc.5 lfc.10 lfc.20 lfc.30 lfc.45#> <chr> <chr> <dbl> <dbl> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>#> 1 AT1G01910.1 AT1G01… 0 0 micv 0 1.01 1.70 2.38 2.60 2.73#> 2 AT1G01910.2 AT1G01… 0 10 cv 0 0 0 0.553 0.775 0.790#> 3 AT1G01910.5 AT1G01… 0 10 cx 0 0 0 -3.20 -4.57 -4.82#> 4 AT1G02610.1 AT1G02… 0 45 mdcx 0 0 0 0 0 0#> 5 AT1G02610.2 AT1G02… 0 10 cx 0 0 0 -0.645 -1.16 -1.71#> 6 AT1G02610.3 AT1G02… 0 10 mdcx 0 0 0 -1.48 -2.11 -2.25#> 7 AT1G04080.1 AT1G04… 0 10 cv 0 0 0 2.75 3.85 3.97#> 8 AT1G04080.2 AT1G04… 0 45 micv 0 0 0 0 0 0#> 9 AT1G04080.3 AT1G04… 0 0 micv 0 0.268 0.445 0.603 0.638 0.656#> 10 AT1G04080.5 AT1G04… 0 10 cx 0 0 0 -2.17 -3.04 -3.10#> # ℹ 15,269 more rows#> # ℹ 14 more variables: lfc.60 <dbl>, lfc.90 <dbl>, lfc.180 <dbl>,#> # lfc.240 <dbl>, counts.0 <dbl>, counts.5 <dbl>, counts.10 <dbl>,#> # counts.20 <dbl>, counts.30 <dbl>, counts.45 <dbl>, counts.60 <dbl>,#> # counts.90 <dbl>, counts.180 <dbl>, counts.240 <dbl>For a quick-to-run introductory example, we have provided a smallsimulated data set as part of the package.
The following two tutorials use real-world data to demonstrate thecapabilities of thecpam package. In addition, theyprovide code to reproduce the results for the case studies presented inthemanuscriptaccompanying thecpam package.
This work was supported by the Australian Research Council, Centre ofExcellence for Plant Success in Nature and Agriculture(CE200100015).
![]()